Solving Molecular Dynamics Energy Minimization Failures: A Troubleshooting Guide for Computational Researchers
This article provides a comprehensive framework for understanding, troubleshooting, and resolving common failures in molecular dynamics (MD) energy minimization.
Solving Molecular Dynamics Energy Minimization Failures: A Troubleshooting Guide for Computational Researchers
Abstract
This article provides a comprehensive framework for understanding, troubleshooting, and resolving common failures in molecular dynamics (MD) energy minimization. Tailored for researchers, scientists, and drug development professionals, it covers foundational principles of minimization algorithms, practical application of methods like steepest descent and conjugate gradients, systematic diagnosis of convergence problems and force field errors, and validation techniques to ensure result reliability. By integrating traditional approaches with emerging machine learning solutions, this guide aims to enhance simulation stability and accuracy in biomedical research, from drug discovery to protein folding studies.
Understanding Energy Minimization: Core Principles and Common Pitfalls
The Critical Role of Energy Minimization in Stable MD Simulations
Energy minimization is a foundational step in molecular dynamics (MD) simulations, crucial for relieving atomic clashes and preparing a stable system for production runs. Without proper minimization, simulations can fail due to unphysically high forces that lead to numerical instabilities, integration errors, or complete simulation collapse. This technical guide addresses the critical role of energy minimization and provides targeted troubleshooting for common failure scenarios encountered by researchers.
Troubleshooting Guide: Common Energy Minimization Failures
Diagnostic Table of Common Errors
The table below summarizes frequent energy minimization issues, their symptoms, and initial diagnostic steps.
| Error Symptom | Potential Root Cause | Initial Diagnostic Action |
|---|---|---|
| Infinite forces (Fmax = inf) and "atoms are overlapping" message [1] | Severe atomic clashes; Topology-coordinate mismatch [1] | Check atom identities in topology vs. coordinate files [1] |
| Forces not converging to requested Fmax despite machine precision convergence [2] | Systematically poor initial structure; Inadequate minimization parameters [2] | Verify initial structure quality; Consider multi-stage minimization [3] |
| Segmentation fault during minimization or subsequent equilibration [2] | Critical software error; Possible hardware/GPU incompatibility [2] | Run on CPU only; Check for system corruption; Verify installation |
| "Residue not found in topology database" during topology generation [4] | Missing force field parameters for molecule/residue [4] | Verify residue naming; Use external tools for parameterization [4] |
| "Long bonds and/or missing atoms" [4] | Incomplete molecular structure in initial coordinates [4] | Inspect PDB file for REMARK 465/470 (missing atoms); Model missing atoms |
Resolving Atomic Overlaps and Infinite Forces
Atomic overlaps creating infinite forces represent one of the most common and critical failures in energy minimization.
Problem Analysis: When atoms are placed impossibly close in the initial configuration, the potential energy and forces calculated by the force field can become astronomically high (e.g., ~10¹⁹ kJ/mol) [1], causing the minimizer to fail immediately. This often manifests with warnings like "the force on at least one atom is not finite" [1].
Solution Protocol:
- Inspect the Offending Atom: The minimization log identifies the problematic atom (e.g., "Maximum force = inf on atom 1251") [1]. Visualize this atom and its local environment using molecular visualization software.
- Verify Topology-Coordinate Consistency: A frequent cause is a mismatch between the atom names or types in the structure file (.gro, .pdb) and the topology file (.top). Meticulously check that the ligand or residue in question matches the definition in the force field's residue topology database (rtp) [1] [4].
- Rebuild Problematic Fragments: If the initial model was built with missing atoms (often noted in REMARK 465/470 of PDB files), use modeling software to reconstruct the missing portions before minimization [4].
- Use a Multi-Stage Minimization Approach: Start with a gentle minimizer like steepest descent for a few steps to relieve the worst clashes, then switch to a more efficient algorithm like conjugate gradient for finer convergence [3].
Addressing Force Convergence Failures
Sometimes minimization stops without achieving the desired force tolerance (Fmax), reporting convergence only to machine precision.
Problem Analysis: This indicates the minimizer cannot find a direction to move atoms that further lowers the energy, given the numerical constraints of the algorithm and the system's current configuration. This does not necessarily mean the system is stable enough for dynamics [2].
