GROMACS vs AMBER vs NAMD: A 2025 Comparative Guide for Molecular Dynamics Simulations
This article provides a comprehensive, up-to-date comparison of the three leading molecular dynamics software packages—GROMACS, AMBER, and NAMD—tailored for researchers, scientists, and drug development professionals.
GROMACS vs AMBER vs NAMD: A 2025 Comparative Guide for Molecular Dynamics Simulations
Abstract
This article provides a comprehensive, up-to-date comparison of the three leading molecular dynamics software packages—GROMACS, AMBER, and NAMD—tailored for researchers, scientists, and drug development professionals. It explores their foundational philosophies, licensing, and usability; details methodological applications and specialized use cases like membrane protein simulations; offers performance benchmarks and hardware optimization strategies for 2025; and critically examines validation protocols and reproducibility. By synthesizing performance data, best practices, and comparative insights, this guide empowers scientists to select the optimal software and hardware configuration to efficiently advance their computational research in biophysics, drug discovery, and materials science.
Understanding the Core Philosophies: GROMACS, AMBER, and NAMD for Beginners
Molecular dynamics (MD) simulations serve as a critical tool for researchers studying the physical movements of atoms and molecules over time. The choice of software fundamentally shapes the research process and outcomes. Among the many available packages, GROMACS, AMBER, and NAMD have emerged as leading tools, each with a distinct philosophical approach prioritizing different aspects of simulation. This guide provides an objective comparison of these three software packages, focusing on their core differences in speed, accuracy, and visualization, supported by experimental data and practical implementation protocols.
Philosophical Foundations and Core Differences
The design and development of GROMACS, AMBER, and NAMD have been guided by different priorities, leading to unique strengths and specializations.
GROMACS: The High-Performance Workhorse
GROMACS was conceived with a primary focus on raw speed and efficiency in high-performance computing (HPC) environments. Its philosophy centers on enabling high-throughput simulation, allowing researchers to sample more conformational space or run more replicas in less time. It is engineered from the ground up for optimal parallelization on both CPUs and GPUs, making it a "total workhorse" [1]. This makes it particularly suited for large biomolecular complexes, membrane proteins, and studies requiring extensive sampling [2].
AMBER: The Force Field Specialist
AMBER's development has been driven by an emphasis on accuracy and refinement of force fields. Its philosophy is rooted in providing highly precise and reliable energy evaluations for biomolecular systems, particularly proteins and nucleic acids. The AMBER suite is renowned for its force fields (e.g., ff14SB, ff19SB), often considered a gold standard in the field [2]. This focus makes AMBER a preferred choice for studies where force field precision is critical, such as protein-ligand interactions, nucleic acid dynamics, and advanced calculations like free energy perturbation (FEP) and hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) [2].
NAMD: The Scalable Visualizer
NAMD is designed with scalability and integration with visualization tools as its core principle. Its architecture is built to efficiently scale across massive numbers of processors, making it exceptionally capable for simulating very large systems, such as viral capsids or entire cellular compartments [1]. A key part of its philosophy is seamless integration with the visual molecular dynamics (VMD) software, also developed by the same group. This tight coupling provides "much better support for visual analysis" [3], making NAMD a powerful tool for researchers who require strong visualization and analysis capabilities throughout their simulation workflow.
Table: Philosophical and Technical Focus of GROMACS, AMBER, and NAMD
| Feature | GROMACS | AMBER | NAMD |
|---|---|---|---|
| Primary Philosophy | High-speed performance and throughput | Force field accuracy and refinement | Scalability on HPC and visualization integration |
| Key Strength | Exceptional speed on CPUs & GPUs | Accurate force fields, free energy calculations | Handling massive systems, integration with VMD |
| Typical Use Case | Large-scale simulations, high-throughput studies | Detailed protein-ligand studies, nucleic acids | Massive complexes (e.g., viruses), interactive visualization |
| Community & Support | Large, active community, extensive tutorials [2] | Specialized, knowledgeable community [2] | Strong support through VMD/NAMD community |
Performance and Accuracy: An Experimental Perspective
Empirical benchmarking and validation studies are crucial for understanding the real-world performance and accuracy of these software packages.
Simulation Speed and Hardware Utilization
The performance of MD software is highly dependent on the underlying hardware, particularly the use of GPUs.
Table: GPU Recommendations and Performance Characteristics
| Software | Recommended GPUs | Performance Notes |
|---|---|---|
| GROMACS | NVIDIA RTX 4090, RTX 6000 Ada [4] | Excels in parallel computations; highly optimized for both CPU and GPU. Ideal for computationally intensive simulations [4] [2]. |
| AMBER | NVIDIA RTX 6000 Ada, RTX 4090 [4] | Recent versions (AMBER GPU) have made significant strides in GPU acceleration. The RTX 6000 Ada is ideal for large-scale simulations [4] [2]. |
| NAMD | NVIDIA RTX 4090, RTX 6000 Ada [4] | Highly optimized for NVIDIA GPUs; can efficiently distribute computation across multiple GPUs, making it ideal for large system sizes [4]. |
All three packages have "mature GPU paths" and can effectively leverage consumer or workstation GPUs like the RTX 4090 for significant acceleration, using mixed precision to maintain accuracy while boosting speed [5]. For multi-GPU setups, GROMACS, AMBER, and NAMD all support parallel execution across multiple GPUs, dramatically enhancing computational efficiency and decreasing simulation times for large, complex systems [4].
Accuracy and Validation Against Experimental Data
A critical study compared four MD packages (including AMBER, GROMACS, and NAMD) by validating them against experimental data for two proteins, the Engrailed homeodomain and RNase H [6]. The findings are nuanced:
- Overall Agreement: At room temperature, the different software packages reproduced a variety of experimental observables "equally well overall" [6].
- Subtle Differences: Despite overall agreement, there were "subtle differences in the underlying conformational distributions and the extent of conformational sampling obtained" [6]. This indicates that multiple conformational ensembles might produce averages consistent with experiment, leading to ambiguity about which results are correct.
- Divergence in Large Motions: The differences between packages became more pronounced when simulating larger amplitude motions, such as thermal unfolding. Some packages failed to allow the protein to unfold at high temperature or produced results at odds with experiment [6].
This study highlights that the outcome of a simulation depends not only on the force field but also on the software itself, including its "algorithms that constrain motion, how atomic interactions are handled, and the simulation ensemble employed" [6].
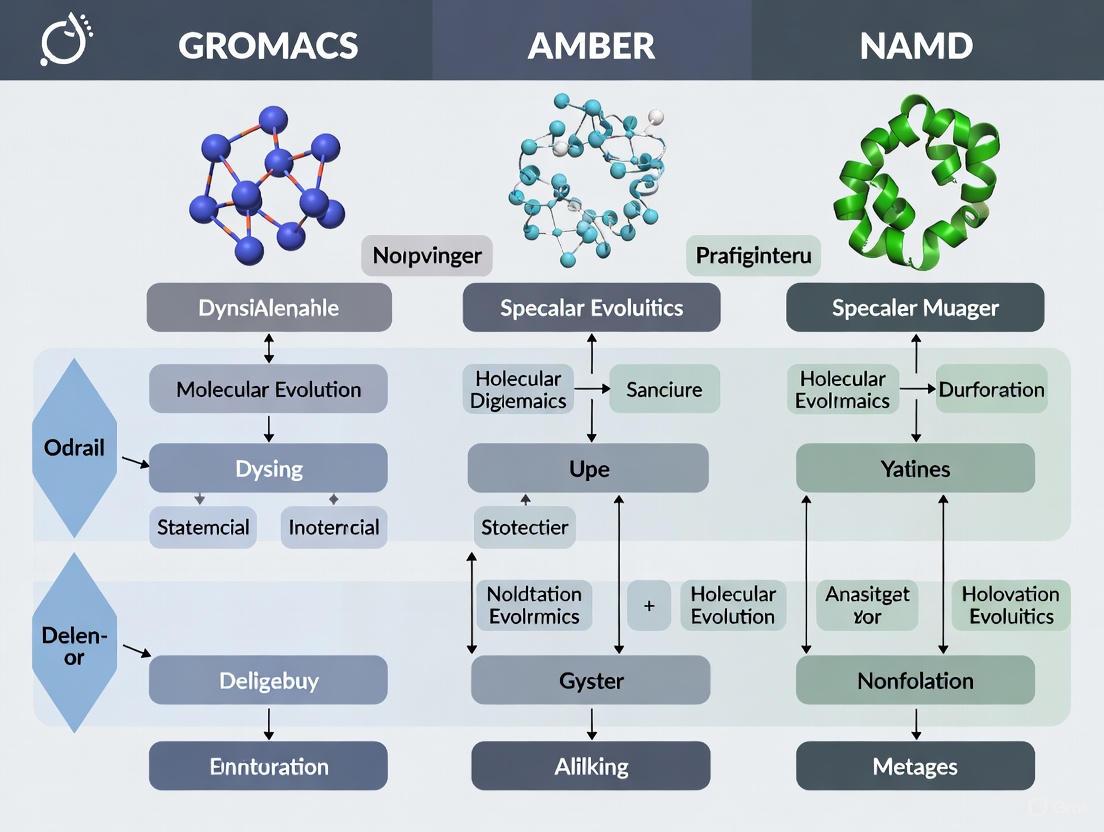
Diagram: A General MD Simulation Workflow. This core protocol is common across GROMACS, AMBER, and NAMD, though specific implementation commands differ.
Visualization and Analysis Capabilities
The ability to visualize and analyze simulation trajectories is as important as the simulation itself.
NAMD and VMD: NAMD stands out due to its deep integration with the visualization package VMD. This integration offers "much better support for visual analysis" compared to GROMACS [3]. The coupling allows for seamless setup, on-the-fly visualization, and sophisticated analysis of simulations, which is a significant advantage for researchers who prioritize visual interrogation of their systems.
GROMACS and AMBER: These packages have their own powerful suites of analysis tools. However, they are generally command-line driven. While they can export data for visualization in other programs (like VMD or PyMOL), they do not offer the same native, integrated visual environment as the NAMD/VMD combination. GROMACS is noted for its extensive and user-friendly tools for pre- and post-processing data [2].
Implementation and Best Practices
Example Simulation Protocols
Detailed below are example job submission scripts for running simulations on HPC clusters, illustrating differences in setup.
GROMACS GPU Script (Slurm)
This script shows GROMACS configured to offload non-bonded forces (nb), Particle Mesh Ewald (pme), and coordinate updates (update) to the GPU [7].
AMBER GPU Script (Slurm)
This script uses the pmemd.cuda module of AMBER for execution on a single GPU [7]. Note that for a single simulation, AMBER typically does not scale beyond 1 GPU [7].
The Scientist's Toolkit: Essential Research Reagents
A successful MD simulation relies on several key "reagents" and tools beyond the simulation engine itself.
Table: Essential Materials and Tools for Molecular Dynamics Simulations
| Item | Function | Notes |
|---|---|---|
| Force Field | Defines the potential energy function and parameters for atoms. | Choice is critical for accuracy. AMBER force fields are renowned for proteins/nucleic acids, but GROMACS supports AMBER, CHARMM, and OPLS [2] [8]. |
| Solvent Model | Represents the water and ion environment. | Common models include TIP3P, TIP4P. The choice of water model can influence simulation outcome [6]. |
| HPC Cluster/Workstation | Provides the computational power to run simulations. | GPU-accelerated systems are essential for performance. BIZON and other vendors offer custom workstations [4]. |
| Visualization Software (VMD) | For visualizing initial structures, trajectories, and analysis. | Deeply integrated with NAMD, but also used with GROMACS/AMBER trajectories [3] [1]. |
| Parameterization Tool (parmed) | For generating parameters for non-standard molecules (e.g., drugs). | Often used with AMBER tools (antechamber) and GROMACS for preparing complex systems [7] [1]. |
Diagram: The Core Components of an MD Simulation Stack. The force field, software engine, and hardware are interdependent components that determine the success of a project.
The choice between GROMACS, AMBER, and NAMD is not about finding the universally "best" software, but rather the most appropriate tool for a specific research question, computational environment, and user expertise.
Choose GROMACS when your primary need is speed and efficiency for large-scale simulations or high-throughput studies. Its exceptional performance, versatility in force fields, and strong community support make it an excellent general-purpose tool, especially for beginners and for simulating large biomolecular complexes [1] [2].
Choose AMBER when your research demands the highest accuracy in force fields and specialized tools for biomolecular systems. It is the preferred choice for detailed studies of protein-ligand interactions, nucleic acid dynamics, and for advanced methods like free energy calculations and QM/MM simulations [2].
Choose NAMD when you need to simulate exceptionally large systems (millions of atoms) or when tight integration with visualization is a priority. Its scalability on large HPC clusters and seamless workflow with VMD are its defining strengths [3] [1].
Ultimately, all three packages are powerful, well-validated tools that continue to evolve. The philosophical differences that guide their development provide the scientific community with a diverse and complementary set of options for exploring the dynamics of molecular worlds.
The selection of molecular dynamics (MD) software is a critical decision that hinges on both computational performance and institutional resources, with licensing and cost being pivotal factors. For researchers, scientists, and drug development professionals, this choice can shape project timelines, methodological approaches, and budget allocations. This guide provides an objective comparison of two leading MD tools—open-source GROMACS and commercially licensed AMBER—situating their licensing and cost structures within the broader ecosystem of computational research. By integrating experimental data and practical protocols, this analysis aims to deliver a foundational resource for making an informed decision that aligns with both scientific goals and operational constraints.
Understanding the Licensing Models
The fundamental distinction between GROMACS and AMBER lies in their software distribution and licensing philosophies. These models directly influence their accessibility, cost of use, and the nature of user support available.
GROMACS (GROningen MAchine for Chemical Simulations): GROMACS is free and open-source software, licensed under the GNU Lesser General Public License (LGPL) [9]. This license grants users the freedom to use, modify, and distribute the software and its source code with minimal restrictions. Its open-source nature fosters a large and active community that contributes to its development, provides support through forums, and creates extensive tutorials [2] [10]. The software is cross-platform, meaning it can be installed and run on various operating systems without licensing fees [10].
AMBER (Assisted Model Building with Energy Refinement): AMBER operates on a commercial, closed-source model. While a subset of utilities, known as AmberTools, is freely available, the core simulation engine required for running production MD simulations is a licensed product [9]. The license fee is tiered based on the user's institution: it is approximately $400 for academic, non-profit, or government entities, but can rise to $20,000 for new industrial (for-profit) licensees [9]. The software is primarily Unix-based, and updates and official support are managed through a consortium [10].
Table 1: Summary of Licensing and Cost Models
| Feature | GROMACS | AMBER |
|---|---|---|
| License Type | Open-Source (LGPL) [9] | Commercial, Closed-Source [10] |
| Cost (Academic) | Free [10] | ~$400 [9] |
| Cost (Industrial) | Free | Up to ~$20,000 [9] |
| Accessibility | High; cross-platform [10] | Medium; primarily Unix-based [10] |
| Community Support | Large and active community [2] [10] | Smaller, more specialized community [10] |
Performance and Cost-Efficiency Analysis
Beyond initial acquisition cost, the performance and hardware efficiency of MD software are critical determinants of its total cost of ownership. Experimental benchmarks provide objective data on how these packages utilize computational resources.
Experimental Protocols for Performance Benchmarking
To ensure fairness and reproducibility, performance comparisons follow standardized protocols. Key benchmarks often use well-defined molecular systems like the Satellite Tobacco Mosaic Virus (STMV) or a double-stranded DNA solvated in saltwater (benchPEP-h), which are designed to stress-test both CPU and GPU performance [7] [11].
A typical benchmarking workflow involves:
- System Preparation: A stable, pre-equilibrated molecular system is used as the starting point. The simulation is extended for a fixed number of steps (e.g., 10,000 steps) to ensure consistent measurement [7].
- Hardware Configuration: Tests are run on controlled hardware, often high-performance computing (HPC) clusters. The following are example submission scripts for GPU-accelerated benchmarks:
- GROMACS GPU Benchmarking Script: This script configures GROMACS to offload non-bonded interactions, Particle Mesh Ewald (PME), and coordinate updates to the GPU, while handling bonded interactions on the CPU [7].
- AMBER GPU Benchmarking Script:
This script uses the
pmemd.cudaexecutable to run a simulation on a single GPU [7].
- Data Collection: The primary metric is simulation throughput, reported in nanoseconds per day (ns/day). Researchers typically scan performance across different CPU core counts and GPU types to identify the optimal hardware configuration for a given system size [11].
Comparative Performance Data
Independent benchmarks reveal a clear performance dichotomy between GROMACS and AMBER. The data below, sourced from community-driven tests on consumer and HPC-grade hardware, illustrate this relationship [11].
Table 2: Performance Benchmark Summary on Select GPUs (STMV System)
| Software | NVIDIA RTX 4090 (ns/day) | NVIDIA RTX 4080 (ns/day) | AMD RX 7900 XTX (ns/day) | Notes |
|---|---|---|---|---|
| GROMACS 2023.2 | ~120 [11] | ~90 [11] | ~65 [11] | SYCL backend; performance varies with CPU core count [11]. |
| AMBER 22 | ~70 (Baseline) [11] | N/A | ~119 (70% faster than RTX 4090) [11] | HIP patch; shows superior scaling on AMD hardware for large systems [11]. |
The data indicates that GROMACS generally excels in raw speed on NVIDIA consumer GPUs like the RTX 4090, making it a high-throughput tool for many standard simulations [2] [11]. In contrast, AMBER demonstrates exceptional scalability on high-end and AMD GPUs, with the RX 7900 XTX significantly outperforming other cards in its class on large systems like the STMV [11]. This suggests that AMBER's architecture is highly optimized for parallel efficiency on capable hardware.
Furthermore, hardware selection is crucial for cost-efficiency. For GROMACS, which is highly dependent on single-core CPU performance to feed data to the GPU, a CPU with high clock speeds (e.g., AMD Ryzen Threadripper PRO or Intel Xeon Scalable) is recommended to avoid bottlenecks [12]. For AMBER, investing in GPUs with high memory bandwidth and double-precision performance can yield significant returns in simulation speed [12] [11].
Decision Framework: Choosing the Right Tool
The choice between GROMACS and AMBER is not a matter of which is universally better, but which is more appropriate for a specific research context. The following diagram outlines the logical decision-making process based on project requirements and resources.