Solution Protocol:
- Adjust Minimization Parameters: In your molecular dynamics parameter (.mdp) file, consider increasing the maximum number of steps (
nsteps). For steepest descent, slightly increasing the initial step size (emstep) can sometimes help, but caution is required to maintain stability [5] [2]. - Switch Minimization Algorithms: If steepest descent fails to converge, try the conjugate gradient (
integrator = cg) or L-BFGS (integrator = l-bfgs) algorithms, which are more efficient for finding minima in complex landscapes [5]. - Consider Double Precision: While single precision is often sufficient, for systems with particularly difficult energy landscapes, compiling and running GROMACS in double precision can provide the necessary numerical accuracy to achieve convergence [1].
- Iterative Minimization and Equilibration: It is sometimes effective to run a short, gentle minimization, followed by a brief position-restrained MD (NVT) simulation, and then another round of minimization. This can help "nudge" the system out of a difficult configuration.
Handling Segmentation Faults and Crashes
Segmentation faults during or immediately after minimization are often related to deeper software or system issues.
Problem Analysis: A segmentation fault indicates the program tried to access memory it was not permitted to, leading to a crash. This can be caused by corrupted input files, bugs in the code, or hardware incompatibilities, especially with GPU acceleration [2].
Solution Protocol:
- Test on CPU: Run the same simulation using only CPU resources (e.g., remove flags like
-nb gpu -pme gpu). If it runs, the issue is likely with the GPU setup or code paths [2]. - Check Input File Integrity: Ensure all input files (.tpr, .gro, .top) are complete and uncorrupted. Try regenerating the .tpr file with
gmx grompp. - Verify System Components: Scrutinize the topology for any unusual molecules, parameters, or inclusions that might trigger a software bug. Pay special attention to non-standard residues and metal ions.
Energy Minimization Workflow and Best Practices
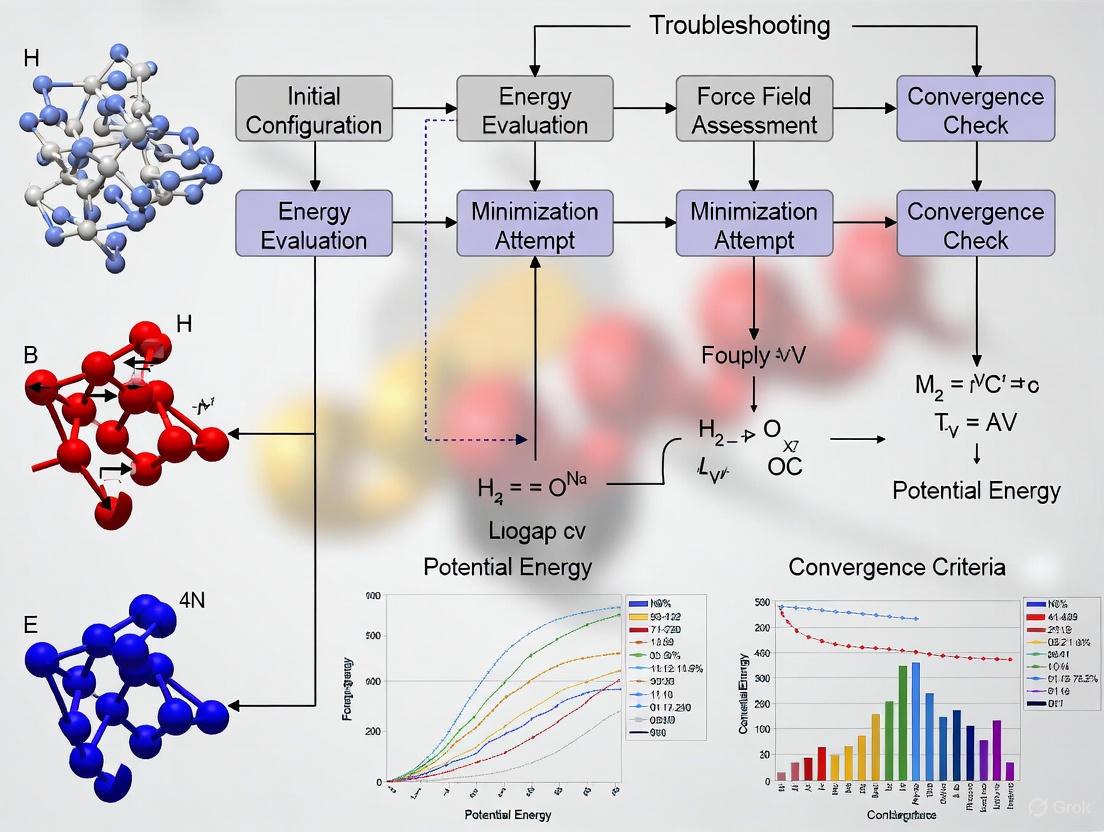
The following diagram illustrates a robust, decision-based workflow for successful energy minimization, integrating the troubleshooting steps outlined above.
Diagram Title: Energy Minimization Troubleshooting Workflow
Step-by-Step Minimization Protocol
- Initial System Preparation: Before minimization, ensure your system's topology and coordinates are consistent. Use tools like
pdb2gmxor external parameterization software correctly, especially for non-standard molecules [4]. - Stage 1: Steepest Descent: This algorithm is robust for relieving severe clashes and should be used first. A typical parameterization in a GROMACS
.mdpfile is [5] [2]: - Stage 2: Conjugate Gradient: After the largest forces are eliminated, switch to a more efficient algorithm for final convergence.
- Validation: Before moving to equilibration, verify the potential energy of the system has dropped to a reasonable, negative value (e.g., for a solvated protein system, typically on the order of -10⁵ to -10⁶ kJ/mol), indicating favorable non-bonded interactions.
The Scientist's Toolkit: Essential Research Reagents
The table below lists critical "research reagents" – software tools, parameters, and methods – essential for successful energy minimization.
| Tool / Reagent | Function & Purpose | Key Considerations |
|---|---|---|
| Steepest Descents Algorithm [5] | Initial "coarse" minimization; Highly effective at relieving severe atomic clashes. | Robust but inefficient for fine convergence; Use first. |
| Conjugate Gradient / L-BFGS [5] [6] | Subsequent "fine" minimization; More efficiently finds local minima after clashes are relieved. | Use after steepest descent; L-BFGS is often faster but may have compatibility constraints [5]. |
| Force Fields (e.g., AMBER ff14SB, GAFF) [3] | Provides the functional form and parameters (bonds, angles, charges) to calculate potential energy. | Choice is system-dependent; Non-standard residues require parameterization (e.g., with GAFF) [3]. |
| Molecular Viewers (e.g., Chimera, VMD) | Visual inspection of atomic overlaps, missing atoms, and post-minimization structure. | Critical for diagnosing atom-specific errors identified in log files [1]. |
| Antechamber [3] | Automates assignment of force field parameters and charges for non-standard residues and small molecules. | Necessary for generating topologies for ligands not in standard force field databases [3]. |
| Position Restraints | Harmonically restraints heavy atoms or specific groups during initial minimization/equilibration. | Allows solvent to relax around a fixed solute, preventing unrealistic protein backbone movements. |
Frequently Asked Questions (FAQs)
Q1: My minimization fails with "atoms are overlapping," but my structure looks fine visually. What should I do?
The problem is often not a visual overlap but a mismatch between your coordinate file and topology. The energy calculation expects specific atom names and types defined in the force field's residue database. An atom named "CB1" in your coordinate file will not be recognized if the topology expects "CB". Carefully check the atom names in your input structure against the names in the force field's residue topology (.rtp) file [1] [4].
Q2: What is the difference between "steepest descent" and "conjugate gradient," and when should I use each?
- Steepest Descent: This algorithm always moves atoms in the direction of the negative gradient (steepest energy decrease). It is very robust and effective at quickly reducing large forces and energies from severe clashes. It should be used for the initial stage of minimization [3].
- Conjugate Gradient: This method uses information from previous steps to choose a more efficient search direction. It converges much faster than steepest descent once the system is near a minimum but can be less stable with large initial forces. Use it for the second stage of minimization after the worst clashes are resolved [5] [3].
Q3: How do I know if my energy minimization was successful?
A successful minimization is typically judged by two criteria:
- Convergence: The maximum force (Fmax) in the system falls below your specified tolerance (
emtol). This is the primary success metric [5]. - Reasonable Potential Energy: The total potential energy should be a large, negative value, indicating favorable van der Waals and electrostatic interactions. A positive or extremely high negative energy suggests persistent problems [2].
Q4: Can I use energy minimization to find the global minimum of my protein structure?
No. Energy minimization locates the nearest local minimum on the potential energy surface. It does not cross energy barriers. To search for a global minimum or sample different conformations, you must use methods like molecular dynamics simulation at finite temperature, replica-exchange MD, or other enhanced sampling techniques [7] [3]. Minimization is for "relaxing" a given structure, not for folding or extensive conformational searching.