This decision flow is guided by the core strengths and constraints of each package:
Choose GROMACS if: Your project operates with limited funds or requires the ability to inspect and modify the source code. It is also the superior choice for high-throughput screening and when your primary hardware consists of consumer-grade NVIDIA GPUs, where it delivers exceptional performance [2] [10] [11]. Its versatility in supporting multiple force fields (AMBER, CHARMM, OPLS) also makes it ideal for simulating diverse molecular systems, including membrane proteins and large complexes [2] [13].
Choose AMBER if: Your research demands the highest accuracy in biomolecular force fields, particularly for proteins and nucleic acids, and your institution can support the licensing cost. AMBER is also preferable for studies requiring advanced specialized capabilities like free energy perturbation (FEP) or hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) simulations [2] [3]. Furthermore, it shows remarkable scaling on high-performance GPUs, making it a powerful option for large, long-timescale biomolecular simulations where its force field precision is critical [2] [11].
The Scientist's Toolkit: Essential Research Reagents and Materials
Beyond the software itself, conducting successful MD simulations requires a suite of supporting tools and resources. The following table details key "research reagents" essential for working with GROMACS and AMBER.
Table 3: Essential Tools and Resources for Molecular Dynamics Simulations
| Item | Function | Relevance to GROMACS & AMBER |
|---|---|---|
| AmberTools | A suite of free programs for system preparation (e.g., tleap) and trajectory analysis [9]. |
Crucial for preparing topologies and parameters for both AMBER and GROMACS (when using AMBER force fields) [9] [3]. |
| Force Field Parameters | Pre-defined mathematical functions and constants describing interatomic interactions (e.g., ff14SB, GAFF) [2]. | AMBER is renowned for its own highly accurate force fields. GROMACS can use AMBER, CHARMM, and OPLS force fields, offering greater flexibility [2] [10]. |
| High-Performance GPU | Hardware accelerator for computationally intensive MD calculations. | NVIDIA RTX 4090/6000 Ada are top performers. AMBER shows exceptional results on AMD RX 7900 XTX for large systems, while GROMACS leads on NVIDIA consumer cards [12] [11]. |
| Visualization Software (VMD) | Molecular visualization and analysis program. | Often used alongside NAMD for visualization, but is equally critical for analyzing and visualizing trajectories from both GROMACS and AMBER simulations [3]. |
| Community Forums | Online platforms for user support and troubleshooting. | GROMACS has extensive, active community forums. AMBER support is more specialized but detailed, often provided via its consortium [2] [10]. |
The comparison between GROMACS and AMBER reveals a trade-off between accessibility and specialized power. GROMACS, as a free and open-source tool, provides unparalleled accessibility, a gentle learning curve, and leading-edge performance on common hardware, making it an excellent choice for high-throughput studies and researchers with budget constraints. In contrast, AMBER, with its commercial licensing, offers exceptional force field accuracy for biomolecules, robust specialized methods, and impressive scalability on high-end computing resources, justifying its cost for research where precision and specific advanced features are paramount. There is no single "best" software; the optimal choice is a strategic decision that aligns the tool's strengths with the project's scientific objectives, technical requirements, and financial resources.
For researchers in drug development, selecting a molecular dynamics (MD) software involves a critical trade-off between raw performance and usability. This guide objectively compares the learning curves of GROMACS, AMBER, and NAMD, analyzing the tutorial availability and community support that can accelerate or hinder your research.
Software Usability and Support at a Glance
The table below summarizes the key usability factors for GROMACS, AMBER, and NAMD to help you evaluate which platform best aligns with your team's expertise and support needs.
| Feature | GROMACS | AMBER | NAMD |
|---|---|---|---|
| Ease of Use & Learning Curve | User-friendly; easier to learn with extensive documentation and tutorials [2]. Known for a less intuitive interface and a steeper learning curve, especially for beginners [2]. | Integrates well with VMD for visualization, but the core software has its own learning curve [3]. | |
| Community Support | Large, active community with extensive forums, tutorials, and resources [2]. | Strong but more niche community; support is highly specialized [2]. | Benefits from strong integration with VMD and its community [3] [1]. |
| Tutorial Availability | Excellent; offers great tutorials and workflows that are beginner-friendly [3]. | Extensive documentation available [2]. | Often praised for visualization and tutorial resources when paired with VMD [3]. |
| Notable Tools & Interfaces | Has packages like MolDy for GUI-based automation [3]. Third-party web tools like VisualDynamics provide a graphical interface [14]. | Tight coupling with VMD visualization software simplifies setup and analysis [3] [1]. |
Essential Research Reagent Solutions
The "research reagents" in computational studies are the software tools, hardware, and datasets that form the foundation of reproducible MD experiments. The following table details these essential components.
| Item | Function |
|---|---|
| GROMACS | Open-source MD software; the core engine for running simulations, known for high speed and versatility [3] [2]. |
| AMBER | A suite of MD programs and force fields; particularly renowned for its high accuracy for biomolecules [3] [2]. |
| NAMD | MD software designed for high parallel scalability, especially on GPU-based systems; excels with very large molecular systems [15] [1]. |
| VMD | Visualization software; used for visualizing trajectories, setting up simulations, and analyzing results. Often used with NAMD [3]. |
| VisualDynamics | A web application that automates GROMACS simulations via a graphical interface, simplifying the process for non-specialists [14]. |
| NVIDIA RTX GPUs | Graphics processing units (e.g., RTX 4090, RTX 6000 Ada) that dramatically accelerate MD simulations in all three packages [15] [5]. |
| Benchmarking Datasets | Experimental datasets (e.g., from NMR, crystallography) used to validate and benchmark the accuracy of simulation methods and force fields [16]. |
Experimental Protocols for Performance Benchmarking
To make an informed choice, you should run a standardized benchmark on your own hardware. The following protocol, based on high-performance computing practices, allows for a fair comparison of simulation speed and efficiency.
Methodology for MD Software Benchmarking
- System Preparation: A well-defined, standardized system must be used. A common choice is a solvated protein-ligand complex, such as Lysozyme with an inhibitor, prepared and parameterized using consistent force fields (e.g., AMBER's ff14SB for the protein and GAFF for the ligand) across all software packages [2] [16].
- Simulation Parameters: All simulations are run using identical conditions. Key parameters include:
- Integration Time Step: 2 femtoseconds (fs).
- Long-Range Electrostatics: Particle Mesh Ewald (PME) method.
- Thermostat: Langevin dynamics or Nose-Hoover.
- Barostat: Parrinello-Rahman pressure coupling.
- Simulation Length: A production run of 10,000 steps is used for the benchmark to ensure comparable results without excessive computational cost [7].
- Hardware Configuration: Tests are performed on a dedicated compute node. A typical modern setup includes:
- Software Versions: All software must be pinned to specific, up-to-date versions (e.g., GROMACS 2023.2, AMBER 20.12-20.15, NAMD 3.0b3) to ensure reproducibility [7] [5].
- Performance Metrics: The primary metric is simulation throughput, measured in nanoseconds per day (ns/day). This is calculated by measuring the wall-clock time taken to complete the 10,000-step simulation and converting it to a daily rate. A higher ns/day value indicates faster performance [7].
Example Submission Scripts
The following scripts illustrate how to run a 10,000-step benchmark on a single GPU for each software, adapted from high-performance computing guidelines [7].
GROMACS
AMBER (pmemd)
NAMD
Workflow for Evaluating MD Software
The diagram below outlines a logical decision pathway to guide researchers in selecting and testing the most suitable MD software for their project.
Key Takeaways for Drug Development Professionals
- For Most Teams: GROMACS offers the most balanced combination of performance, usability, and support. Its beginner-friendly tutorials and active community can significantly reduce the startup time for new researchers [3] [2].
- For Specialized Biomolecular Studies: AMBER remains the gold standard where force field accuracy is paramount, such as in detailed protein-ligand interaction studies or nucleic acid dynamics. However, budget for a steeper learning curve and be aware of potential licensing costs for commercial use [3] [2].
- For Large Complexes and HPC: NAMD's architecture is designed for scalability on high-performance computing systems, making it a strong candidate for massive simulations, such as large viral capsids or membrane complexes [1]. Its deep integration with VMD is a significant advantage for visualization-centric workflows [3].
Force Field Strengths and Native Compatibility
Force Field Strengths and Native Compatibility
Selecting the right molecular dynamics (MD) software is a critical decision that can directly impact the efficiency and reliability of research simulations. For researchers, scientists, and drug development professionals, understanding the core strengths and native compatibility of popular packages like GROMACS, AMBER, and NAMD is essential for aligning software capabilities with project goals. This guide provides an objective comparison based on performance benchmarks and experimental data, focusing on their force field implementations and operational workflows.
Core Software Profiles and Force Field Specialization
Each major MD package has evolved with distinct philosophical approaches to force fields and biomolecular simulation.
| Software | Native Force Field Strengths | Cross-Compatibility | Primary Simulation Focus |
|---|---|---|---|
| AMBER | AMBER (ff14SB, ff19SB) - considered gold standard for proteins/nucleic acids [2] | Can be used in other packages (e.g., GROMACS) [2] [6] | High-accuracy biomolecular simulations [2] |
| GROMACS | GROMOS; OPLS-AA [2] | Highly versatile; natively supports AMBER, CHARMM, OPLS [2] | High-performance, scalable MD [2] |
| NAMD | CHARMM [6] [17] | CHARMM force field is standard; inputs can be generated via CHARMM-GUI [17] | Large biomolecular complexes [6] |
AMBER is renowned for its highly accurate and specialized force fields, particularly for biomolecular systems. Its AMBER family of force fields (e.g., ff14SB, ff19SB) is often considered the gold standard for simulating proteins and nucleic acids, having been extensively validated by the scientific community [2]. While it excels with its native force fields, AMBER can be less flexible for non-standard simulations [2].
GROMACS distinguishes itself with exceptional versatility in force field support. It natively supports a wide range of force fields, including AMBER, CHARMM, and OPLS, allowing researchers to select the most appropriate model for their specific system [2]. This flexibility makes GROMACS adaptable to a broad spectrum of research needs, from small molecules to large biomolecular complexes.
NAMD works most naturally with the CHARMM force field. The CHARMM-GUI input generator facilitates the preparation of simulation inputs for NAMD using the CHARMM36 additive force field, ensuring proper treatment of nonbonded interactions which is crucial for simulation accuracy [17].
Quantitative Performance and Benchmarking Data
Performance and scalability are critical for executing simulations efficiently, especially for large systems.
| Performance Aspect | GROMACS | AMBER | NAMD |
|---|---|---|---|
| Raw Speed & Scalability | Exceptional; highly optimized for CPU & GPU [2] | Good; significant GPU acceleration improvements [2] | Good; optimized for parallelization [3] |
| Optimal Use Case | Large-scale simulations & high-throughput studies [2] | Detailed studies of protein-ligand interactions [2] | Large biomolecular complexes [6] |
| Multi-GPU Scaling | Excellent scaling with multiple GPUs [18] | Primarily for multi-replica methods (e.g., REMD) [7] | Efficient distribution across multiple GPUs [18] |
Experimental Validation from Comparative Studies
A rigorous study compared four MD packages (AMBER, GROMACS, NAMD, and ilmm) by simulating two proteins, the Engrailed homeodomain (EnHD) and RNase H, using best-practice parameters for 200 ns each [6]. The findings provide critical insights for researchers:
- Overall, at room temperature, all packages reproduced a variety of experimental observables equally well. This suggests that for simulating native state conditions, the choice of software may be less critical [6].
- Subtle differences in underlying conformational distributions and sampling were observed. This can lead to ambiguity about which results are most correct, as experiments cannot always provide the atomistic detail to distinguish between ensembles [6].
- Differences became more pronounced during larger amplitude motions, such as thermal unfolding. Some packages failed to allow the protein to unfold at high temperature or produced results inconsistent with experiment, highlighting that outcomes can diverge significantly when simulating non-equilibrium or highly dynamic processes [6].
- Differences are not solely due to force fields. Other factors including the water model, algorithms constraining motion, handling of atomic interactions, and the simulation ensemble significantly influence the outcome. It is incorrect to place all blame for deviations on the force field alone [6].
Another key study focusing on energy comparisons found that with careful parameter choices, different MD engines (including GROMACS and AMBER) can achieve energy agreement of better than 0.1% for all energy components [19]. However, one of the largest sources of discrepancy was the use of different values for Coulomb's constant between programs [19].
Experimental Protocols and Workflows
Reproducibility is a cornerstone of scientific computing. Below is a generalized workflow for setting up and running an MD simulation, with software-specific details.
Diagram 1: A generalized molecular dynamics simulation workflow, showing the common stages from structure preparation to analysis, with software-specific tools at key stages.
Detailed Methodology for Key Steps
1. Structure Preparation and Checking
- Initial Check: Before simulation, PDB files must be rigorously checked for common problems, including the presence of non-protein molecules (crystallographic waters, ligands), alternate conformations, missing side-chain atoms, and atomic clashes [20].
- Tool Implementation: The
check_structureutility from the BioBB project can automate this process [20]. For example, to check a structure and remove all ligands:check_structure -i input.pdb -o protein.pdb ligands --remove all[20]. - Handling Alternate Conformations: Structures often contain residues with multiple conformers (e.g., Asp20 with conformers A and B). These must be resolved by selecting one conformer per residue for simulation [20].
2. System Building and Topology Generation
- AMBER: Uses the
tLEaPorxLEaPutilities to load coordinates, apply force field parameters, solvate the system (e.g., in a TIP4P-EW water box), and generate the necessary topology (.prmtop) and coordinate (.inpcrd) files [20]. - GROMACS: Employs tools like
pdb2gmxto generate topology andgromppto process files into a run input (.tpr). - NAMD: Often leverages CHARMM-GUI, a web-based platform that generates all necessary input files (including structure file PSF and configuration file) for NAMD simulations using the CHARMM36 force field [17].
3. Simulation Execution and Performance Optimization
- Performance Optimization: A key technique to increase simulation speed is to use a 4 fs time step with hydrogen mass repartitioning. This involves increasing hydrogen masses and correspondingly decreasing the masses of atoms to which they are bonded, which can be done automatically with the
parmedutility [7]. - Hardware Utilization: The submission scripts in section 4 highlight how each software leverages CPUs and GPUs. GROMACS, for instance, can efficiently use multiple GPUs per node for a single simulation, while AMBER's multi-GPU
pmemdis primarily designed for running multiple simultaneous simulations like replica exchange [7].
Computational Hardware and Research Reagents
Optimizing computational resources is essential for achieving efficient simulation throughput.
| Component | Recommended Specifications | Function in MD Simulations |
|---|---|---|
| GPU | NVIDIA RTX 6000 Ada (48 GB VRAM) / RTX 4090 (24 GB VRAM) [18] | Accelerates computationally intensive non-bonded force calculations; essential for large systems. |
| CPU | AMD Threadripper PRO (high clock speed) [18] | Manages simulation control, data I/O, and directs GPU computations; high clock speed is prioritized. |
| RAM | 4-8 GB per CPU core [7] | Holds atom coordinates, velocities, forces, and topology data for rapid access during integration. |
| Storage | High-speed NVMe SSD | Writes large trajectory files (often terabytes) generated during production runs. |
Example Job Submission Scripts
GROMACS on a Single GPU [7]:
AMBER on a Single GPU [7]:
NAMD on Multiple GPUs [7]:
The choice between GROMACS, AMBER, and NAMD is not about finding the "best" software in absolute terms, but rather selecting the right tool for a specific research question and resource context.
- For maximum accuracy and specialization in biomolecular studies, particularly for protein-ligand interactions, nucleic acid dynamics, and advanced methods like free energy calculations or QM/MM, AMBER is the preferred choice, leveraging its refined force fields [2].
- For high-throughput simulations, large-scale systems, and general-purpose MD where computational speed, scalability, and force field flexibility are paramount, GROMACS is unparalleled [2] [21].
- For studying complex systems like membrane proteins and for researchers who prefer the integrated environment of CHARMM-GUI, NAMD paired with the CHARMM force field is a robust and well-validated option [17].
Ultimately, researchers should be aware that while these tools are powerful, outcomes can vary not just due to the software or force field, but also because of specific simulation protocols, water models, and treatment of long-range interactions [6] [19]. Careful setup and validation against experimental data, when possible, remain crucial for generating meaningful and reliable results.
Setting Up Simulations: A Practical Guide to File Formats and Specialized Systems
For researchers in drug development and computational biophysics, leveraging existing AMBER files (prmtop, inpcrd, parm7, rst7) in NAMD or GROMACS can maximize workflow flexibility and computational efficiency. This guide provides an objective, data-driven comparison of the file compatibility and resulting performance across these molecular dynamics software.
Software-Specific AMBER File Handling Mechanisms
The process and underlying mechanisms for reading AMBER files differ significantly between NAMD and GROMACS.
NAMD's Direct AMBER Interface
NAMD features a direct interface for AMBER files, allowing it to natively read the parm7 (or prmtop) topology file and coordinate files. This direct read capability means NAMD uses the complete topology and parameter information from the AMBER force field as provided [22].
Key configuration parameters for NAMD include:
amber on: Must be set to specify the use of the AMBER force field [22].parmfile: Defines the input AMBER format PARM file [22].ambercoor: Specifies the AMBER format coordinate file. Alternatively, thecoordinatesparameter can be used for a PDB format file [22].exclude scaled1-4: This setting mirrors AMBER's handling of non-bonded interactions [22].oneFourScaling: Should be set to the inverse of the SCEE value used in AMBER (e.g., 0.833333 for SCEE=1.2) [22].
A critical consideration is the oldParmReader option. It should be set to off for modern force fields like ff19SB that include CMAP terms, as the old reader does not support them [22].
GROMACS's Indirect Conversion Pathway
In contrast, GROMACS typically relies on an indirect conversion pathway. The most common method involves using the parmed tool (from AmberTools) to convert the AMBER prmtop file into a GROMACS-compatible format (.top file), while the coordinate file (e.g., inpcrd) can often be used directly [7] [2].
An alternative method leverages VMD plug-ins. If GROMACS is built with shared library support and a VMD installation is available, GROMACS tools can use VMD's plug-ins to read non-native trajectory formats directly [23]. This capability can also assist with file format interoperability at the system setup stage.
Performance Benchmark Comparisons
The different handling mechanisms and underlying codebases lead to distinct performance profiles. The following tables summarize performance data from various benchmarks for different system sizes.