PES Fundamentals FAQ
What is a Potential Energy Surface (PES)? A Potential Energy Surface describes the energy of a system, especially a collection of atoms, in terms of certain parameters, normally the positions of the atoms. The PES is a conceptual tool for analyzing molecular geometry and chemical reaction dynamics, creating a smooth energy "landscape" where chemistry can be viewed from a topology perspective of particles evolving over "valleys" and "passes" [8] [9]. For a system with two degrees of freedom, the value of the energy is analogous to the height of the land as a function of two coordinates [9].
What are stationary points on a PES? Stationary points are points on the PES where the gradient (first derivative of energy with respect to position) is zero [8]. The two most important types of stationary points are:
- Minima: Correspond to the most stable structures of a molecule, such as reaction intermediates or stable products [8]. At a minimum, the potential energy surface curves upwards [8].
- Saddle Points: Correspond to transition states—the highest energy point on the lowest energy pathway connecting two minima [9]. These are maxima along the reaction coordinate but minima in all other directions [9].
What is the mathematical foundation of a PES? The PES originates from solving the time-independent Schrödinger equation, $\hat H \Psi(r, R) = E \Psi(r, R)$, under the Born-Oppenheimer approximation [8]. This approximation separates nuclear and electronic motion, allowing the wavefunction to be approximated as $\Psi(r, R) \approx \Psi(r)\Phi(R)$ [8]. The total energy for a given nuclear configuration $R$ is then $E{tot \; R} = E{el \; R} + E_{nuc \; R}$ [8].
What are internal coordinates and why are they important? Internal coordinates describe molecular geometry in terms of bond lengths, angles, and dihedral angles, eliminating the "external coordinates" related to translation and rotation [8]. For a system of N atoms, this typically results in $3N - 6$ internal coordinates (or $3N - 5$ for linear molecules) [8]. This representation is computationally more efficient than Cartesian coordinates and directly relevant to chemical bonding [8].
Troubleshooting Energy Minimization Failures
Why does my energy minimization stop abruptly without force convergence? This common error in molecular dynamics simulations occurs when:
- The minimization algorithm attempts steps that are too small to significantly reduce forces [2] [10]
- There is no change in energy between consecutive steps despite persistent high forces [2] [10]
- The system contains severe atomic clashes generating excessively high potential energies and forces [10]
Table 1: Common Energy Minimization Error Messages and Causes
| Error Message | Possible Causes | System Context |
|---|---|---|
| "Energy minimization has stopped, but the forces have not converged..." [2] | - Severe atomic clashes [10]- Incorrect position restraints [10]- Inappropriate constraint settings [2] | Protein-DNA complex [2] |
| "Potential Energy = 1.3146015e+32 Maximum force = 1.3916486e+11" [10] | - Initial structure with overlapping atoms [10]- Incorrect system building [10] | Phospholipid membrane simulation [10] |
| Segmentation fault during minimization [2] | - Software/hardware compatibility issues- Problematic position restraint files | After minimization during equilibration [2] |
How can I resolve atomic clashes causing minimization failures?
- Visualize the system: Identify atoms with extremely high forces and check for steric clashes [10]
- Check system building procedures: Ensure the initial structure was built correctly without overlapping atoms [10]
- Remove unnecessary restraints: Position restraints (
define = -DPOSRES) may interfere with minimization and should be temporarily removed [10]
What algorithmic adjustments can improve minimization?
- Switch algorithms: If steepest descent fails, try conjugate gradient methods [2]
- Adjust parameters: Modify the maximum force tolerance (
emtol) or step size (emstep) [2] - Multiple minimization rounds: Perform successive minimizations with different parameters [2]
Experimental Protocols & Methodologies
PES Stationary Point Identification Protocol
Objective: Systematically identify and characterize stationary points on a PES [11]
Methodology:
- Energy Calculation: Compute the energy for atomic arrangements using quantum chemical methods (e.g., Density Functional Theory) [11]
- Gradient Evaluation: Calculate the first derivatives of energy with respect to nuclear coordinates [8]
- Geometry Optimization: Iteratively adjust nuclear coordinates until the gradient approaches zero [8]
- Hessian Analysis: Compute second derivatives to characterize the nature of stationary points [11]
High-Index Saddle Dynamics (HiSD) Method: Recent advanced approaches use improved HiSD schemes combined with downward and upward searches to systematically enumerate stationary points and their connections [11]. This method introduces generalized coordinates to encode crystals that satisfy translational and rotational invariance [11].