Table 1: Performance on Large Systems (>100,000 atoms)
| Software | System Description | System Size (Atoms) | Hardware | Performance (ns/day) |
|---|---|---|---|---|
| AMBER (pmemd.cuda) | STMV (NPT) [24] | 1,067,095 | NVIDIA RTX 5090 | 109.75 |
| AMBER (pmemd.cuda) | Cellulose (NVE) [24] | 408,609 | NVIDIA RTX 5090 | 169.45 |
| GROMACS | Not specified in sources | ~1,000,000 | Modern GPU | Excellent multi-node scaling [25] |
| NAMD | Not specified in sources | ~1,000,000 | Modern GPU | Efficient multi-GPU execution [26] |
Table 2: Performance on Medium Systems (~20,000-100,000 atoms)
| Software | System Description | System Size (Atoms) | Hardware | Performance (ns/day) |
|---|---|---|---|---|
| AMBER (pmemd.cuda) | FactorIX (NPT) [24] | 90,906 | NVIDIA RTX 5090 | 494.45 |
| AMBER (pmemd.cuda) | JAC (DHFR, NPT) [24] | 23,558 | NVIDIA RTX 5090 | 1632.97 |
| GROMACS | DHFR [25] | ~23,000 | Single High-End GPU | Extremely high throughput [25] |
| NAMD | Not specified in sources | ~25,000-90,000 | 2x NVIDIA A100 | Fast simulation times [26] |
Table 3: Performance on Small Systems & Implicit Solvent
| Software | System Description | System Size (Atoms) | Hardware | Performance (ns/day) |
|---|---|---|---|---|
| AMBER (pmemd.cuda) | Nucleosome (GB) [24] | 25,095 | NVIDIA RTX 5090 | 58.61 |
| AMBER (pmemd.cuda) | Myoglobin (GB) [24] | 2,492 | NVIDIA RTX 5090 | 1151.95 |
| GROMACS | Solvated Protein [25] | ~23,000 | Single High-End GPU | ~1,700 [25] |
Performance Analysis
- AMBER: Demonstrates strong single-GPU performance, particularly on biomolecular systems of small to medium size. Its efficiency for a single simulation on one GPU is a recognized strength [2] [25].
- GROMACS: Consistently benchmarks as one of the fastest MD engines, excelling in raw throughput and parallel scalability across multiple CPUs and GPUs, especially for large systems [2] [25].
- NAMD: Also shows high performance and is optimized for parallel execution, including multi-GPU setups [26]. It is recognized for superior performance on high-performance GPUs and mature features like collective variables [3].
Experimental Protocols for Performance Evaluation
The performance data cited in this guide are derived from standardized benchmarking suites and real-world simulation workflows.
AMBER GPU Benchmarking Protocol
The AMBER 24 benchmark data is generated using the software's built-in benchmark suite [24].
- Systems: Pre-defined test cases (e.g., STMV, Cellulose, DHFR, FactorIX) covering a range of sizes and simulation types (explicit solvent NPT/NVE, implicit solvent GB) [24].
- Workflow: The standard simulation workflow involves energy minimization, heating, equilibration, and production, as implemented in the benchmark suite [27].
- Measurement: The key output metric is simulation throughput, reported in nanoseconds per day (ns/day) [24].
- Hardware: All benchmarks are performed on a single GPU, even in multi-GPU systems, as AMBER primarily leverages multiple GPUs for running independent simulations in parallel [24].
GROMACS and NAMD Performance Assessment
Performance data for GROMACS and NAMD are gathered from published benchmark studies and hardware recommendation guides [26] [25].
- Methodology: These studies typically involve running production-level simulations of standardized systems (e.g., DHFR, membrane proteins, large viral capsids) on controlled hardware configurations [25].
- Key Metrics: The primary evaluation criteria are simulation throughput (ns/day) and parallel scaling efficiency—how performance changes with increasing CPU cores or GPUs [7] [25].
- Hardware Consideration: Studies emphasize that optimal performance requires matching the hardware to the software. GROMACS and NAMD can scale across multiple nodes, while AMBER's strength for a single calculation often lies on a single GPU [26] [25].
The diagram below illustrates the general workflow for setting up and running a simulation with AMBER files in NAMD or GROMACS, incorporating performance benchmarking.
Simulation Setup and Benchmarking Workflow
The Scientist's Toolkit: Essential Research Reagents and Solutions
This table details key software and hardware tools essential for working with AMBER files across different simulation platforms.
Table 4: Essential Research Tools and Materials
| Item Name | Function/Benefit | Relevance to AMBER File Compatibility |
|---|---|---|
| AmberTools | A suite of programs for molecular mechanics and dynamics, including parmed and LEaP. [27] |
Crucial for preparing and modifying AMBER parameter/topology (prmtop) files and for file conversion for GROMACS. [7] |
| VMD | A visualization and analysis program for biomolecular systems. [23] | Its plug-ins enable GROMACS to read AMBER format trajectories directly. Essential for visualization and analysis post-simulation. [23] |
| parmed | A parameter file editor included in AmberTools. [7] | The primary tool for converting AMBER prmtop files to GROMACS-compatible .top files and for applying hydrogen mass repartitioning. [7] |
| High-End NVIDIA GPUs (e.g., RTX 5090, A100, H100) | Accelerate MD calculations dramatically. [24] [26] | AMBER (pmemd.cuda), GROMACS, and NAMD all leverage CUDA for GPU acceleration, making modern NVIDIA GPUs critical for high performance. [24] [26] |
| SLURM Workload Manager | Manages and schedules computational jobs on HPC clusters. [7] | Used to submit simulation jobs for all three packages with specified computational resources (CPUs, GPUs, memory). [7] |
Choosing the right software for using AMBER files involves a trade-off between implementation ease, performance needs, and project goals.
- NAMD offers the most straightforward path for direct use of AMBER files with good single- and multi-GPU performance.
- GROMACS requires an extra conversion step but often delivers superior throughput and scalability for very large systems on extensive computing resources.
- AMBER itself remains a competitive choice, especially for simulations run on a single GPU where its specialized algorithms and force field accuracy are paramount.
Researchers are advised to base their decision on the specific size of their system, available computational resources, and the importance of maximum simulation throughput versus workflow simplicity.
This guide provides a detailed, objective comparison of molecular dynamics (MD) software—GROMACS, AMBER, and NAMD—with a specific focus on simulating membrane proteins. For researchers in drug development and structural biology, selecting the appropriate MD engine is crucial for balancing computational efficiency, force field accuracy, and workflow practicality.
Molecular dynamics simulations of membrane proteins are computationally demanding. The choice of software significantly impacts project timelines and resource allocation. The table below summarizes the core characteristics and performance metrics of GROMACS, AMBER, and NAMD.
Table 1: Core Features and Performance Comparison of GROMACS, AMBER, and NAMD
| Feature | GROMACS | AMBER | NAMD |
|---|---|---|---|
| Primary Strength | High throughput & parallel scaling on CPUs/GPUs [25] | Accurate force fields & rigorous free-energy calculations [25] [3] | Excellent visualization integration & scalable parallelism [3] |
| Typical Performance (Single GPU) | Among the highest of MD codes [25] | ~1.7 μs/day for a 23,000-atom system [25] | Good performance on high-end GPUs [3] |
| Multi-GPU Scaling | Excellent, with GPU decomposition for PME [25] | Limited; 1 GPU often saturates performance for a single simulation [25] | Good, especially for very large systems [25] |
| Key Membrane Protein Feature | Comprehensive tutorial for membrane-embedded proteins [28] [29] | Integrated with PACKMOL-Memgen for system building [30] | Tight integration with VMD for setup and analysis [3] |
| Licensing | Open-source (GPL/LGPL) [25] | AmberTools (free), full suite requires license [25] | Free for non-commercial use [25] |
Quantitative performance data from independent benchmarks on HPC clusters provide critical insights for resource planning [7]. The following table summarizes key performance metrics for the three software packages.
Table 2: Quantitative Performance Benchmarking Data [7]
| Software | Hardware Configuration | Reported Performance | Key Benchmarking Insight |
|---|---|---|---|
| GROMACS | 1 node, 1 GPU, 12 CPU cores | 403 ns/day | Efficient use of a single GPU with moderate CPU core count. |
| GROMACS | 1 node, 2 GPUs, 2x12 CPU cores | 644 ns/day | Good multi-GPU scaling on a single node. |
| AMBER (PMEMD) | 1 node, 1 GPU, 1 CPU core | 275 ns/day | Highly efficient on a single GPU with minimal CPU requirement. |
| NAMD 3 | 1 node, 2 A100 GPUs, 2 CPU cores | 257 ns/day | Effective leverage of multiple high-end GPUs. |
Detailed Methodologies and Protocols
Specialized Protocol for Membrane Proteins in GROMACS
Simulating a membrane protein in GROMACS requires careful system setup and equilibration. The established protocol consists of several key stages [28]:
- System Setup: Choose a consistent force field for both the protein and lipids. Insert the protein into a pre-formed bilayer using a tool like
g_membed, or through coarse-grained self-assembly followed by conversion to an atomistic representation [28]. - Solvation and Ions: Solvate the system and add ions to neutralize any excess charge and achieve a physiologically relevant ion concentration [28].
- Energy Minimization: Perform energy minimization to remove any steric clashes or unrealistic geometry in the initial structure [28].
- Membrane Adjustment: Run a short (~5-10 ns) MD simulation with strong restraints (e.g., 1000 kJ/(mol nm²)) on the heavy atoms of the protein. This allows the lipid membrane to adapt to the presence of the protein without the protein structure distorting [28].
- Equilibration and Production: Conduct equilibration runs with the restraints progressively released before starting a final, unrestrained production MD simulation [28].
The following diagram illustrates this multi-stage workflow.
A common challenge during solvation is the placement of water molecules into hydrophobic regions of the membrane. This can be addressed by [28]:
- Letting a short MD run expel the waters via the hydrophobic effect.
- Using the
-radiusoption ingmx solvateto increase the water exclusion radius. - Modifying the
vdwradii.datfile to increase the atomic radii of lipid atoms, preventingsolvatefrom identifying small interstices as suitable for water.
AMBER Protocol for a GPCR Membrane Protein
A typical AMBER workflow for a complex membrane protein, such as a GPCR, leverages different tools for system building and follows a careful equilibration protocol [30]:
- System Building with PACKMOL-Memgen: The process often begins with a protein structure from the OPM database, which is pre-aligned for membrane embedding. After protein and ligand preparation, PACKMOL-Memgen is used to construct a mixed lipid bilayer (e.g., POPC/Cholesterol at a 9:1 ratio), solvate the system, and add ions around the pre-oriented protein [30].
- Topology Building with tleap: The coordinates for the protein, ligand, and membrane box are combined in
tleapto generate the topology (prmtop) and coordinate (inpcrd) files using the appropriate force fields (e.g., Lipid21) [30]. - Staged Equilibration: The system is equilibrated through a series of restrained simulations:
- Minimization: One or more rounds of energy minimization, often starting with a short CPU minimization to resolve severe lipid clashes [30].
- Heating: The system is heated to the target temperature (e.g., 303 K) with restraints on the protein, ligand, and lipid head groups [30].
- Backbone and Side-Chain Relaxation: Short (e.g., 1 ns) NPT simulations are run with restraints first on the protein backbone atoms, and then only on the C-alpha atoms, allowing the side chains to relax [30].
- Production: Finally, all restraints are removed for an extended production run [30].
This protocol is visualized in the following workflow.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful membrane protein simulations rely on a suite of software tools and resources. The following table details key components of a modern computational researcher's toolkit.
Table 3: Essential Tools and Resources for Membrane Protein Simulations
| Tool/Resource | Function | Relevance to Membrane Simulations |
|---|---|---|
| CHARMM-GUI [31] | A web-based platform for building complex molecular systems and generating input files. | Streamlines the setup of membrane-protein systems for various MD engines (GROMACS, AMBER, NAMD), providing pre-equilibrated lipid bilayers of different compositions. |
| OPM Database | (Orientations of Proteins in Membranes) provides spatially-oriented structures of membrane proteins. | Supplies protein structures pre-aligned in the lipid bilayer, defining the membrane boundaries, which is a critical starting point for system building [30]. |
| Lipid21 Force Field | The AMBER force field for lipids. | A modern, comprehensive set of parameters for various lipids, compatible with the AMBER protein force fields, enabling accurate simulations of complex membrane compositions [30]. |
| PACKMOL-Memgen | A program for building membrane-protein systems within the AMBER ecosystem. | Automates the construction of a lipid bilayer around an inserted protein, followed by solvation and ion addition, simplifying a traditionally complex and error-prone process [30]. |
| VMD | A molecular visualization and analysis program. | Tightly integrated with NAMD, it is extensively used for trajectory analysis, visualization, and initial system setup for membrane simulations [3]. |
| BioExcel Tutorials | A suite of advanced GROMACS tutorials. | Includes a dedicated tutorial "KALP15 in DPPC" designed to teach users how to simulate membrane proteins and understand force field structure and modification [29] [32]. |
The choice between GROMACS, AMBER, and NAMD for membrane protein simulations involves clear trade-offs. GROMACS excels in raw speed and strong parallel scaling on HPC resources, making it ideal for high-throughput simulations. AMBER is distinguished by its highly validated force fields and robust free-energy calculation methods, which are critical for drug discovery applications like binding affinity prediction. NAMD, with its deep integration to VMD, offers a powerful environment for simulating massive systems and for researchers who prioritize extensive visual analysis. The decision should be guided by the specific research goals, available computational infrastructure, and the need for specific force fields or analysis features.
The accuracy of any molecular dynamics (MD) simulation is fundamentally constrained by the quality of the force field parameters that describe the interactions between atoms. While modern MD software packages like GROMACS, AMBER, and NAMD have reached impressive levels of performance and sophistication, enabling simulations on the microsecond to millisecond timescales [33], the challenge of generating reliable parameters for novel chemical entities remains a significant bottleneck, particularly in fields like drug discovery [34]. This parameterization problem is acute because the chemical space of potential small molecules is astronomically large, estimated at 10¹⁸ to 10²⁰⁰ compounds, compared to the ~25,000 proteins encoded in the human genome [34]. The inability to rapidly generate accurate and robust parameters for these novel molecules severely limits the application of MD simulations to many biological systems of interest [34].
This guide provides an objective comparison of parameterization methodologies across three leading MD software packages, detailing best practices, common pitfalls, and evidence-based protocols for developing reliable parameters for novel molecules. By synthesizing information from experimental benchmarks and developer documentation, we aim to equip researchers with the knowledge to navigate the complexities of force field development for their specific systems.
Force Field Philosophies and Software-Specific Implementations
Foundational Concepts and Terminology
Force fields are mathematical models that calculate the potential energy of a system of atoms. The total energy is typically a sum of bonded terms (bonds, angles, dihedrals) and non-bonded terms (electrostatic and van der Waals interactions). Parameterization is the process of determining the numerical constants (the "parameters") in these equations that best reproduce experimental data or high-level quantum mechanical calculations.
The concept of transferability—where a single set of parameters for a given atom type accurately describes its behavior in various chemical contexts—is central to force field design. While this works well for the modular building blocks of biopolymers, it becomes challenging for the diverse and exotic structures often found in small molecules, such as engineered drug-like compounds with complex fused aromatic scaffolds and specialized functional groups [34].
Comparative Analysis of AMBER, CHARMM, and GAFF
Different force fields follow distinct philosophies for deriving parameters, particularly for partial atomic charges, which is a key distinguishing aspect.
AMBER Force Fields: The AMBER family, including the widely used General AMBER Force Field (GAFF) for small molecules, typically derives partial atomic charges by fitting to the electrostatic potential (ESP) surrounding the molecule, often using the Restricted Electrostatic Potential (RESP) fitting procedure [34]. The
antechambertool is the primary utility for generating GAFF parameters. A notable characteristic of the AMBER force field is that it treats dihedrals and impropers with the same mathematical form [35].CHARMM Force Fields: In contrast to AMBER, the CHARMM force field and its general version, CGenFF, derive partial atomic charges from water-interaction profiles [34]. This method involves optimizing charges to reproduce quantum mechanical interaction energies and distances between the target molecule and water molecules. The Force Field Toolkit (ffTK), a VMD plugin, is designed specifically to facilitate this CHARMM-compatible parameterization workflow.
GROMACS and Force Field Agnosticism: GROMACS is itself a simulation engine that supports multiple force fields. A researcher can choose to use AMBER, CHARMM, GROMOS, or other force fields within GROMACS [36]. Its preparation tools, like
gmx pdb2gmx, can generate topologies for different force fields, and it can interface with external parameterization resources such as the SwissParam server (for CHARMM force fields) or the Automated Topology Builder (ATB) (for GROMOS96 53A6) [36].
Table 1: Comparison of Force Field Philosophies and Parameterization Tools
| Feature | AMBER/GAFF | CHARMM/CGenFF | GROMACS (Engine) |
|---|---|---|---|
| Charge Derivation Method | Electrostatic Potential (ESP) fitting (e.g., RESP) [34] | Water-interaction energy profiles [34] | Agnostic (depends on selected force field) |
| Primary Parameterization Tool | antechamber |
Force Field Toolkit (ffTK), ParamChem [34] | gmx pdb2gmx, SwissParam, ATB, manual editing [36] |
| Small Molecule Force Field | GAFF | CGenFF | Varies (e.g., GAFF, CGenFF via import) |
| Treatment of Dihedrals/Impropers | Same functional form [35] | Distinct treatment | Agnostic (depends on selected force field) |
Best Practices and Workflows for Parameterizing Novel Molecules
A Generalized Parameterization Workflow
Regardless of the specific force field, a systematic and careful workflow is essential for developing high-quality parameters. The following diagram, generated from a synthesis of the cited methodologies, outlines the key stages in a robust parameterization pipeline, highlighting critical validation steps.
Diagram 1: The Parameterization Workflow for Novel Molecules. This flowchart outlines the iterative process of developing and validating force field parameters, from initial structure preparation to final production simulation.
Detailed Workflow Stages and Software-Specific Protocols
Stage 1: System Preparation and Initial Setup
The process begins with obtaining or generating a high-quality initial 3D structure for the novel molecule. The key step here is assigning preliminary atom types, which form the basis for all subsequent parameters. For CHARMM/CGenFF, the ParamChem web server provides excellent automated atom-typing functionality [34]. For AMBER/GAFF, antechamber performs this role. It is critical to note that these automated assignments are only a starting point; the associated penalty scores (in ParamChem) must be carefully reviewed to identify atoms with poor analogy to the existing force field, as these will be priorities for optimization [34].
Stage 2: Generating Quantum Mechanical (QM) Target Data Meaningful atomistic MD simulations require accurate potential energy functions, which are calibrated against QM target data [37] [34]. Essential QM calculations include:
- Geometry Optimization: To find the molecule's minimum energy structure.
- Charge Derivation Data: For CHARMM, this involves calculating water-interaction profiles; for AMBER, it involves computing the electrostatic potential around the molecule.