Diagram Title: PES Exploration Workflow
Stationary-Point Network Construction
Objective: Create a comprehensive representation of PES topology using a stationary-point network (SPN) [11]
Procedure:
- Stationary Point Identification: Locate all minima and saddle points where the gradient vanishes [11]
- Connection Mapping: Establish pathways between connected stationary points [11]
- Network Construction: Build a graph with nodes representing stationary points and edges representing their connections [11]
- Visualization: Create SPN diagrams to visualize and analyze complex PES terrains [11]
Application Example: For crystalline materials like GaN and Si, this approach has discovered hidden transition pathways with lower enthalpy barriers than previously known [11].
Research Reagent Solutions
Table 2: Essential Computational Tools for PES Exploration
| Tool/Software | Function | Application Context |
|---|---|---|
| GROMACS [2] [12] | Molecular dynamics simulation package | Energy minimization, force field applications [2] |
| xtb Software [8] | Quantum chemical geometry optimization | Fast optimization for structures under 100 atoms [8] |
| VASP Package [11] | First-principles density functional theory calculations | Energy and force calculations for crystalline systems [11] |
| AMBER Force Fields (ff19SB) [2] | Protein force field parameters | Protein-DNA complex simulations [2] |
| pdb2gmx [12] | Topology generation from PDB files | Building block assembly and topology creation [12] |
Advanced PES Topology Visualization
Diagram Title: PES Topology with Stationary Points
The Stationary-Point Network (SPN) provides a powerful framework for visualizing complex PESs, where nodes represent stationary points and edges represent their connections [11]. This approach enables researchers to discover hidden transition pathways with low energy barriers that might be missed with conventional sampling methods [11].
In molecular dynamics (MD) simulations, energy minimization is a critical first step used to find the stable, low-energy configuration of a molecular system before beginning a production run. It involves adjusting atomic coordinates to reduce the net forces on atoms until a minimum on the potential energy surface is reached. This process helps avoid unrealistic high-energy configurations that could lead to simulation instability or failure. The three most common algorithms for this task are Steepest Descent, Conjugate Gradient, and L-BFGS, each with distinct mathematical foundations and performance characteristics that make them suitable for different scenarios in computational drug development and research.
Algorithm Mechanics and Mathematical Foundations
Steepest Descent Method
The Steepest Descent method is one of the simplest optimization algorithms. It relies exclusively on first-derivative information (the gradient) to find the minimum of a function.
Mathematical Formulation: The core update formula for Steepest Descent is: [ x{new} = x{old} - \gamma \nabla E(x{old}) ] where ( \nabla E(x{old}) ) is the gradient of the potential energy function at the current geometry, and ( \gamma ) is a constant step size parameter [13].
Mechanism: The algorithm moves in the direction opposite to the largest gradient (i.e., the steepest descent) at the current point. Once a minimum in that direction is found, a new minimization begins from that point in the new steepest direction. This process continues until a minimum is reached in all directions within a specified tolerance [13].
Key Characteristics:
- Uses only gradient information (first derivative)
- Assumes the second derivative is constant
- Generally requires more steps than higher-order methods
- Often exhibits oscillatory behavior when approaching the minimum
Conjugate Gradient Method
The Conjugate Gradient (CG) method improves upon Steepest Descent by incorporating information from previous search directions to achieve more efficient convergence.
Mathematical Formulation:
CG uses the following iterative process:
[
x{k+1} = xk + \alphak pk
]
[
pk = rk - \sum{i
Conjugacy Principle: Two vectors ( u ) and ( v ) are conjugate with respect to a positive definite matrix ( A ) if: [ u^T A v = 0 ] This conjugacy condition ensures that each step minimizes the function in a direction that does not spoil the minimization achieved in previous directions [14] [15].
Mechanism: Unlike Steepest Descent, CG mixes in a component of the previous search direction when determining the new direction. This approach prevents the oscillatory behavior typical of Steepest Descent and typically converges in at most n steps for an n-dimensional quadratic problem [13] [15].
L-BFGS Algorithm
The Limited-memory Broyden-Fletcher-Goldfarb-Shanno (L-BFGS) algorithm is a quasi-Newton method that approximates the second derivative (Hessian) information without explicitly storing the full matrix.
Mathematical Foundation: L-BFGS builds upon the BFGS update formula: [ B{k+1} = Bk - \frac{Bk sk sk^T Bk^T}{sk^T Bk sk} + \frac{yk yk^T}{yk^T sk} ] where ( sk = x{k+1} - xk ) is the change in parameters and ( yk = \nabla f{k+1} - \nabla f_k ) is the change in the gradient [16].
Limited-Memory Approach: Instead of storing the complete Hessian approximation (which would require O(n²) memory), L-BFGS stores only the last m vector pairs (typically m = 3-20), significantly reducing memory usage to O(n·m) [17] [16] [18].
Two-Loop Recursion: L-BFGS uses an efficient two-loop recursion to compute the search direction:
- First Loop (Forward): Computes intermediate quantities using recent vector pairs
- Middle Step: Applies initial Hessian approximation
- Second Loop (Backward): Refines the search direction using stored vectors [16]
This approach allows L-BFGS to maintain curvature information without the computational burden of storing and manipulating large matrices.
Performance Comparison and Quantitative Analysis
Table 1: Algorithm Characteristics Comparison
| Characteristic | Steepest Descent | Conjugate Gradient | L-BFGS |
|---|---|---|---|
| Derivative Order | First-order | First-order | Quasi-Second-order |
| Memory Requirements | O(n) | O(n) | O(n·m) |
| Convergence Rate | Linear | Superlinear (theoretical) | Superlinear |
| Computational Cost per Step | Low | Moderate | Moderate to High |
| Typical Use Cases | Initial minimization, very rough surfaces | Medium to large systems, good initial guess | Large systems, expensive gradients |
Table 2: Empirical Performance on Rosenbrock Function
| Algorithm | Iterations | Function Evaluations | Time (seconds) | Solution Accuracy |
|---|---|---|---|---|
| L-BFGS | 24 | 24 | 0.0046 | 0.00000 |
| Conjugate Gradient | 2129 | ~3000 | 0.0131 | 0.00001 |
| Steepest Descent | ~5000* | ~5000* | ~0.0300* | 0.00001* |
Note: Steepest Descent values are estimates based on typical performance; actual values may vary [17] [18].
Table 3: GROMACS Implementation Comparison
| Algorithm | GROMACS Integrator | Best For | Limitations |
|---|---|---|---|
| Steepest Descent | steep |
Initial stages of minimization | Slow convergence near minimum |
| Conjugate Gradient | cg |
Pre-normal mode analysis | Requires occasional steepest descent steps |
| L-BFGS | l-bfgs |
Fast convergence for large systems | Not yet parallelized in GROMACS |
Troubleshooting Common Energy Minimization Failures
Convergence Problems
Problem: Minimization fails to converge within the allowed steps.
- Cause 1: Poor initial structure with severe steric clashes.
- Solution: Use stronger minimization parameters initially (e.g., Steepest Descent with large initial step size) before switching to more advanced algorithms.
- Cause 2: Incorrect convergence criteria.
- Solution: Adjust the force tolerance (
emtolin GROMACS). For stable minimization, the maximum force should typically be < 1000 kJ/mol/nm initially, and < 10-50 kJ/mol/nm for final minimization [5].
- Solution: Adjust the force tolerance (
- Cause 3: Incompatible force field parameters.
- Solution: Verify bond lengths, angles, and dihedral parameters match your molecular system.
Problem: Convergence is unacceptably slow.
- Cause 1: Using Steepest Descent near the minimum.
- Solution: Switch to Conjugate Gradient or L-BFGS after initial rough minimization.
- Cause 2: Poorly conditioned problem with variables of vastly different scales.
- Solution: Implement variable scaling or preconditioning [18].
- Cause 3: Memory parameter too small in L-BFGS.
- Solution: Increase the number of stored vector pairs (typically to 10-15).
Numerical Instabilities
Problem: Energy becomes NaN during minimization.
- Cause 1: Extremely high forces from atomic overlaps.
- Solution: Reduce initial step size; use Steepest Descent with small steps initially.
- Cause 2: Incorrect charge assignments or missing parameters.
- Solution: Check topology for missing parameters or unrealistic charge distributions.
- Cause 3: Numerical overflow in force calculations.
- Solution: Verify cut-off schemes and neighbor searching parameters.
Problem: Forces oscillate without decreasing.
- Cause 1: Step size too large for the potential energy surface.