- Dihedral Scans: Performing constrained optimizations by rotating key dihedral angles to map the rotational energy profile, which is used for fitting dihedral parameters [34].
Stage 3 & 4: Parameter Assignment and Optimization This is the core iterative stage. Tools like the Force Field Toolkit (ffTK) for CHARMM significantly reduce the barrier to parameterization by automating tasks such as setting up optimization routines and scoring the fit of molecular mechanics (MM) properties to the QM target data [34]. A best practice is to optimize parameters in a specific order:
- Bonds and Angles: Fit force constants and equilibrium values to reproduce QM potential energy surfaces of small distortions from the optimized geometry.
- Dihedrals: Fit the amplitudes and phases of dihedral terms to match the QM dihedral scan profiles. A common pitfall is to use excessively large force constants; it is often necessary to scale down the barrier heights from gas-phase QM scans to account for condensed-phase effects [35]. For example, a QM scan might suggest a dihedral barrier of 40.5 kcal/mol, but a more transferable parameter for the condensed phase might be much lower, consistent with the values found in established force fields like GAFF where major barriers rarely exceed 6 kcal/mol [35].
- Charges: Optimize partial atomic charges to match the chosen target (ESP or water-interaction energies).
Stage 5: Validation against Experimental Data The final, crucial step is to validate the complete parameter set against available experimental data. This tests the parameters in a realistic, condensed-phase environment. Key validation metrics include:
- Pure-Solvent Properties: Density and enthalpy of vaporization should typically be within <15% error from experiment [34].
- Free Energy of Solvation: This is a stringent test; well-parameterized molecules should reproduce experimental solvation free energies within ±0.5 kcal/mol [34].
Table 2: Key Validation Metrics for Parameterized Molecules
| Validation Metric | Target Accuracy | Experimental Reference |
|---|---|---|
| Density (ρ) | < 15% error | Measured pure-solvent density |
| Enthalpy of Vaporization (ΔHvap) | < 15% error | Thermodynamic measurements |
| Free Energy of Solvation (ΔGsolv) | ± 0.5 kcal/mol | Experimental solvation free energies |
Performance Benchmarks and Experimental Data
Software Performance and Scaling
The choice of MD software can significantly impact computational efficiency and the feasibility of long time-scale simulations. Performance benchmarks on high-performance computing clusters provide critical data for resource planning.
- GROMACS is widely recognized for its computational speed, especially on GPUs [3] [7]. It is often the fastest engine for running standard atomistic simulations on a single GPU or multiple GPUs.
- AMBER's
pmemd.cudais highly optimized for single-GPU simulations. It is important to note that the multiple-GPU PMEMD version is designed primarily for running multiple simultaneous simulations (e.g., replica exchange), as a single simulation generally does not scale beyond one GPU [7]. - NAMD 3 demonstrates strong performance on high-performance GPUs, with some users reporting superior performance compared to GROMACS in certain hardware configurations [3].
Force Field Accuracy and Convergence in Real-World Applications
The accuracy of the underlying force field is as important as software performance. Extensive validation studies have been conducted, particularly for biomolecules.
A landmark study assessing AMBER force fields for DNA aggregated over 14 milliseconds of simulation time across five test systems [33]. The study compared the bsc1 and OL15 force field modifications, which were developed to correct artifacts observed in earlier versions like parm99 and bsc0. The key finding was that both bsc1 and OL15 are "a remarkable improvement," with average structures deviating less than 1 Å from experimental NMR and X-ray structures [33]. This level of exhaustive sampling—including a single trajectory of the Drew-Dickerson dodecamer concatenated to 1 ms for each force field/water model combination—demonstrates the time scales required to properly converge and validate conformational ensembles [33].
Table 3: Essential Software Tools for Parameterization and Simulation
| Tool Name | Function | Compatible Force Field/Software |
|---|---|---|
| Force Field Toolkit (ffTK) [34] | A VMD plugin that provides a GUI for the complete CHARMM parameterization workflow, from QM data generation to parameter optimization. | CHARMM, CGenFF, NAMD |
| Antechamber [34] | Automates the process of generating force field parameters for most organic molecules for use with AMBER. | AMBER, GAFF |
| ParamChem Web Server [34] | Provides initial parameter assignments for CGenFF based on molecular analogy, including all-important penalty scores. | CHARMM, CGenFF |
| Automated Topology Builder (ATB) [34] [36] | A web server that generates topologies and parameters for molecules, compatible with the GROMOS force field and others. | GROMOS, GROMACS |
| SwissParam [34] [36] | A web service that provides topologies and parameters for small molecules for use with the CHARMM force field. | CHARMM, GROMACS |
| gmx pdb2gmx [36] | A core GROMACS tool that generates topologies from a coordinate file, selecting from a range of built-in force fields. | GROMACS (multiple force fields) |
| Parmed [7] | A versatile program for manipulating molecular topology and parameter files, notably used for hydrogen mass repartitioning to enable 4 fs time steps. | AMBER |
Parameterizing novel molecules remains a complex but manageable challenge. Success hinges on selecting an appropriate force field philosophy (e.g., AMBER's ESP charges vs. CHARMM's water-interaction profiles), following a rigorous and iterative workflow grounded in QM target data, and employing robust validation against experimental observables. Software tools like ffTK, Antechamber, and ParamChem have dramatically reduced the practical barriers to performing these tasks correctly.
The ongoing development of force fields, as evidenced by the incremental improvements in the AMBER DNA parameters [33], shows that this is a dynamic field. As MD simulations are increasingly used to support and interpret experimental findings in areas like surfactant research [38], the demand for reliable parameters for exotic molecules will only grow. By adhering to the best practices and leveraging the tools outlined in this guide, researchers can generate parameters that ensure their simulations of novel molecules are both accurate and scientifically insightful.
This guide provides an objective comparison of three major molecular dynamics (MD) software packages—GROMACS, AMBER, and NAMD—focusing on their integration into a complete research workflow, from initial system setup to production simulation.
The table below summarizes the core characteristics of GROMACS, AMBER, and NAMD to help researchers make an initial selection.
Table 1: High-Level Comparison of GROMACS, AMBER, and NAMD
| Feature | GROMACS | AMBER | NAMD |
|---|---|---|---|
| Primary Strength | Raw speed for GPU-accelerated simulations on a single node [3] | Accurate force fields, particularly for biomolecules; strong support for advanced free energy calculations [3] | Excellent parallel scaling and visualization integration; robust collective variables [3] |
| License & Cost | Free, open-source (GNU GPL) [39] | Proprietary; requires a license for commercial use [3] [39] | Free for academic use [39] |
| Ease of Use | Great tutorials and workflows for beginners [3] | Steeper learning curve; some tools require a license [3] | Easier visual analysis, especially when paired with VMD [3] |
| Force Fields | Supports AMBER, CHARMM, GROMOS, etc. [39] | Known for its own accurate and well-validated force fields [3] | Often used with CHARMM force fields [39] |
| GPU Support | Excellent; highly optimized CUDA support, with a growing HIP port for AMD GPUs [40] [41] | Excellent; optimized CUDA support via PMEMD [24] | Excellent; CUDA-accelerated [39] |
| Multi-GPU Scaling | Good; supports single-node multi-GPU simulation [7] | Limited; primarily for running multiple independent simulations (task-level parallelism) [24] [7] | Excellent; efficient distribution across multiple GPUs [42] [7] |
Quantitative Performance Benchmarking
Performance is a critical factor in software selection. The following data, gathered from published benchmarks, provides a comparison of simulation throughput.
Table 2: Performance Benchmarking on NVIDIA GPUs (Simulation Speed in ns/day)
| System Description (Atoms) | Software | NVIDIA RTX 5090 | NVIDIA RTX 6000 Ada | NVIDIA GH200 Superchip (GPU only) |
|---|---|---|---|---|
| STMV (1,067,095 atoms) | AMBER 24 [24] | 109.75 | 70.97 | 101.31 |
| Cellulose (408,609 atoms) | AMBER 24 [24] | 169.45 | 123.98 | 167.20 |
| Factor IX (90,906 atoms) | AMBER 24 [24] | 529.22 | 489.93 | 191.85 |
| DHFR (23,558 atoms) | AMBER 24 [24] | 1655.19 | 1697.34 | 1323.31 |
These results highlight several key trends. For large systems (over 1 million atoms), the NVIDIA RTX 5090 and data center GPUs like the GH200 show leading performance with AMBER [24]. In mid-sized systems, the RTX 6000 Ada is highly competitive, sometimes even outperforming the RTX 5090 [24]. It is crucial to note that performance is system-dependent; the GH200, for example, shows lower performance on the Factor IX system despite its capability with larger systems [24]. While this data is for AMBER, GROMACS is widely recognized for its superior raw speed on a single GPU, while NAMD excels in multi-node, multi-GPU parallelism [3] [42].
Experimental Protocols for Production Simulations
This section provides standard protocols for running production simulations on high-performance computing (HPC) clusters, a common environment for researchers.
GROMACS Production Run
GROMACS is highly efficient for single-node, GPU-accelerated simulations. The protocol below is for a production run on a single GPU [7].
Key Parameters:
-nb gpu: Offloads non-bonded calculations to the GPU.-pme gpu: Offloads Particle Mesh Ewald (PME) calculations to the GPU.-update gpu: Offloads coordinate and velocity updates to the GPU (a more recent feature) [40].-bonded cpu: Calculates bonded interactions on the CPU (can also be set togpu).
AMBER Production Run
AMBER's GPU-accelerated engine (pmemd.cuda) is optimized for single-GPU simulations. A single simulation does not scale beyond one GPU [7].
NAMD Production Run
NAMD is designed to leverage multiple GPUs across nodes. The following script is an example for a multi-GPU simulation [7].
Performance Optimization Technique
A universal technique to improve simulation speed across all packages is hydrogen mass repartitioning, which allows for a 4-fs time step. This can be done using the parmed tool available in AmberTools [7].
Workflow Integration and Decision Pathway
The following diagram illustrates the decision process for selecting and integrating an MD software into a research workflow, based on the project's primary requirements.
MD Software Selection Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Beyond software, a successful MD simulation requires a suite of tools and hardware.
Table 3: Essential Research Reagents and Computational Solutions
| Item Name | Function / Purpose | Example / Note |
|---|---|---|
| Structure Prediction | Generates 3D protein structures from sequence. | AlphaFold2, Robetta, trRosetta, I-TASSER [43] |
| Structure Preparation & Visualization | Builds, edits, and visualizes molecular systems. | VMD (with NAMD), MOE, AmberTools (antechamber), Avogadro [39] |
| Force Field Parameters | Defines energy terms for atoms and molecules. | AMBER FF (via AMBER), CHARMM36 (via NAMD/CHARMM), GROMOS (via GROMACS) [3] [39] |
| High-Performance GPU | Accelerates compute-intensive MD calculations. | NVIDIA RTX 5090 (cost-effective), RTX 6000 Ada (memory capacity) [24] [42] |
| Workstation/Server | Hosts hardware for local simulations. | Custom-built systems (e.g., BIZON) for optimal GPU configuration and cooling [42] |
| Benchmarking Dataset | Standardized systems for performance testing. | Public datasets (e.g., STMV, Cellulose) or custom sets from ACGui [24] [44] |
Maximizing Performance: Hardware Selection and Benchmarking for 2025
For molecular dynamics (MD) researchers selecting hardware in 2025, the new NVIDIA GeForce RTX 50 Series GPUs, based on the Blackwell architecture, represent a significant performance leap, particularly for AI-accelerated workloads and memory-bound simulations. This guide objectively compares the new RTX 50 Series against previous generations and evaluates their performance within the context of the three dominant MD software packages: GROMACS, NAMD, and AMBER. The analysis confirms that while all three software packages benefit from GPU acceleration, GROMACS often leads in raw speed for classical MD on NVIDIA hardware, NAMD excels in scalability and visualization integration, and AMBER is renowned for its accurate force fields, though with potential licensing considerations. The experimental data and structured tables below will help researchers and drug development professionals make an informed hardware decision tailored to their specific simulation needs.
Hardware Landscape: NVIDIA RTX 50 Series (Blackwell)
The NVIDIA Blackwell architecture brings key innovations that are highly relevant to computational molecular dynamics.
RTX 50 Series Specifications & Pricing
The initial release of the GeForce RTX 50 series in January 2025 includes several models suited for different tiers of research computing. Table 1 summarizes the key specifications for the newly announced models [45] [46].
Table 1: Specifications of the Announced NVIDIA GeForce RTX 50 Series GPUs
| Graphics Card | RTX 5090 | RTX 5080 | RTX 5070 Ti | RTX 5070 |
|---|---|---|---|---|
| Architecture | GB202 (Blackwell) | GB203 (Blackwell) | GB203 (Blackwell) | GB205 (Blackwell) |
| GPU Shaders (ALUs) | 21,760 | 10,752 | 8,960 | 6,144 |
| Boost Clock (MHz) | 2,407 | 2,617 | 2,452 | 2,512 |
| VRAM (GB) | 32 | 16 | 16 | 12 |
| VRAM Bus Width | 512-bit | 256-bit | 256-bit | 192-bit |
| VRAM Speed (Gbps) | 28 | 30 | 28 | 28 |
| Memory Bandwidth | 1,792 GB/s | 960 GB/s | 896 GB/s | 672 GB/s |
| L2 Cache | 96 MB | 64 MB | 48 MB | 48 MB |
| Tensor Cores | 5th Gen | 5th Gen | 5th Gen | 5th Gen |
| TBP (watts) | 575 | 360 | 300 | 250 |
| Launch Price | $1,999 | $999 | $749 | $549 |
Key Architectural Advances for MD
- GDDR7 Memory: The shift to GDDR7 memory provides a substantial ~30% increase in memory bandwidth over the previous generation (RTX 40-series) [47]. This is critical for handling large biological systems and reducing bottlenecks in data transfer between the GPU and its memory.
- Fifth-Gen Tensor Cores & AI Performance: Blackwell introduces native FP4 precision support, massively increasing AI throughput [45] [48]. While directly beneficial for AI-driven research, this also powers new AI-based graphics technologies like Neural Texture Compression (NTC), which could reduce texture VRAM requirements by about one-third in visualization-heavy tasks [46].
- Blackwell in Data Center vs. GeForce: It is important to distinguish the consumer GeForce RTX 50 series from the data-center-grade Blackwell Ultra chips. The latter, featured in MLPerf benchmarks, demonstrated a 5x higher throughput per GPU compared to a Hopper-based system on the DeepSeek-R1 AI benchmark [48]. This performance trend in professional AI workloads strongly suggests significant generational gains for MD simulations that leverage similar computational principles.
MD Software Ecosystem: GROMACS, AMBER, and NAMD
The choice of MD software is as critical as the choice of hardware. The "big three" packages have different strengths, licensing models, and hardware optimization. Table 2 provides a high-level comparison [3] [39].
Table 2: Comparison of GROMACS, AMBER, and NAMD for Molecular Dynamics Simulations
| Feature | GROMACS | NAMD | AMBER |
|---|---|---|---|
| Primary Strength | Raw speed for classical MD on GPUs | Excellent parallel scalability & integration with VMD | Accurate force fields, especially for biomolecules |
| GPU Support | Excellent (CUDA, OpenCL, HIP) | Excellent (CUDA) | Yes |
| Licensing | Free Open Source (GPL) | Free for academic use | Proprietary; requires a license for commercial use |
| Learning Curve | Beginner-friendly tutorials and workflows [3] | Steeper | Steeper |
| Visualization | Weak; requires external tools | Strong (tightly integrated with VMD) [3] | Requires external tools |
| Notable Features | High performance, open-source, active community | Handles very large systems well, robust collective variable methods [3] | Suite of analysis tools (e.g., MMPBSA, FEP) is more user-friendly [3] |
Community and Expert Insights
Informal consensus among researchers highlights these practical nuances:
- GROMACS is frequently praised for its speed, versatility, and open-source nature but is noted for its weaker visualization capabilities [3].
- NAMD is recognized for its superior performance on high-performance GPUs and more mature, robust implementation of certain methods like collective variables (colvar) [3].
- AMBER is often the choice for its accurate force fields, though some users find calculations like MMGBSA and MMPBSA to be more user-friendly in AMBER/Desmond compared to GROMACS [3].
Performance Analysis & Experimental Data
Projected Performance of RTX 50 Series for MD
While specific MD benchmarks for the consumer RTX 50 series are not yet published, performance can be projected from architectural improvements and data center results.
- AI and Inference Gains: The Blackwell Ultra architecture's demonstrated 4x training and up to 30x inference performance gains for large AI models on the data center side signals a major architectural efficiency improvement [48] [49]. MD simulations that incorporate machine learning potentials will directly benefit from these advances.
- GROMACS on AMD vs. NVIDIA: A 2025 benchmark study using the SCALE platform to run CUDA-based GROMACS on AMD GPUs showed that performance is "broadly comparable" to GROMACS's native HIP port, with some kernels even performing faster [41]. This confirms that GROMACS is highly optimized for NVIDIA's CUDA architecture, and the new RTX 50 series, with its increased memory bandwidth and compute, will extend this performance lead.
Experimental Protocol for MD Benchmarking
To ensure consistent and reproducible performance measurements across different hardware and software, the following experimental protocol is recommended. This workflow is standardized by initiatives like the Max Planck Institute Benchmarks for GROMACS [41].
Detailed Methodology:
- System Preparation: A standardized system, such as DHFR (dihydrofolate reductase, ~23,000 atoms), is prepared. It is placed in a solvation box with water molecules and ions to neutralize charge. The system undergoes energy minimization until the maximum force is below a set threshold (e.g., 1000 kJ/mol/nm).
- System Equilibration: The system is equilibrated in two phases:
- NVT Ensemble: The number of particles, volume, and temperature are held constant. The system is relaxed for 100-500 ps while restraining solute positions.
- NPT Ensemble: The number of particles, pressure, and temperature are held constant. The system is further equilibrated for 100-500 ps to achieve correct density without restraints.
- Production Run: A multi-step production simulation is performed without restraints. The performance metric is the simulation speed measured in nanoseconds per day (ns/day). The wall-clock time to complete a fixed simulation length (e.g., 10 ns) is also recorded. This step should be repeated multiple times to account for performance variability.
- Performance Analysis: Data from the production run is analyzed. The key metrics are:
- ns/day: A higher value indicates faster simulation speed.
- Wall-time: The actual time taken to complete the simulation.
- These metrics should be collected for different hardware (e.g., RTX 5090 vs. RTX 4090) and different software (GROMACS vs. NAMD vs. AMBER) using the same input system and parameters.