- Solution: Reduce step size parameter; for L-BFGS, ensure line search is enabled.
- Cause 2: Incorrect gradient calculations.
- Solution: Verify analytical gradients against numerical approximations if possible.
Algorithm-Specific Issues
L-BFGS Problems:
- Hessian approximation becomes inaccurate: Reset the Hessian approximation or switch to Steepest Descent briefly [19] [16].
- Memory issues with large systems: Reduce the number of stored vectors (m) or switch to Conjugate Gradient.
Conjugate Gradient Problems:
- Loss of conjugacy due to round-off error: Periodically reset the search direction to the steepest descent direction [5] [15].
- Slow convergence on ill-conditioned problems: Implement preconditioning using diagonal Hessian approximations.
Frequently Asked Questions (FAQs)
Q1: Which algorithm should I choose for my biomolecular system?
- A: Use Steepest Descent for the first 50-100 steps to eliminate large forces, then switch to L-BFGS for faster convergence. For very large systems (>100,000 atoms) with memory constraints, use Conjugate Gradient [5] [18].
Q2: How do I know if my minimization has converged properly?
- A: Check that the maximum force is below your tolerance (typically 10-100 kJ/mol/nm for biomolecular systems) and that the energy has stabilized. Also verify that the energy decrease per step approaches zero [19] [5].
Q3: Why does L-BFGS sometimes perform worse than Conjugate Gradient?
- A: For computationally inexpensive functions, L-BFGS's overhead per iteration may make it slower than CG despite fewer iterations. Also, on extremely ill-conditioned problems, L-BFGS can degenerate to Steepest Descent behavior [18].
Q4: How do I set the optimal memory parameter for L-BFGS?
- A: Start with m=5-10 for small systems (<10,000 atoms) and m=10-15 for larger systems. Increase if convergence is slow, decrease if memory is limited [16] [18].
Q5: Can I restart a minimization from a previous run?
- A: Yes, most MD packages support restart capabilities. For L-BFGS and BFGS, it's crucial to also save the Hessian approximation or trajectory to maintain convergence properties [19].
Q6: How do I handle minimization failures with membrane proteins or complex systems?
- A: Use multi-stage minimization: (1) Steepest Descent with position restraints on protein backbone, (2) Steepest Descent on all atoms, (3) Conjugate Gradient or L-BFGS for final convergence.
Experimental Protocols and Methodologies
Standard Energy Minimization Protocol
Diagram 1: Energy Minimization Workflow
Force Tolerance Selection Guidelines
For molecular dynamics simulations, the appropriate force tolerance depends on your subsequent simulation type:
- Equilibration MD: 100-1000 kJ/mol/nm
- Production MD: 10-100 kJ/mol/nm
- Normal Mode Analysis: < 1 kJ/mol/nm (requires double precision)
Algorithm Switching Protocol
When transitioning between algorithms:
- From Steepest Descent to CG/L-BFGS: Wait until energy decrease per step becomes gradual (not oscillatory)
- Between CG variants: Possible without loss of information
- To/from L-BFGS: Save trajectory information to preserve Hessian approximation
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Software Tools for Energy Minimization
| Tool | Function | Application Context |
|---|---|---|
| GROMACS | MD simulation package with multiple minimizers | Biomolecular systems, high performance |
| ASE (Atomic Simulation Environment) | Python package for structure optimization | Materials science, surface chemistry |
| ALGLIB | Numerical optimization library | Custom optimization implementations |
| AMBER | MD package with minimization capabilities | Biomolecular simulations, drug design |
| NAMD | Scalable MD software | Large biomolecular complexes |
Table 5: Critical Parameters and Their Functions
| Parameter | Function | Typical Values |
|---|---|---|
| Force Tolerance (emtol) | Determines convergence criterion | 10-1000 kJ/mol/nm |
| Step Size (emstep) | Maximum displacement per step | 0.001-0.1 nm |
| L-BFGS Memory (m) | Number of stored vector pairs | 3-20 |
| CG Steepest Descent Frequency | How often to reset to steepest descent | Every 10-100 CG steps |
| Update Frequency | How often to update neighbor lists | Every 10-20 steps |
Advanced Optimization Strategies
Preconditioning Techniques
Preconditioning transforms the optimization problem to improve its conditioning and accelerate convergence:
Diagonal Preconditioning:
- Scales variables based on their magnitudes
- Essential when atomic masses vary significantly
- Implemented via variable scaling functions in optimization packages [18]
Incomplete Cholesky Factorization:
- Approximates the Hessian for more effective preconditioning
- Particularly useful for systems with varying stiffness
Hybrid Approaches
For challenging systems, combine multiple algorithms:
- Stage 1: Steepest Descent with position restraints to remove severe clashes
- Stage 2: Pure Steepest Descent to reduce forces below 1000 kJ/mol/nm
- Stage 3: L-BFGS with moderate memory (m=10) for rapid convergence
- Stage 4: Conjugate Gradient for final polishing if needed
Monitoring and Diagnostics
Implement these checks to detect minimization problems early:
- Energy Conservation: Monitor energy decrease per step; sudden increases indicate problems