The Scientist's Toolkit: Essential Research Reagents
Beyond hardware and software, a successful MD simulation relies on a suite of computational "reagents." Table 3 details these essential components.
Table 3: Key Research Reagent Solutions for Molecular Dynamics
| Item | Function / Purpose | Examples |
|---|---|---|
| Force Field | Defines the potential energy function and parameters for atoms and molecules. | CHARMM36, AMBER, OPLS-AA, GROMOS |
| Solvation Model | Simulates the effect of a water environment around the solute. | TIP3P, SPC/E, implicit solvent models (GB/SA) |
| System Preparation Tool | Handles building, solvation, and ionization of the initial simulation system. | CHARMM-GUI, PACKMOL, gmx pdb2gmx (GROMACS) |
| Visualization & Analysis Suite | Used to visualize trajectories, analyze results, and create figures. | VMD (tightly integrated with NAMD), PyMOL, ChimeraX, gmx analysis tools |
| Parameterization Tool | Generates force field parameters for small molecules or non-standard residues. | CGenFF, ACPYPE, Antechamber (AMBER) |
Hardware Selection Workflow
Choosing the right GPU requires balancing budget, software choice, and project scope. The following decision diagram provides a logical pathway for researchers.
The introduction of the NVIDIA Blackwell-based RTX 50 Series GPUs in 2025 provides molecular dynamics researchers with powerful new hardware options. The RTX 5090, with its massive 32 GB of VRAM and high memory bandwidth, is the clear choice for researchers working with the largest systems and aiming for maximum throughput. The RTX 5080 and 5070 Ti offer a compelling balance of performance and cost for most standard simulation projects. The choice between GROMACS, NAMD, and AMBER remains dependent on specific research needs: raw speed and open-source access (GROMACS), scalability for massive systems (NAMD), or well-validated force fields and analysis suites (AMBER). By aligning their software selection with the appropriate tier of the new RTX 50 series hardware, researchers can significantly accelerate their discovery timeline in computational drug development and biomolecular research.
Understanding how molecular dynamics (MD) software performs across different molecular systems is crucial for selecting the right tools and computational resources. This guide objectively compares the performance of three major MD software packages—GROMACS, AMBER, and NAMD—by examining their simulation throughput, measured in nanoseconds per day (ns/day), across small, medium, and large molecular systems.
Performance Comparison: ns/day Across System Sizes
The tables below summarize performance data (ns/day) for GROMACS, AMBER, and NAMD across different system sizes and hardware. Performance is influenced by hardware, software version, and specific simulation parameters.
Table 1: GROMACS Performance on Consumer GPUs (SaladCloud Benchmark) [50]
| System Size | Example System (Atoms) | GeForce RTX 4090 (ns/day) | GeForce RTX 4080 (ns/day) | GeForce RTX 4070 (ns/day) |
|---|---|---|---|---|
| Small | RNA in water (31,889 atoms) | ~200-250 (est.) | ~175-225 (est.) | ~150-200 (est.) |
| Medium | Protein in membrane (80,289 atoms) | ~100-150 (est.) | ~80-120 (est.) | ~60-100 (est.) |
| Large | Virus protein (1,066,628 atoms) | ~15-25 (est.) | ~10-20 (est.) | ~5-15 (est.) |
Table 2: AMBER 24 Performance on Select NVIDIA GPUs [24]
| GPU Model | Small: DHFR NPT (23,558 atoms) | Medium: FactorIX NPT (90,906 atoms) | Large: STMV NPT (1,067,095 atoms) |
|---|---|---|---|
| RTX 5090 | 1632.97 | 494.45 | 109.75 |
| RTX 5080 | 1468.06 | 365.36 | 63.17 |
| B200 SXM | 1447.75 | 427.26 | 114.16 |
| GH200 Superchip | 1322.17 | 206.06 | 101.31 |
Table 3: NAMD and GROMACS Historical CPU-Based Scaling (NIH HPC Benchmark) [51] [52]
| Number of Cores | NAMD: ApoA1 (92,224 atoms) days/ns [52] | GROMACS: ADH Cubic (est. ~80k atoms) ns/day [51] |
|---|---|---|
| 32 | 0.61 | ~17.80 |
| 64 | 0.31 | ~31.12 |
| 128 | 0.15 | ~44.73 |
Experimental Protocols for MD Benchmarks
Standardized methodologies ensure consistent, comparable benchmark results.
GROMACS Benchmarking Protocol
A typical GROMACS benchmark uses the gmx mdrun command with specific flags to offload computations to the GPU [50] [7].
Example Command:
Key Parameters:
- -nb gpu -pme gpu -bonded gpu -update gpu: Offload non-bonded, Particle Mesh Ewald (PME), bonded, and coordinate update calculations to the GPU [50].
- -ntomp: Sets the number of OpenMP threads per MPI process, crucial for balancing CPU-GPU load [7] [53].
- -nsteps: Defines the number of simulation steps to run [50].
Performance is measured from the log file output, which reports the simulation speed in ns/day [50].
AMBER Benchmarking Protocol
AMBER GPU benchmarks use the pmemd.cuda engine for single-GPU runs [7] [54].
Example Command for a Single GPU:
Example Command for Multiple GPUs:
The mdin input file contains all simulation parameters, such as nstlim (number of steps) and dt (time step) [54]. The performance figure (ns/day) is found in the final mdout output file [54].
NAMD Benchmarking Protocol
NAMD benchmarks, especially on GPUs, require specifying the number of CPU cores and GPUs in the command line [7].
Example Submission Script for a GPU Simulation:
Key Performance Workflow and Relationships
The diagram below illustrates the logical workflow for planning, running, and interpreting an MD benchmark.
The Scientist's Toolkit: Essential Research Reagents & Materials
This table details key components required for running and optimizing MD simulations.
Table 4: Essential Materials and Tools for MD Simulations
| Item | Function & Purpose |
|---|---|
| Molecular System (TPR/PRMTOP Files) | Input files containing the initial atomic coordinates, topology, and force field parameters for the system to be simulated [50] [54]. |
| MD Software (GROMACS/AMBER/NAMD) | The core computational engine that performs the numerical integration of Newton's equations of motion for all atoms in the system [7]. |
| GPU Accelerators | Hardware that drastically speeds up computationally intensive calculations like non-bonded force evaluations and PME [55]. |
| Benchmark Input (MDIN/Configuration Files) | Text files that define all the simulation parameters, including timestep, number of steps, and cutoff schemes [7] [54]. |
| High-Performance Computing (HPC) Cluster | A collection of networked computers that provides the necessary computational power for running large-scale or multiple simultaneous simulations [51] [53]. |
Key Insights for Researchers
- System Size Dictates Hardware Choice: Small systems are often CPU-bound due to CPU-GPU communication overhead, making powerful CPU cores important. Large systems fully utilize high-end GPUs, where performance scales with CUDA cores and memory bandwidth [50] [55].
- Consumer vs. Data Center GPUs: Consumer GPUs like the RTX 4090 can provide comparable performance to data center GPUs (e.g., A100, H100) for many systems at a fraction of the cost, offering superior cost-effectiveness (ns/dollar), especially for large models [50].
- Software-Specific Strengths: AMBER excels on single GPUs across various system sizes [24]. GROMACS shows excellent scaling on hybrid CPU-GPU systems [50] [53]. NAMD efficiently leverages multiple GPUs for large, parallel simulations [55] [52].
- Optimization is Critical: Using 4 fs time steps with hydrogen mass repartitioning can significantly increase ns/day. For GPU runs, dynamically balancing load between CPU and GPU cores (
-ntompsetting) is essential for peak performance [7] [53].
Molecular dynamics (MD) simulations are computationally intensive, and their performance is highly dependent on effective parallelization and judicious resource allocation. This guide objectively compares the parallel scaling approaches and performance of three leading MD software packages—GROMACS, AMBER, and NAMD—to help researchers avoid common performance pitfalls and optimize their simulations.
Parallelization Architectures and Scaling Behavior
The core performance differences between GROMACS, AMBER, and NAMD stem from their fundamental parallelization strategies and how they distribute computational workloads.
GROMACS: Domain Decomposition with Dynamic Load Balancing
GROMACS employs a domain decomposition strategy, where the simulation box is divided into spatial domains, each assigned to a different MPI rank [56]. This approach is highly efficient for short-range interactions due to its intrinsic locality.
- Force Calculation and Communication: The eighth-shell method minimizes communication volume by ensuring that only the necessary coordinates from neighboring domains are communicated before force calculation, with reverse communication for forces [56].
- Dynamic Load Balancing: A critical feature automatically adjusts domain volumes during a simulation if load imbalance exceeds 2%, counteracting performance loss from inhomogeneous particle distribution or interaction costs [56].
- Mixed-Mode Parallelism: Effectively combines MPI with OpenMP threading. Using 2-4 OpenMP threads per MPI rank can reduce communication needs and improve performance, especially on multi-socket nodes [57].
- PME Separation: For Particle-Mesh Ewald electrostatics, dedicating a subset of ranks (e.g., one-quarter to one-half) solely to the long-range PME calculation can significantly enhance performance by reducing communication bottlenecks in the global 3D FFT [57].
AMBER (pmemd.cuda): GPU-Accelerated with Limited Multi-GPU Scaling
AMBER's pmemd.cuda engine is predominantly optimized for single-GPU acceleration. Its parallelization strategy differs markedly from GROMACS.
- Primary GPU Focus: The core simulation runs on a single GPU, leveraging CUDA for acceleration [24].
- Limited Multi-GPU Support: Unlike GROMACS and NAMD, AMBER does not use multi-GPU acceleration for a single simulation. To utilize multiple GPUs, researchers must run independent, concurrent simulations [24].
- CPU Role: CPU and memory resources have minimal impact on simulation throughput once the simulation is loaded onto the GPU, making single-GPU performance the critical bottleneck [24].
NAMD: Charm++ Parallelization for Multi-Node Scaling
NAMD is built on the Charm++ parallel programming model, which is designed for scalable performance on multi-node systems.
- Adaptive Load Balancing: Charm++ enables sophisticated dynamic load balancing, allowing NAMD to efficiently handle inhomogeneous systems [58].
- Hybrid Parallelization: Supports multi-GPU configurations for a single simulation, distributing computation across multiple GPUs to handle larger systems [58].
- Wide Node Support: Its architecture is designed to scale effectively across many nodes in a cluster, making it suitable for very large simulations on supercomputers.
Comparative Performance Data
The following tables synthesize performance data from hardware benchmarks to illustrate how these packages perform on different GPU hardware and problem sizes.
GPU Performance Across System Sizes (ns/day)
| GPU Model | Memory | GROMACS (STMV ~1M atoms) | AMBER (STMV ~1M atoms) | NAMD (STMV ~1M atoms) |
|---|---|---|---|---|
| NVIDIA RTX 5090 | 24 GB GDDR7 | ~110 [58] | 109.75 [24] | Data Unavailable |
| NVIDIA RTX 6000 Ada | 48 GB GDDR6 | ~97 [58] | 70.97 [24] | Excellent [58] |
| NVIDIA RTX 4090 | 24 GB GDDR6X | ~100 [58] | 63.17 [24] | Excellent [58] |
| NVIDIA H100 PCIe | 80 GB HBM2e | ~101 [58] | 74.50 [24] | Data Unavailable |
Note: GROMACS and NAMD values are estimated from relative performance descriptions. AMBER values are from explicit benchmarks [24] [58].
Performance Scaling with Simulation Size on AMBER
| Benchmark System | Atoms | NVIDIA RTX 5090 | NVIDIA RTX 6000 Ada | NVIDIA H100 PCIe |
|---|---|---|---|---|
| STMV (NPT, 4fs) | 1,067,095 | 109.75 | 70.97 | 74.50 |
| Cellulose (NVE, 2fs) | 408,609 | 169.45 | 123.98 | 125.82 |
| FactorIX (NVE, 2fs) | 90,906 | 529.22 | 489.93 | 410.77 |
| DHFR (NVE, 4fs) | 23,558 | 1655.19 | 1697.34 | 1532.08 |
| Myoglobin GB (Implicit) | 2,492 | 1151.95 | 1016.00 | 1094.57 |
All values are simulation throughput in nanoseconds/day (ns/day). Data sourced from AMBER 24 benchmarks [24].
Multi-GPU Scaling Efficiency
| Software | Primary Parallelization Model | Multi-GPU for Single Simulation | Recommended Use Case |
|---|---|---|---|
| GROMACS | Hybrid MPI/OpenMP Domain Decomposition | Yes | High-throughput on mixed CPU/GPU systems |
| AMBER | Single-GPU Acceleration | No (Concurrent runs only) | Fast single-node, single-GPU simulations |
| NAMD | Charm++ | Yes | Large systems on multi-node supercomputers |
Experimental Protocols and Methodologies
The performance data presented relies on standardized benchmark systems and simulation parameters.
Benchmark Systems and Parameters
- STMV (Satellite Tobacco Mosaic Virus): A large, explicit solvent system with 1,067,095 atoms, simulated with a 4-fs time step and NPT ensemble [24].
- Cellulose: A fibrous system with 408,609 atoms, simulated with a 2-fs time step in both NVE and NPT ensembles [24].
- DHFR (Dihydrofolate Reductase): A classic benchmark protein with 23,558 atoms, simulated with a 4-fs time step in NVE and NPT ensembles [24].
- Implicit Solvent Models: Myoglobin and Nucleosome simulations used the Generalized Born (GB) model with 2,492 and 25,095 atoms, respectively [24].
GROMACS Performance Measurement Protocol
Performance tuning in GROMACS follows a logical workflow to identify the optimal run configuration, with a particular emphasis on managing domain decomposition and load balancing.
GROMACS Performance Tuning Workflow
Key performance tuning steps include:
- Domain Decomposition Grid: GROMACS automatically selects a grid, but users can manually specify the number of grid divisions (
-dd) to avoid domains that are too small, which violate the condition (LC \geq \max(r{\mathrm{mb}},r_{\mathrm{con}})) [56]. - Load Imbalance Monitoring: The log file reports load imbalance at each output step. The total performance loss due to imbalance is summarized at the end [56].
- Dynamic Load Balancing: Activated automatically, DLB can be controlled with the
-ddsoption to set the minimum allowed cell scaling factor (default 0.8) [56]. - PME Tuning: For systems around 100,000 atoms, dedicating 25-33% of ranks to PME often yields optimal performance [57].
The Scientist's Toolkit: Essential Research Reagents and Hardware
Selecting appropriate hardware and software configurations is as crucial as selecting biological reagents for a successful experiment.
Research Reagent Solutions
| Item | Function in MD Simulations | Recommendation |
|---|---|---|
| NVIDIA RTX 5090 | Consumer-grade GPU with high clock speeds for cost-effective performance [24]. | Best for AMBER, GROMACS on a budget [24] [58]. |
| NVIDIA RTX 6000 Ada | Professional workstation GPU with large VRAM for massive systems [58]. | Top for large GROMACS, NAMD simulations [58]. |
| NVIDIA RTX PRO 4500 Blackwell | Mid-range professional GPU with excellent price-to-performance [24]. | Ideal for small-medium AMBER simulations [24]. |
| AMD Threadripper PRO | High-core-count CPU with sufficient PCIe lanes for multi-GPU setups [58]. | Optimal for GROMACS/NAMD CPU parallelism [58]. |
| Dynamic Load Balancing | Automatically adjusts computational domains to balance workload [56]. | Critical for inhomogeneous GROMACS systems [56]. |
| Dedicated PME Ranks | Separates long-range electrostatic calculation to improve scaling [57]. | Use for GROMACS systems >50,000 atoms [57]. |
The performance characteristics and optimal resource requests differ significantly among these MD packages:
- Choose GROMACS for high-throughput simulation of medium to large systems on workstations or small clusters, leveraging its sophisticated domain decomposition and dynamic load balancing [56] [57].
- Choose AMBER for rapid turnaround on single-GPU workstations, especially when running multiple concurrent simulations of small to medium systems [24].
- Choose NAMD for extremely large systems requiring scaling across many nodes of a supercomputer, utilizing its Charm++ parallelization model [58].
Avoiding the common mistake of applying a one-size-fits-all resource template is crucial. By understanding each software's parallelization strategy and leveraging the performance data and tuning guidelines presented, researchers can significantly enhance simulation efficiency and accelerate scientific discovery.
Molecular dynamics (MD) simulations are indispensable in computational chemistry, biophysics, and drug discovery, enabling the study of atomic-level interactions in complex biological systems. The computational intensity of these simulations necessitates high-performance computing hardware, with modern workflows increasingly leveraging multiple graphics processing units (GPUs) to accelerate time to solution. The effective implementation of multi-GPU configurations, however, is highly dependent on the specific MD software application and its underlying parallelization algorithms.
This guide objectively compares the multi-GPU support and performance characteristics of three leading MD software packages: GROMACS, AMBER, and NAMD. We synthesize experimental benchmark data, outline best-practice methodologies, and provide hardware recommendations to help researchers optimize computational resources for specific scientific workloads.
Comparative Analysis of Multi-GPU Support
The three major MD software packages exhibit fundamentally different approaches to leveraging multiple GPUs, which directly impacts their efficiency and optimal use cases.
GROMACS: Domain Decomposition for Single Simulations
GROMACS features sophisticated multi-level parallelization and can effectively use multiple GPUs to accelerate a single, large simulation. Its performance relies on a domain decomposition algorithm, which divides the simulation box into spatial domains, each assigned to a different MPI rank. The Particle-Mesh Ewald (PME) method for long-range electrostatics can be offloaded to a subset of ranks (potentially using dedicated GPUs) for optimal load balancing [59].
For ensembles of multiple, independent simulations, GROMACS throughput can be dramatically increased by running multiple simulations per physical GPU using technologies like NVIDIA's Multi-Process Service (MPS). Benchmarks demonstrate this approach can achieve up to 1.8X improvement in overall throughput for smaller systems like the 24K-atom RNAse, and a 1.3X improvement for larger systems like the 96K-atom ADH on an NVIDIA A100 GPU [60].
AMBER: Focused Multi-GPU Applications
AMBER's approach to multiple GPUs is more specialized. The core pmemd.cuda engine is primarily optimized for single-GPU execution, and the general recommendation from developers is to "stick with single GPU runs since GPUs are now so fast that the communication between them is too slow to be effective" [61].
The multi-GPU implementation (pmemd.cuda.MPI) is recommended only for specific use cases:
- Replica exchange simulations where individual replicas run on separate GPUs
- Very large implicit solvent GB simulations (>5000 atoms)
- Thermodynamic integration calculations where different lambda windows run on different GPUs [61]
For most standard explicit solvent simulations, running independent simulations on multiple GPUs yields better overall throughput than using multiple GPUs for a single simulation [7].
NAMD: Strong Scaling Across Multiple GPUs
NAMD is designed from the ground up for parallel execution and demonstrates excellent strong scaling across multiple GPUs for single simulations. It uses a dynamic load balancing system that distributes computation—including both nonbonded forces and PME calculations—across available resources [62].
Benchmarks show NAMD can effectively utilize 2-4 GPUs for medium to large systems, though scaling efficiency decreases as more GPUs are added. For a 456K-atom Her1-Her1 membrane simulation, performance increases from approximately 21 ns/day on one RTX 6000 Ada GPU to about 65 ns/day on four GPUs, representing roughly 77% parallel efficiency [62].
Table 1: Multi-GPU Support Comparison Across MD Software Packages
| Software | Primary Multi-GPU Approach | Optimal Use Cases | Key Considerations |
|---|---|---|---|
| GROMACS | Domain decomposition for single simulations; Multiple simulations per GPU for ensembles | Large systems (>100,000 atoms); High-throughput screening | PME ranks should be ~1/4 to 1/2 of total ranks; MPS can significantly boost throughput for small systems |
| AMBER | Multiple independent simulations; Specialized methods (REMD, TI) | Replica exchange; Thermodynamic integration; Large implicit solvent | Single simulations generally do not scale beyond 1 GPU; Multi-GPU recommended only for specific algorithms |
| NAMD | Strong scaling across multiple GPUs for single simulations | Medium to large biomolecular systems; Membrane proteins | Shows good scaling up to 4 GPUs; Dynamic load balancing adapts to system heterogeneity |
Quantitative Performance Benchmarks
Synthesized benchmark data reveals how each application performs across different hardware configurations and system sizes.
AMBER Performance Across GPU Architectures
Recent AMBER 24 benchmarks across various NVIDIA GPU architectures show performance characteristics for different simulation sizes [24]:
Table 2: AMBER 24 Performance (ns/day) on Select NVIDIA GPUs
| GPU Model | STMV (1.06M atoms) | Cellulose (408K atoms) | Factor IX (90K atoms) | DHFR (23K atoms) | Myoglobin GB (2.5K atoms) |
|---|---|---|---|---|---|
| RTX 5090 | 109.75 | 169.45 | 529.22 | 1655.19 | 1151.95 |
| RTX 6000 Ada | 70.97 | 123.98 | 489.93 | 1697.34 | 1016.00 |
| RTX 5000 Ada | 55.30 | 95.91 | 406.98 | 1562.48 | 841.93 |
| B200 SXM | 114.16 | 182.32 | 473.74 | 1513.28 | 1020.24 |
| GH200 Superchip | 101.31 | 167.20 | 191.85 | 1323.31 | 1159.35 |
The benchmarks confirm that for AMBER, running multiple independent simulations—each on a single GPU—typically yields better aggregate throughput than using multiple GPUs for a single simulation [24].
NAMD Multi-GPU Scaling Performance
NAMD benchmarks demonstrate its ability to effectively leverage multiple GPUs for single simulations [62]:
Table 3: NAMD Multi-GPU Scaling on Intel Xeon W9-3495X with RTX 6000 Ada GPUs
| Number of GPUs | Performance (ns/day) | Scaling Efficiency |
|---|---|---|
| 1 GPU | 21.21 | 100% |
| 2 GPUs | 38.15 | 90% |
| 4 GPUs | 65.40 | 77% |
These results were obtained using a 456K-atom membrane protein system, representing a typical large biomolecular simulation where multi-GPU parallelization becomes beneficial.
GROMACS Multi-Simulation Throughput
GROMACS exhibits remarkable throughput when running multiple simulations per GPU, particularly for smaller systems [60]:
Table 4: GROMACS Aggregate Throughput on 8-GPU DGX A100 Server
| Simulations per GPU | RNAse (24K atoms) ns/day | ADH (96K atoms) ns/day |
|---|---|---|
| 1 | 8,664 | 3,024 |
| 4 | 12,096 | 3,528 |
| 16 | 15,120 | 3,780 |
| 28 | 15,552 | 3,456 |
The peak throughput improvement for RNAse reaches 1.8X with 28 simulations per GPU (using MIG + MPS), while the larger ADH system shows a 1.3X improvement with 16 simulations per GPU (using MPS alone).
Experimental Protocols and Methodologies
GROMACS Multi-Simulation Configuration
To achieve optimal multi-GPU performance with GROMACS, specific configuration protocols are recommended:
For single simulations across multiple GPUs:
This configuration uses 2 MPI ranks (one per GPU) with 12 OpenMP threads each, offloading nonbonded, PME, and update/constraints to the GPU [7].
For multiple simulations per GPU using MPS: The NVIDIA Multi-Process Service must be enabled to allow multiple processes to share a single GPU concurrently. The optimal number of simulations per GPU depends on system size and available GPU memory, typically ranging from 2-8 for current generation GPUs [60].
AMBER Multi-GPU Setup
For AMBER, the multi-GPU configuration is only recommended for specific algorithms like replica exchange:
The explicit note from AMBER developers bears repeating: "The general recommendation is if you have 4 GPUs it's better to run 4 independent simulations than try to run a single slightly longer simulation on all 4 GPUs" [61].
NAMD Multi-GPU Execution
NAMD's multi-GPU configuration utilizes a different approach:
NAMD automatically detects available GPUs and distributes computation across them, with performance tuning primarily involving the allocation of CPU cores to manage the GPU workloads [7].
Conceptual Workflow and Parallelization Strategies
The following diagram illustrates the fundamental multi-GPU approaches employed by GROMACS, AMBER, and NAMD:
Diagram 1: Multi-GPU Parallelization Approaches in MD Software
Essential Research Toolkit
Hardware Recommendations
Based on comprehensive benchmarking, the following GPU configurations are recommended for multi-GPU MD workflows:
Table 5: Recommended GPU Configurations for Multi-GPU MD Simulations
| Use Case | Recommended GPU(s) | Key Features | Rationale |
|---|---|---|---|
| Cost-Effective GROMACS/NAMD | 4x RTX A4500 | 20GB VRAM, Moderate Power | Best performance per dollar, high scalability in multi-GPU servers [62] |
| High-Throughput AMBER | 2-4x RTX 5090 | 32GB GDDR7, High Clock Speed | Excellent single-GPU performance for multiple independent simulations [24] |
| Large System GROMACS/NAMD | 4x RTX 6000 Ada | 48GB VRAM, ECC Memory | Large memory capacity for massive systems, professional driver support [63] |
| Mixed Workload Server | 8x RTX PRO 4500 Blackwell | 24GB VRAM, Efficient Cooling | Balanced performance and density for heterogeneous research workloads [24] |
Software and Environment Configuration
- NVIDIA MPS (Multi-Process Service): Essential for running multiple GROMACS simulations per GPU, enabling up to 1.8X throughput improvement for small systems [60]
- NVIDIA MIG (Multi-Instance GPU): Useful for partitioning large GPUs (A100, H100) among multiple users or workload types [60]
- CUDA 12.0+: Required for optimal performance on Ada Lovelace and Blackwell architecture GPUs [24] [62]
- OpenMPI 4.0.3+: Provides optimal performance for multi-node multi-GPU communication in GROMACS and NAMD [7]
The optimal multi-GPU configuration for molecular dynamics simulations depends critically on the specific software application and research objectives. GROMACS offers the most flexible approach, efficiently utilizing multiple GPUs for both single large simulations and high-throughput ensembles. NAMD demonstrates excellent strong scaling across multiple GPUs for single simulations of medium to large biomolecular systems. AMBER benefits least from traditional multi-GPU parallelization for single simulations, instead achieving maximum throughput by running independent simulations on separate GPUs, with specialized multi-GPU support reserved for replica exchange and thermodynamic integration.
Researchers should carefully consider their primary workflow—whether it involves few large systems or many smaller systems—when selecting both software and hardware configurations. The benchmark data and methodologies presented here provide a foundation for making informed decisions that maximize research productivity and computational efficiency.
Ensuring Scientific Rigor: Reproducibility, Validation, and Future Trends
Validating Simulations Against Experimental Observables
Molecular dynamics (MD) simulations provide atomic-level insights into biological processes, but their predictive power hinges on the ability to validate results against experimental observables. For researchers selecting between major MD software packages—GROMACS, AMBER, and NAMD—understanding their performance characteristics, specialized capabilities, and validation methodologies is crucial for generating reliable, reproducible data. This guide objectively compares these tools through current benchmark data and experimental protocols.
Raw performance, measured in nanoseconds of simulation completed per day (ns/day), directly impacts research throughput. The following tables summarize performance data across different hardware and system sizes.
Table 1: AMBER 24 Performance on Select NVIDIA GPUs (Simulation Size: ns/day) [24]
| GPU Model | STMV (1M atoms) | Cellulose (408K atoms) | Factor IX (90K atoms) | DHFR (23K atoms) |
|---|---|---|---|---|
| NVIDIA RTX 5090 | 109.75 | 169.45 | 529.22 | 1655.19 |
| NVIDIA RTX 5080 | 63.17 | 105.96 | 394.81 | 1513.55 |
| NVIDIA GH200 Superchip | 101.31 | 167.20 | 191.85 | 1323.31 |
| NVIDIA B200 SXM | 114.16 | 182.32 | 473.74 | 1513.28 |
| NVIDIA H100 PCIe | 74.50 | 125.82 | 410.77 | 1532.08 |
Table 2: Relative Performance and Characteristics of MD Software
| Software | Primary Performance Strength | Key Hardware Consideration | Scalability |
|---|---|---|---|
| GROMACS | High speed for most biomolecular simulations on CPUs and GPUs [3] | AMD GPU support via HIP port or SCALE platform (for CUDA code) [41] | Highly scalable across CPU cores; multi-GPU support for single simulations [40] |
| AMBER | Optimized for single-GPU performance; efficient for multiple concurrent simulations [24] | Best performance with latest NVIDIA GPUs (e.g., RTX 50-series); does not use multi-GPU for a single calculation [24] | Run multiple independent simulations in parallel on multiple GPUs [24] |
| NAMD | Designed for high-performance simulation of large biomolecular systems [64] | Charm++ parallel objects enable scaling to hundreds of thousands of CPU cores [64] | Excellent strong scaling for very large systems on CPU clusters [64] |
For GROMACS, independent benchmarks show that performance varies significantly with the CPU architecture and core count. For instance, high-end server CPUs can achieve performance ranging from under 1 ns/day to over 18 ns/day on the water_GMX50 benchmark, with performance generally scaling well with increasing core counts [65].
Specialized Capabilities for Validation
Beyond raw speed, the specialized features of each package directly impact the types of experimental observables that can be effectively validated.
Force Field Accuracy and Development (AMBER): AMBER is renowned for its accurate and well-validated force fields [3]. Its framework is frequently used to develop and validate new force fields for specific systems, such as metals in proteins. For example, a 2025 study developed a new polarized force field for cadmium-binding proteins involving cysteine and histidine, which was validated against quantum mechanics/molecular mechanics (QM/MM) calculations and showed strong agreement with experimental crystal structures by preserving tetra-coordination geometry with mean distances under 0.3 Å from reference data [66].
Advanced Sampling and Enhanced Visualization (NAMD): NAMD integrates robust support for collective variable (colvar) methods, which are crucial for studying processes like protein folding or ligand binding. These methods are considered more mature and robust in NAMD compared to GROMACS [3]. Furthermore, NAMD's deep integration with the visualization program VMD provides a superior environment for setting up simulations, analyzing trajectories, and visually comparing simulation results with experimental structures [3] [64]. This tight workflow was instrumental in a study of staph infection adhesins, where GPU-accelerated NAMD simulations combined with atomic force microscopy explained the resilience of pathogen adhesion under force [64].
Performance and Accessibility (GROMACS): GROMACS stands out for its computational speed, open-source nature, and extensive tutorials, making it beginner-friendly and highly efficient for standard simulations [3]. Recent versions have focused on performance optimizations, such as offloading more calculations (including update and constraints) to the GPU and fusing bonded kernels, which can lead to performance improvements of up to a factor of 2.5 for specific non-bonded free-energy calculations [40].
Experimental Protocols for Validation
The following workflow diagrams and detailed protocols illustrate how these tools are used in practice to validate simulations against experimental data.
Diagram 1: The standard workflow for running and validating an MD simulation. The critical validation step involves comparing simulation-derived observables with experimental data.
Protocol: Validating a Metal-Binding Site with AMBER
This protocol is based on a 2025 study that developed a force field for cadmium-binding proteins [66].
- Objective: To validate the structure and dynamics of a cadmium(II)-binding site in a protein against known crystal structures.
- System Setup:
- Initial Structure: Obtain a PDB file of a cysteine and histidine cadmium-binding protein.
- Force Field: Apply the newly developed AMBER force field parameters for cadmium(II), cysteine, and histidine, which include polarized atomic charges derived from QM calculations [66].
- Solvation: Solvate the protein in a TIP3P water box with a minimum 10 Å distance from the protein.
- Neutralization: Add counterions (e.g., Na⁺ or Cl⁻) to neutralize the system's charge.
- Simulation Parameters:
- QM/MM MD: Perform quantum mechanics/molecular dynamics (QM/MM) simulations, treating the metal center and its immediate ligands with QM and the rest of the system with MM [66].
- Periodic Boundary Conditions: Use periodic boundary conditions.
- Electrostatics: Use the Particle Mesh Ewald (PME) method for long-range electrostatics.
- Temperature & Pressure: Maintain temperature at 300 K using a Langevin thermostat and pressure at 1 bar using a Berendsen barostat.
- Production Run: Run the simulation for a sufficient time to ensure stability (e.g., >100 ns).
- Validation Metrics:
- Geometry: Calculate the mean distance between the cadmium ion and the coordinating atoms (N and S). A successful validation shows a mean distance of less than 0.3 Å from the crystal structure reference [66].
- Stability: Monitor the root-mean-square deviation (RMSD) of the metal-binding site to ensure the tetra-coordination is preserved throughout the simulation.
Protocol: Validating a Protein-Ligand Interaction with NAMD and VMD
This protocol leverages NAMD's strengths in visualization and analysis [3] [64].
- Objective: To validate the binding mode and stability of a ligand within a protein's active site.
- System Setup:
- Initial Structure: Obtain a protein-ligand complex from a crystal structure or docking study.
- Force Field: Use the CHARMM36 force field for the protein and the CGenFF tool to generate parameters for the ligand.
- Solvation and Neutralization: Follow similar steps as in the AMBER protocol.
- Simulation Parameters:
- Enhanced Sampling: If studying binding/unbinding, use the colvars module in NAMD to set up metadynamics or umbrella sampling [3].
- Standard MD: Otherwise, run a conventional MD simulation with PME and controls for temperature and pressure.
- Production Run: Run one or more replicas of the simulation.
- Validation Metrics (Analyzed in VMD):
- Binding Pose: Visualize the trajectory to see if the ligand remains in the crystallographic pose. Calculate the ligand RMSD relative to the starting structure.
- Interaction Analysis: Use VMD's built-in tools to measure specific protein-ligand interactions (hydrogen bonds, salt bridges, hydrophobic contacts) over time and compare the prevalence to the crystal structure.
- MM/PBSA Calculations: Perform Molecular Mechanics/Poisson-Boltzmann Surface Area calculations to estimate the binding free energy, which can be compared with experimental values (e.g., from ITC or SPR).
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for MD Validation
| Item | Function in Validation |
|---|---|
| Benchmark Suite (e.g., AMBER 24 Suite, Max Planck Institute GROMACS benchmarks) | Provides standardized test cases (e.g., STMV, DHFR) to compare software performance and hardware efficiency objectively [24]. |
| Visualization Software (VMD) | Crucial for visual analysis of trajectories, setting up simulations, and directly comparing simulation snapshots with experimental structures [3] [64]. |
| Force Field Parameterization Tools (e.g., CGenFF, Antechamber) | Generates necessary force field parameters for non-standard molecules like novel drug ligands, which is a prerequisite for accurate simulation [3]. |
| Collective Variable Module (colvars in NAMD) | Enables advanced sampling techniques to study rare events (e.g., ligand unbinding) and compute free energies, providing data to compare with kinetic experiments [3] [64]. |
| QM/MM Software (e.g., included in AMBER tools) | Allows for more accurate treatment of metal ions or chemical reactions in a biological context, providing a higher level of theory for validating metalloprotein structures [66]. |
Diagram 2: A conceptual map linking common types of experimental data with the corresponding observables that can be calculated from an MD simulation for validation purposes. AFM: Atomic Force Microscopy; ITC: Isothermal Titration Calorimetry; SPR: Surface Plasmon Resonance.
The choice between GROMACS, AMBER, and NAMD for validating simulations is not a matter of which is universally best, but which is most appropriate for the specific research problem. GROMACS offers top-tier performance and accessibility for standard simulations. AMBER provides highly validated force fields and a robust environment for specialized parameterization, particularly for non-standard residues and metal ions. NAMD excels at simulating massive systems on large CPU clusters and, coupled with VMD, offers an unparalleled workflow for visual analysis and advanced sampling. A well-validated study will select the software whose strengths align with the system's size, the required force field, the necessary sampling methods, and the experimental observables being targeted for comparison.
Comparative Analysis of Conformational Sampling and Unfolding Behavior
Molecular dynamics (MD) simulation serves as a "virtual molecular microscope," enabling researchers to observe protein dynamics with atomistic detail. This computational technique has become indispensable for studying fundamental biological processes, including protein folding, conformational changes, and molecular recognition—events that are often difficult to capture with traditional biophysical methods. Within this field, three software packages have emerged as prominent tools: GROMACS, AMBER, and NAMD. Each offers distinct approaches to simulating biomolecular systems, with particular strengths and limitations in conformational sampling and characterizing unfolding behavior.
Understanding the relative performance of these packages is critical for researchers investigating protein dynamics, especially those in drug development who rely on accurate predictions of molecular behavior. This guide provides an objective comparison of GROMACS, AMBER, and NAMD, drawing on experimental data and established best practices to inform software selection for specific research scenarios. The analysis focuses particularly on two challenging aspects of biomolecular simulation: efficiently sampling conformational space and accurately modeling unfolding processes, both essential for advancing computational structural biology.
Each MD software package has evolved with different philosophical approaches, optimization strategies, and target applications, leading to distinctive performance characteristics.
AMBER (Assisted Model Building with Energy Refinement) has been developed since the late 1970s with a strong focus on accuracy in biomolecular simulations, particularly for proteins and nucleic acids. It is renowned for its sophisticated force fields (e.g., ff14SB, GAFF) that are often considered the gold standard for biological systems. AMBER has historically been optimized for CPU architecture, though recent versions have made significant strides in GPU acceleration [2].
GROMACS (GROningen MAchine for Chemical Simulations) emerged in the early 1990s and has become celebrated for its exceptional speed and efficiency in parallel computations. Its optimization for both CPU and GPU architectures makes it one of the fastest MD engines available, particularly advantageous for large-scale simulations and high-throughput studies. GROMACS distinguishes itself with support for multiple force fields (AMBER, CHARMM, OPLS) and strong community support with extensive documentation [2].
NAMD (Nanoscale Molecular Dynamics), not as extensively covered in the search results but mentioned in comparative studies, is recognized for its strong parallel scaling capabilities and excellent integration with the visualization tool VMD. It is particularly effective for large, complex systems including membrane proteins and massive biomolecular complexes [3] [67].
Table 1: Core Characteristics of MD Software Packages
| Feature | AMBER | GROMACS | NAMD |
|---|---|---|---|
| Primary Strength | High-accuracy force fields | Speed and scalability | Parallel scaling and visualization |
| Force Field Specialization | AMBER family (ff14SB, GAFF) | Multi-force field support | CHARMM force fields |
| Hardware Optimization | CPU, with recent GPU advances | CPU and GPU | High-performance GPUs |
| Learning Curve | Steeper | More beginner-friendly | Moderate |
| Licensing | Some tools require license | Open-source | Free for non-commercial use |
| Best For | Detailed biomolecular studies | Large systems & high-throughput | Large complexes & membrane proteins |
Performance Comparison in Conformational Sampling
The ability to efficiently and accurately sample protein conformational space varies significantly across MD packages due to differences in their underlying algorithms, integration methods, and handling of non-bonded interactions.
Sampling Efficiency and Convergence
A critical validation study compared four MD packages (AMBER, GROMACS, NAMD, and ilmm) using best practices established by their respective developers. The research employed three different protein force fields and water models to simulate two structurally distinct proteins: the Engrailed homeodomain (EnHD) and Ribonuclease H (RNase H). The findings revealed that while all packages reproduced experimental observables equally well at room temperature overall, subtle differences emerged in underlying conformational distributions and sampling extent [6] [67].
GROMACS consistently demonstrates superior performance in raw simulation speed, making it particularly advantageous for projects requiring extensive sampling or high-throughput simulations. This performance advantage becomes especially pronounced for larger systems (>100,000 atoms) where parallel efficiency is critical. AMBER, while historically optimized for single-node CPU performance, has narrowed this gap with its GPU implementations, though may still lag behind GROMACS for very large systems [2].
Accuracy of Conformational Ensembles
Despite using the same force field (AMBER ff99SB-ILDN), simulations run in different packages produced variations in conformational sampling. These observations highlight that force fields alone do not determine simulation outcomes; algorithmic differences in handling constraints, water models, and integration methods significantly influence results [6].
In studies of intrinsically disordered proteins and unfolded states, AMBER force fields have shown a slightly higher β-sheet propensity compared to CHARMM equivalents, which may influence the observed conformational ensembles in sampling simulations. This suggests that force field selection interacts with software-specific implementations to determine ultimate accuracy [68].
Table 2: Quantitative Performance Metrics in Conformational Sampling
| Metric | AMBER | GROMACS | NAMD |
|---|---|---|---|
| Simulation Speed (ns/day) | Moderate to High (GPU-dependent) | Very High | High (scales with cores) |
| Strong Scaling Efficiency | Good | Excellent | Excellent |
| Memory Usage | Moderate | Low to Moderate | Moderate to High |
| Sampling Breadth | High accuracy for biomolecules | Broad, force field dependent | Broad, CHARMM optimized |
| Unfolded State Dynamics | Slight β-preference | Force field dependent | Balanced with CHARMM36 |
| Community Resources | Specialized, detailed | Extensive, beginner-friendly | VMD integration |
Unfolding Behavior and Enhanced Sampling
Protein unfolding simulations present particular challenges due to the high energy barriers and slow timescales involved. The performance differences between MD packages become more pronounced when simulating these large-amplitude motions.
Thermal Unfolding Simulations
In comparative studies of thermal unfolding at 498K, results diverged more significantly across packages than in room-temperature simulations. Some packages failed to allow proteins to unfold properly at high temperatures or produced results inconsistent with experimental expectations. This suggests that differences in temperature coupling methods and integration algorithms significantly impact the simulation of extreme conformational changes [6].
Enhanced Sampling Methods
Each package offers specialized approaches for accelerating rare events like unfolding transitions:
GROMACS includes robust implementations of enhanced sampling methods, including umbrella sampling (discussed in detail below) and temperature replica-exchange MD (T-REMD). Its pull code provides versatile options for defining reaction coordinates and applying biasing potentials [69].
AMBER excels in advanced sampling techniques like free energy calculations and hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) simulations. These capabilities make it particularly strong for studying chemical reactions in biological contexts and detailed binding interactions [2].
NAMD offers mature collective variable (colvar) implementations with significant robustness and flexibility, allowing complex reaction coordinates for studying unfolding pathways [3].
Diagram 1: Enhanced Sampling Workflow for Unfolding Studies
Experimental Protocols and Methodologies
To ensure reproducible and comparable results across different MD packages, researchers must follow standardized protocols while respecting software-specific requirements.
System Preparation and Equilibration
A typical protocol for unfolding studies involves these key stages, adapted from comparative studies [6] [68]:
Initial Structure Preparation: Obtain coordinates from protein data banks, add hydrogen atoms appropriate for physiological pH, and assign protonation states.
Solvation: Solvate the protein in an explicit water box (e.g., TIP3P, TIP4P-EW) with a minimum 10-12 Å buffer between the protein and box edge.
Neutralization: Add counterions (e.g., Na+, Cl-) to achieve system electroneutrality.
Energy Minimization: Perform steepest descent or conjugate gradient minimization until forces fall below a threshold (typically 1000 kJ/mol/nm).
Equilibration:
- NVT equilibration (100-500 ps) with position restraints on protein heavy atoms
- NPT equilibration (500 ps - 2 ns) without restraints to achieve proper density
Production Simulation: Run extended simulations (typically 100 ns - 1 μs) using appropriate thermodynamic ensembles.
Umbrella Sampling Protocol
For quantifying free energy changes during unfolding, umbrella sampling provides a rigorous approach. The GROMACS implementation exemplifies this method [69]:
Reaction Coordinate Selection: Define a suitable collective variable (e.g., distance between molecular centers of mass, radius of gyration).
Steered MD Simulation: Perform a non-equilibrium "pulling" simulation to generate configurations along the reaction coordinate.
Window Selection: Choose 10-50 overlapping windows along the reaction coordinate to ensure sufficient overlap in sampled distributions.
Umbrella Simulations: Run independent simulations at each window with harmonic restraints (typically 1000-5000 kJ/mol/nm²) to maintain position.
WHAM Analysis: Use the Weighted Histogram Analysis Method to reconstruct the potential of mean force from all window data.
Diagram 2: Umbrella Sampling Methodology
Research Reagent Solutions
Successful MD simulations require both software tools and specialized "research reagents" - the force fields, water models, and analysis tools that form the foundation of computational experiments.
Table 3: Essential Research Reagents for MD Simulations
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Protein Force Fields | AMBER ff19SB, CHARMM36, OPLS-AA/M | Define atomic interactions and potentials governing protein dynamics |
| Nucleic Acid Force Fields | AMBER OL3, CHARMM36 | Specialized parameters for DNA/RNA simulations |
| Water Models | TIP3P, TIP4P-EW, SPC/E | Represent solvation effects with different accuracy/computational cost |
| Ion Parameters | Joung-Cheatham, Aqvist | Models for physiological ion behavior in solution |
| Enhanced Sampling Algorithms | Umbrella Sampling, Metadynamics, REPLICA | Accelerate rare events and improve conformational sampling |
| Analysis Tools | MDAnalysis, VMD, CPPTRAJ | Process trajectory data and calculate experimental observables |
The comparative analysis of GROMACS, AMBER, and NAMD reveals a nuanced landscape where software selection should be guided by specific research priorities rather than absolute performance rankings.
For investigations requiring the highest accuracy in biomolecular force fields and specialized simulations such as protein-ligand interactions, nucleic acid dynamics, and QM/MM studies, AMBER remains the preferred choice. Its carefully validated parameters and focus on biological systems provide confidence in results, particularly for publishing force field developments or detailed mechanistic studies.
When computational throughput, scalability, and efficiency are paramount, particularly for large systems or high-throughput screening, GROMACS offers superior performance. Its versatility in supporting multiple force fields, strong community support, and exceptional parallel efficiency make it ideal for researchers needing to maximize sampling within limited computational resources.
NAMD excels in scenarios requiring advanced visualization capabilities and robust collective variable implementations, particularly for complex systems like membrane proteins and large biomolecular complexes where its integration with VMD provides significant workflow advantages.
Ultimately, all three packages can produce scientifically valid results when used with appropriate best practices. The convergence of results across packages when using similar force fields and simulation parameters should increase confidence in MD simulations as a robust tool for exploring protein conformational landscapes and unfolding behavior. Future developments in force fields, enhanced sampling algorithms, and hardware acceleration will continue to refine these tools, pushing the boundaries of what can be simulated at atomistic resolution.
Market Trends and Growing Adoption in Pharmaceutical R&D
Molecular dynamics (MD) simulations have become indispensable in pharmaceutical R&D, enabling researchers to study drug-target interactions, predict binding affinities, and elucidate biological mechanisms at an atomic level. Among the numerous MD software available, GROMACS, AMBER, and NAMD stand out as the most widely adopted in the industry. This guide provides an objective, data-driven comparison of their performance, supported by experimental data and detailed methodologies, to inform researchers and drug development professionals.
The table below summarizes the primary characteristics, strengths, and predominant use cases for GROMACS, AMBER, and NAMD within pharmaceutical research.
Table 1: Core Software Characteristics and Pharmaceutical R&D Applications
| Software | Primary Strength | Licensing Model | Key Pharmaceutical R&D Use Cases |
|---|---|---|---|
| GROMACS | High simulation speed, superior parallelization, and cost-efficiency [3] | Open-source [3] | High-throughput virtual screening, protein-ligand dynamics, membrane protein simulations [70] |
| AMBER | Superior force field accuracy for biomolecules, specialized advanced free energy methods [3] | Requires a license for the full suite (commercial use) [3] | Binding free energy calculations (MM/GBSA, MMPBSA), lead optimization, FEP studies [3] |
| NAMD | Exceptional scalability for massive systems, superior visualization integration [3] | Free for non-commercial use | Simulation of large complexes (viral capsids, ribosomes), cellular-scale models [62] |
Performance Benchmarking and Experimental Data
Performance varies significantly based on the simulation size, hardware configuration, and specific algorithms used. The following data, gathered from public benchmarks and hardware vendors, provides a comparative overview.
Throughput Performance on Standard Benchmark Systems
The performance metric "nanoseconds per day" (ns/day) indicates how much simulated time a software can compute in a 24-hour period, with higher values being better. The data below shows performance across different system sizes and hardware.
Table 2: Performance Benchmark (ns/day) on STMV System (~1 Million Atoms) [24] [71]
| GPU Model | AMBER 24 (pmemd.cuda) | NAMD 3.0 |
|---|---|---|
| NVIDIA RTX 5090 | 109.75 ns/day | Data Pending |
| NVIDIA RTX 6000 Ada | 70.97 ns/day | 21.21 ns/day [62] |
| NVIDIA RTX 4090 | Data Pending | 19.87 ns/day [62] |
| NVIDIA H100 PCIe | 74.50 ns/day | 17.06 ns/day [62] |
Table 3: Performance Benchmark (ns/day) on Mid-Sized Systems (20k-90k Atoms) [24]
| GPU Model | AMBER 24 (FactorIX ~91k atoms) | AMBER 24 (DHFR ~24k atoms) |
|---|---|---|
| NVIDIA RTX 5090 | 529.22 ns/day (NVE) | 1655.19 ns/day (NVE) |
| NVIDIA RTX 6000 Ada | 489.93 ns/day (NVE) | 1697.34 ns/day (NVE) |
| NVIDIA RTX 5000 Ada | 406.98 ns/day (NVE) | 1562.48 ns/day (NVE) |
Performance per Dollar Analysis
For research groups operating with budget constraints, the cost-to-performance ratio is a critical factor.
Table 4: NAMD Performance per Dollar Analysis (Single GPU, 456k Atom Simulation) [62]
| GPU Model | Performance (ns/day) | Approximate MSRP | Performance per Dollar (ns/day/$) |
|---|---|---|---|
| RTX 4080 | 19.82 | $1,200 | 0.0165 |
| RTX A4500 | 13.00 | $1,000 | 0.0130 |
| RTX 4090 | 19.87 | $1,599 | 0.0124 |
| RTX A5500 | 16.39 | $2,500 | 0.0066 |
| RTX 6000 Ada | 21.21 | $6,800 | 0.0031 |
Experimental Protocols and Methodologies
To ensure the reproducibility of simulations and the validity of comparative benchmarks, standardized protocols are essential. The following section outlines common methodologies for performance evaluation.
Standardized Benchmarking Workflow
The diagram below illustrates a generalized workflow for setting up, running, and analyzing an MD simulation benchmark, applicable to all three software packages.
General MD Benchmarking Workflow
Detailed Software-Specific Execution Protocols
The execution commands and resource allocation differ significantly between packages. The protocols below are derived from real-world cluster submission scripts [7].
GROMACS Execution Protocol
- Use Case: High-throughput production simulation on a single GPU.
- Key Flags:
-nb gpu -pme gpu -update gpuoffloads non-bonded, Particle Mesh Ewald, and coordinate update tasks to the GPU for maximum performance [40] [7]. - Example Slurm Script:
AMBER Execution Protocol
- Use Case: Standard production simulation on a single GPU.
- Key Note: AMBER's multi-GPU version (
pmemd.cuda.MPI) is designed for running multiple independent simulations (e.g., replica exchange), not for accelerating a single simulation [7] [24]. - Example Slurm Script:
NAMD Execution Protocol
- Use Case: Leveraging multiple GPUs for a single, large simulation.
- Key Note: NAMD efficiently distributes computation across multiple GPUs, which is crucial for scaling to very large system sizes [62] [7].
- Example Slurm Script:
Hardware Selection and Optimization
Choosing the right hardware is critical for maximizing simulation throughput. The following diagram guides the selection of an optimal configuration based on research needs and constraints.
Hardware and Software Selection Guide
CPU and GPU Recommendations
- CPU: Prioritize processors with high clock speeds over extreme core counts. Mid-tier workstation CPUs like the AMD Threadripper PRO 5995WX offer a good balance of cores and speed. Single-CPU configurations are generally recommended to avoid performance bottlenecks from dual-CPU interconnects [62] [70].
- GPU: The choice depends on the software and simulation size.
- AMBER: For large simulations, the NVIDIA RTX 6000 Ada (48 GB VRAM) is ideal. For cost-effective performance on smaller systems, the NVIDIA RTX 5090 is excellent [70] [24].
- GROMACS: The NVIDIA RTX 4090/5090 offers high CUDA core counts and excellent price-to-performance for computationally intensive simulations [70].
- NAMD: The NVIDIA RTX 6000 Ada provides peak speed, while a 4x RTX A4500 setup offers the best performance per dollar for parallel simulations [62].
The Scientist's Toolkit: Essential Research Reagent Solutions
Beyond the simulation engine itself, a successful MD study relies on a suite of ancillary tools and "reagents" for system preparation and analysis.
Table 5: Essential Tools and Resources for Molecular Dynamics
| Tool/Resource | Function | Common Examples |
|---|---|---|
| Force Fields | Mathematical parameters defining interatomic potentials. | AMBER [3], CHARMM36 [3], OPLS-AA |
| Visualization Software | Graphical analysis of trajectories and molecular structures. | VMD (tightly integrated with NAMD) [3], PyMol, ChimeraX |
| System Preparation Tools | Adds solvent, ions, and parameterizes small molecules/ligands. | AmberTools (for AMBER) [7], CHARMM-GUI, pdb2gmx (GROMACS) |
| Topology Generators | Creates molecular topology and parameter files for ligands. | ACPYPE, CGenFF (for CHARMM) [3], tleap (AMBER) |
| Accelerated Computing Hardware | Drastically reduces simulation time from months/weeks to days/hours. | NVIDIA GPUs (RTX 4090, RTX 6000 Ada) [70], NVLink [40] |
The Impact of Machine Learning and AI on Future MD Workflows
Molecular dynamics (MD) simulations stand as a cornerstone in computational chemistry, biophysics, and drug development, enabling the study of physical movements of atoms and molecules over time. The predictive capacity of traditional MD methodology, however, is fundamentally limited by the large timescale gap between the complex processes of interest and the short simulation periods accessible, largely due to rough energy landscapes characterized by numerous hard-to-cross energy barriers [72]. Artificial Intelligence and Machine Learning are fundamentally transforming these workflows by providing a systematic means to differentiate signal from noise in simulation data, thereby discovering relevant collective variables and reaction coordinates to accelerate sampling dramatically [72]. This evolution is transitioning MD from a purely simulation-based technique to an intelligent, automated, and predictive framework that can guide its own sampling process, promising to unlock new frontiers in the study of protein folding, ligand binding, and materials science.
Comparative Performance Analysis of MD Software
Performance and Scalability Characteristics
The effective integration of AI and ML techniques into future MD workflows will build upon the established performance profiles of the major MD software packages. Understanding their current computational efficiency, scaling behavior, and hardware utilization is paramount for selecting the appropriate platform for AI-augmented simulations.
Table 1: Comparative Performance Characteristics of GROMACS, AMBER, and NAMD
| Feature | GROMACS | AMBER | NAMD |
|---|---|---|---|
| Computational Speed | High performance, particularly on GPU hardware [3] | Strong performance, especially with its GPU-optimized PMEMD [7] | Competitive performance, with some users reporting superior performance on high-end GPUs [3] |
| GPU Utilization | Excellent; mature mixed-precision CUDA path with flags -nb gpu -pme gpu -update gpu [5] |
Optimized for NVIDIA GPUs via PMEMD.CUDA; multi-GPU typically for replica exchange only [7] | Efficient GPU use; supports multi-GPU setups via Charm++ parallel programming model [7] [73] |
| Multi-GPU Scaling | Good scaling with multiple GPUs [7] [73] | Limited; a single simulation typically does not scale beyond 1 GPU [7] | Excellent distribution across multiple GPUs [73] |
| Force Fields | Compatible with various force fields; often used with CHARMM36 [3] | Particularly known for its accurate force fields (e.g., ff19SB) [3] | Supports common force fields; often used with CHARMM [3] |
| Learning Curve | Beginner-friendly with great tutorials and workflows [3] | Steeper learning curve; some tools require licenses [3] | Intermediate; benefits from strong VMD integration [3] |
| Key Strength | Speed, versatility, and open-source nature [3] | Force field accuracy and well-validated methods [3] | Strong visualization integration and scalable architecture [3] |
AI and Enhanced Sampling Readiness
Each MD package presents different advantages for integration with AI methodologies. GROMACS's open-source nature and extensive community facilitate the rapid implementation and testing of new AI algorithms [3]. AMBER's well-established force fields provide an excellent foundation for generating high-quality training data for ML potentials [3]. NAMD's robust collective variable (colvar) implementation and integration with visualization tools like VMD offer superior capabilities for analyzing and interpreting AI-derived reaction coordinates [3]. Christopher Stepke notes that "the implementation of collective variable methods in GROMACS is relatively recent, while their utilization in NAMD is considerably more robust and mature" [3], highlighting a crucial consideration for AI-enhanced sampling workflows that depend heavily on accurate collective variable definition.
AI-Driven Enhanced Sampling Methodologies
The Data Sparsity Challenge in MD
Traditional AI applications thrive in data-rich environments, but MD simulations per construction suffer from limited sampling and thus limited data [72]. This creates a fundamental problem where AI optimization can get stuck in spurious regimes, leading to incorrect characterization of the reaction coordinate. When such an incorrect RC is used to perform additional simulations, researchers can progressively deviate from the ground truth [72]. This dangerous situation is analogous to a self-driving car miscategorizing a "STOP" sign, resulting in catastrophic failure of the intended function [72].
Spectral Gap Optimization Framework
To address the challenge of spurious AI solutions, a novel automated algorithm using ideas from statistical mechanics has been developed [72]. This approach is based on the notion that a more reliable AI-solution will be one that maximizes the timescale separation between slow and fast processes—a property known as the spectral gap [72]. The method builds a maximum caliber or path entropy-based model of the unbiased dynamics along different AI-based representations, which then yields spectral gaps along different slow modes obtained from AI trials [72].
Table 2: AI-Enhanced Sampling Experimental Protocol
| Step | Procedure | Purpose | Key Parameters |
|---|---|---|---|
| 1. Initial Sampling | Run initial unbiased MD simulation using chosen MD engine (GROMACS/AMBER/NAMD) | Generate initial trajectory data for AI training | Simulation length: Sufficient to sample some transitions; Order parameters: Generic variables (dihedrals, distances) |
| 2. AI Slow Mode Identification | Apply iterative MD-AI approach (e.g., RAVE - Reweighted Autoencoded Variational Bayes) | Identify low-dimensional RC approximating true slow modes | PIB objective function: L ≡ I(s,χ) - γI(sΔt,χ); Training epochs: Until convergence |
| 3. Spurious Solution Screening | Implement spectral gap optimization (SGOOP) | Screen and rank multiple AI solutions to eliminate spurious RCs | Timescale separation: Maximize slow vs. fast mode gap; Path entropy model: Maximum caliber framework |
| 4. Enhanced Sampling | Perform biased sampling using identified RC | Accelerate configuration space exploration | Biasing method: Metadynamics, ABF; Biasing potential: Adjusted based on RC |
| 5. Iterative Refinement | Use expanded sampling for new AI training | Refine RC estimate and explore new regions | Convergence criterion: Stable free energy estimate |
The following diagram illustrates the iterative workflow of this AI-enhanced sampling protocol, showing how short MD simulations are combined with AI analysis to progressively improve the reaction coordinate and expand sampling:
Benchmarking and Validation
Validating AI-enhanced MD workflows requires careful benchmarking against known systems. The approach has demonstrated applicability for three classic benchmark problems: the conformational dynamics of a model peptide, ligand-unbinding from a protein, and the folding/unfolding energy landscape of the C-terminal domain of protein G (GB1-C16) [72]. For each system, the spectral gap optimization successfully identified spurious solutions and selected RCs that provided maximal timescale separation, leading to more efficient sampling and accurate free energy recovery [72].
Hardware and Computational Infrastructure for AI-MD Workflows
GPU Selection for AI-Augmented Simulations
The hardware landscape for AI-MD workflows requires careful consideration, as both the MD simulation and AI components demand high computational resources.
Table 3: Recommended GPU Hardware for AI-Enhanced MD Simulations
| GPU Model | Memory | CUDA Cores | Suitability for MD | Suitability for AI/ML |
|---|---|---|---|---|
| NVIDIA RTX 4090 | 24 GB GDDR6X | 16,384 | Excellent for GROMACS and most MD simulations [73] | Strong performance for training moderate-sized models |
| NVIDIA RTX 6000 Ada | 48 GB GDDR6 | 18,176 | Ideal for large-scale simulations in AMBER and other memory-intensive workloads [73] | Excellent for larger models with substantial memory requirements |
| NVIDIA A100 | 40/80 GB HBM2e | 6,912 (FP64) | Superior for FP64-dominated calculations [5] | Industry standard for large-scale AI training |
Precision Considerations in AI-MD Workflows
A critical consideration in hardware selection is precision requirements. Many MD codes like GROMACS, AMBER, and NAMD have mature mixed-precision GPU pathways that maintain accuracy while significantly accelerating performance [5]. However, AI-enhanced workflows may have different precision needs:
- MD Force Calculations: Most modern MD packages use mixed precision, performing the bulk of calculations in single precision while maintaining double precision for critical accumulations [5].
- Neural Network Training: AI components typically use single precision (FP32) or half precision (FP16), which aligns well with consumer and workstation GPUs [5].
- Quantum Mechanics/ML Potentials: Hybrid QM/MM simulations with ML potentials may require stronger double precision support, necessitating data-center GPUs [5].
Researchers should verify precision requirements through quick checks: if code defaults to double precision and fails with mixed precision, if published benchmarks specify "double precision only," or if results drift when moving from double to mixed precision [5].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Software and Hardware Solutions for AI-MD Research
| Tool Category | Specific Solutions | Function in AI-MD Workflow |
|---|---|---|
| MD Software | GROMACS, AMBER, NAMD [3] | Core simulation engines providing physical models and integration algorithms |
| Enhanced Sampling Packages | PLUMED, SSAGES | Collective variable-based sampling and AI integration frameworks |
| AI-MD Integration | RAVE [72] | Iterative MD-AI approach for learning reaction coordinates and accelerating sampling |
| Visualization & Analysis | VMD [3] | Visualization of trajectories and analysis of AI-identified reaction coordinates |
| Neural Network Frameworks | PyTorch, TensorFlow | Implementation and training of deep learning models for CV discovery |
| Hardware Platforms | BIZON ZX Series [73] | Purpose-built workstations with multi-GPU configurations for high-throughput simulations |
| Cloud Computing | hiveCompute [5] | Scalable GPU resources for burst capacity and large-scale AI training |
Future Directions and Challenges
The convergence of AI and MD is paving the way for fully automated chemical discovery systems that can autonomously design experiments, simulate outcomes, and refine models [74]. Key emerging trends include the development of neural network potentials that can achieve quantum-mechanical accuracy at classical force field costs, transfer learning approaches that enable pre-trained models to be fine-tuned for specific systems, and active learning frameworks that optimally select which simulations to run next for maximum information gain [72] [74].
However, significant challenges remain. Data quality and quantity continue to limit the generalizability of AI models, particularly for rare events [74]. The black-box nature of many deep learning approaches creates interpretability issues, though methods like spectral gap optimization help address this [72]. Additionally, the computational cost of generating sufficient training data and the need for robust validation frameworks present ongoing hurdles to widespread adoption [72].
As these challenges are addressed, AI-enhanced MD workflows will increasingly become the standard approach in computational chemistry and drug development, enabling researchers to tackle increasingly complex biological questions and accelerate the discovery of novel therapeutics.
Conclusion
Selecting between GROMACS, AMBER, and NAMD is not a one-size-fits-all decision but a strategic choice balanced between raw speed, force field specificity, and application needs. GROMACS excels in performance and open-source accessibility, AMBER is renowned for its rigorous force fields, and NAMD offers superior scalability and visualization integration. The convergence of advanced hardware, robust validation protocols, and growing integration with machine learning is poised to significantly enhance the predictive power and scope of molecular dynamics simulations. This progress will directly accelerate discoveries in drug development, personalized medicine, and materials science, making informed software selection more critical than ever for research efficiency and impact.
References
- 1. Which MD software is preferable for protein simulation? [https://mattermodeling.stackexchange.com/questions/11701/which-md-software-is-preferable-for-protein-simulation]
- 2. AMBER vs GROMACS [https://diphyx.com/stories/amber-vs-gromacs/]
- 3. Choosing the best tool for MD simulations: GROMACS ... [https://www.linkedin.com/posts/omarariasgaguancela_gromacs-vs-amber-vs-namd-which-one-do-you-activity-7339451262129606656-xgBd]
- 4. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOoqRW2PxUIL3qo-5jlr7TsCpkChYPL-lXZ7FhcZDxw_WZSbU3CdW]
- 5. Scientific modeling on cloud GPUs: fit guide for 2025 [https://compute.hivenet.com/post/scientific-modeling-cloud-gpus-fit-guide]
- 6. Validating Molecular Dynamics Simulations Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420231/]
- 7. Running Molecular Dynamics on Alliance clusters with AMBER [https://computecanada.github.io/molmodsim-amber-md-lesson/26-MD_Software_Benchmarks/index.html]
- 8. Is There A 'Best' Molecular Dynamics Software? How Is ... [https://www.biostars.org/p/4400/]
- 9. 1. AMBER Package Installation & Configuration [https://mmb.irbbarcelona.org/NAFlex/help.php?id=MDinstallation]
- 10. GROMACS Vs. AMBER for Molecular Dynamics Simulations [https://parssilico.com/blogs/85-gromacs-vs-amber-md-software]
- 11. Performance benchmarks for mainstream molecular dynamics ... [https://blog.enthalpy.space/archives/290]
- 12. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOopvNYgi2ervGmbZgdpl8D_pkB7Lqx2AEz1a9vj1lXtaoMyVEdu2]
- 13. Running membrane simulations in GROMACS [https://manual.gromacs.org/2025-rc/how-to/special.html]
- 14. a WEB application for molecular dynamics simulation using ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05234-y]
- 15. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOoqoJUrPfTcp1u5kjPkQ0P0CP1rHSyxuCOlNgmhrBux1qz6T-9jT]
- 16. Structure-Based Experimental Datasets for Benchmarking ... [https://livecomsjournal.org/index.php/livecoms/article/view/v6i1e3871]
- 17. CHARMM-GUI Input Generator for NAMD, GROMACS ... [https://www.osti.gov/pages/biblio/1261132]
- 18. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOoqtlFGtPgDzCSIARCM2mCUpfumIEv3ruHcRGUtUWnqjR9443KK4]
- 19. Lessons learned from comparing molecular dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5581938/]
- 20. Running Molecular Dynamics on Alliance clusters with AMBER [https://computecanada.github.io/molmodsim-amber-md-lesson/aio/index.html]
- 21. GROMACS: High performance molecular simulations ... [https://www.sciencedirect.com/science/article/pii/S2352711015000059]
- 22. AMBER file and force field support (NAMD Git-2025-10-14 ... [https://www.ks.uiuc.edu/Research/namd/cvs/ug/node13.html]
- 23. Using VMD plug-ins for trajectory file I/O [https://manual.gromacs.org/2024.4/reference-manual/special/vmd-imd.html]
- 24. AMBER 24 NVIDIA GPU Benchmarks | B200, RTX PRO ... [https://www.exxactcorp.com/blog/molecular-dynamics/amber-molecular-dynamics-nvidia-gpu-benchmarks]
- 25. A Technical Overview of Molecular Simulation Software [https://intuitionlabs.ai/articles/molecular-modeling-simulation-software]
- 26. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOopU-4uRp3iRCuZEtpmvCUiVq5ys7m9gdkMmteNrlxOyOlGFHL-C]
- 27. Running Molecular Dynamics Simulations with AMBER [https://computecanada.github.io/molmodsim-amber-md-lesson/13-Running_Simulations/index.html]
- 28. Running membrane simulations in GROMACS [https://manual.gromacs.org/2025.1/how-to/special.html]
- 29. GROMACS Tutorials [http://www.mdtutorials.com/gmx/]
- 30. callumjd/AMBER-Membrane_protein_tutorial [https://github.com/callumjd/AMBER-Membrane_protein_tutorial]
- 31. CHARMM-GUI [https://www.charmm-gui.org/]
- 32. New to GROMACS. need help to build a membrane protein on ... [https://gromacs.bioexcel.eu/t/new-to-gromacs-need-help-to-build-a-membrane-protein-on-lipid-bilayer/11718]
- 33. Assessing the Current State of Amber Force Field ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4980684/]
- 34. Rapid parameterization of small molecules using the Force ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3874408/]
- 35. Amber Force Field Improper Parameterization Huge Kb ... [https://chemistry.stackexchange.com/questions/109915/amber-force-field-improper-parameterization-huge-kb-problem]
- 36. System preparation - GROMACS 2025.4 documentation [https://manual.gromacs.org/current/user-guide/system-preparation.html]
- 37. Parametrization, molecular dynamics simulation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2766176/]
- 38. Molecular dynamics simulations as support for ... [https://www.sciencedirect.com/science/article/pii/S1359029424000347]
- 39. Comparison of software for molecular mechanics modeling [https://en.wikipedia.org/wiki/Comparison_of_software_for_molecular_mechanics_modeling]
- 40. Performance improvements - GROMACS 2025.1 ... [https://manual.gromacs.org/2025.1/release-notes/2020/major/performance.html]
- 41. SCALE Benchmark case study: GROMACS [https://scale-lang.com/posts/2025-05-19-GROMACS]
- 42. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOooVZ-KBd2GGVGD43GMHCzgXym4C1kRpAYVheknLJf4rhXyxJGCO]
- 43. Comparison, Analysis, and Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10525864/]
- 44. Simulation dataset to benchmark the performance of ... [https://zenodo.org/records/17632513]
- 45. Latest GeForce RTX Cards for PC | 50 Series Graphics NVIDIA [https://www.nvidia.com/en-ph/geforce/graphics-cards/50-series/]
- 46. announces Nvidia 5090 for $1,999, 5070 for... | Tom's Hardware RTX [https://www.tomshardware.com/pc-components/gpus/nvidia-announces-rtx-50-series-at-up-to-usd1-999]
- 47. Nvidia Blackwell and GeForce RTX 50-Series GPUs [https://www.tomshardware.com/pc-components/gpus/nvidia-blackwell-rtx-50-series-gpus-everything-we-know]
- 48. NVIDIA Blackwell Ultra Sets New Inference Records in ... [https://developer.nvidia.com/blog/nvidia-blackwell-ultra-sets-new-inference-records-in-mlperf-debut/]
- 49. Deep dive into NVIDIA Blackwell Benchmarks — where does ... [https://adrianco.medium.com/deep-dive-into-nvidia-blackwell-benchmarks-where-does-the-4x-training-and-30x-inference-0209f1971e71]
- 50. Molecular Simulation: GROMACS Benchmark on 30 GPUs [https://blog.salad.com/gromacs-benchmark/]
- 51. Gromacs Benchmarks on Biowulf [https://hpc.nih.gov/apps/gromacs/]
- 52. NAMD 2.10 Benchmarks [https://hpc.nih.gov/apps/namd/2.10.html]
- 53. Benchmarking Molecular Dynamics Performance Using ... [https://epcced.github.io/2021-03-29-archer2-developers-online/11-GROMACS-benchmark/index.html]
- 54. GPU-Accelerated Amber [https://www.nvidia.com/es-la/data-center/gpu-accelerated-applications/amber/]
- 55. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOoqczTlKonQUvHxZoyyXQEm9vP0kQ1nDVrKMopUIUYbaZuq1G8uL]
- 56. Parallelization - GROMACS 2025.1 documentation [https://manual.gromacs.org/documentation/2025.1/reference-manual/algorithms/parallelization-domain-decomp.html]
- 57. Getting good performance from mdrun [https://manual.gromacs.org/documentation/2025.2/user-guide/mdrun-performance.html]
- 58. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOorgEXsitOoi_qZSxpdCCULBbfQ2gfY9j56Io-OoAtsGWNMdFWYr]
- 59. Getting good performance from mdrun [https://manual.gromacs.org/current/user-guide/mdrun-performance.html]
- 60. Maximizing GROMACS Throughput with Multiple ... [https://developer.nvidia.com/blog/maximizing-gromacs-throughput-with-multiple-simulations-per-gpu-using-mps-and-mig/]
- 61. Re: [AMBER] Multi-GPU Bug in Amber20 [http://archive.ambermd.org/202201/0080.html]
- 62. NAMD GPU Benchmarks and Hardware Recommendations [https://www.exxactcorp.com/blog/benchmarks/namd-gpu-benchmarks-and-hardware-recommendations]
- 63. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOopvoRThXNXGBsxjiu80tJufNrQpSpyD9dP3VUGWW1_1igx31bO9]
- 64. NAMD - Scalable Molecular Dynamics [http://www.ks.uiuc.edu/Research/namd/index.html]
- 65. GROMACS Benchmark [https://openbenchmarking.org/test/pts/gromacs&eval=a0ebf2010808203b4693fb624326fbf35d8d6b1f]
- 66. Development of a New AMBER Force Field for Cysteine ... [https://pubmed.ncbi.nlm.nih.gov/40509949/]
- 67. Validating Molecular Dynamics Simulations against ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/29864281/]
- 68. Conformational dynamics of two natively unfolded fragment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4685472/]
- 69. Umbrella sampling [https://tutorials.gromacs.org/docs/umbrella-sampling.html]
- 70. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOoq0zvLcRFopmr9W5ggUnBzMB4CZRNG53z_KrWaw_9pF8w39TZCX]
- 71. NAMD Benchmark [https://openbenchmarking.org/test/pts/namd]
- 72. Confronting pitfalls of AI-augmented molecular dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7863682/]
- 73. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOorMDrMpHOFEan1CBKYIqGbqVVdQitko43JFUtPrllWAjaEFxL3j]
- 74. Review of the latest progress of AI and Machine Learning ... [https://www.sciencedirect.com/science/article/pii/S2949866X25000620]