- Gradient Norm: Should decrease monotonically in well-behaved minimization
- Parameter Updates: Watch for unusually large coordinate changes
- Convergence History: Track force reduction rates to identify stagnation
Diagram 2: Minimization Troubleshooting Decision Tree
Troubleshooting Guide: Frequently Asked Questions
1. My system's energy and density stabilize quickly, but the pressure remains unstable. Is the simulation converged?
No, your simulation has likely not reached full equilibrium. While energy and density can converge rapidly in the initial stages of a simulation, pressure often requires a significantly longer time to stabilize. [20] Relying solely on energy and density as indicators is insufficient and can lead to inaccurate results. You should monitor additional properties, such as the Radial Distribution Function (RDF), particularly for key molecular components like asphaltenes, which converge much slower and are a better indicator of true system equilibrium. [20]
2. What is a more reliable metric than energy for assessing the equilibrium of my molecular dynamics (MD) system?
The convergence of the Radial Distribution Function (RDF) curve is a more robust indicator of system equilibrium, especially for complex components. [20] In systems such as asphalt, the resin, aromatics, and saturates may converge quickly, but the asphaltene-asphaltene RDF curve converges much slower. The simulation can only be considered truly balanced once this curve has stabilized. [20] Non-converged RDF curves often appear as a superposition of multiple irregular peaks and exhibit considerable fluctuations. [20]
3. How can I simulate bond breaking in my MD study, as my current harmonic force field does not allow it?
You can replace the harmonic bond potentials in your force field with reactive Morse potentials, as implemented in methods like the Reactive INTERFACE Force Field (IFF-R). [21] This substitution allows for bond dissociation while maintaining the accuracy of the original non-reactive force field and is compatible with common force fields like CHARMM, AMBER, and OPLS-AA. [21] This approach is about 30 times faster than complex bond-order potentials like ReaxFF. [21] The key parameters required are the bond dissociation energy ((D{ij})) and the equilibrium bond length ((r{0,ij})), which can be derived from experimental data or quantum mechanical calculations. [21]
4. What are the primary causes of force divergence and structural instability during energy minimization and equilibration?
Force divergence often stems from:
- Excessive repulsive forces in the initial system configuration, which can cause atomic velocities to become too high. [20]
- Incorrect force field parameters for the specific atoms and bonds in your system.
- Insufficient energy minimization before starting the dynamics simulation, failing to relieve high-energy clashes. Structural instability can arise from improper thermostat/barostat settings, an unstable integration time step, or modeling chemical reactions with a non-reactive (harmonic) force field. [21]
5. How can machine learning help troubleshoot issues related to molecular properties?
Machine Learning (ML) can analyze MD-derived properties to predict complex behaviors like solubility, potentially identifying the root cause of simulation instability. [22] For instance, ML models can use properties such as Solvent Accessible Surface Area (SASA), Coulombic and Lennard-Jones interaction energies (LJ), and Root Mean Square Deviation (RMSD) to predict aqueous solubility, which is critical for drug development. [22] If a simulated molecule is predicted to have poor solubility based on these MD properties, it might indicate underlying stability issues in the simulation that reflect real-world physicochemical challenges. [22]
Quantitative Data and Parameter Reference
| System Property | Typical Relative Convergence Time | Notes |
|---|---|---|
| Energy (Potential, Kinetic) | Fast | Stabilizes quickly in initial stages; not a sole indicator of equilibrium. |
| Density | Fast | Stabilizes quickly in initial stages; not a sole indicator of equilibrium. |
| Pressure | Slow | Requires much longer time to equilibrate compared to energy/density. |
| RDF (Aromatics, Resins) | Moderate | Faster than asphaltenes. |
| RDF (Asphaltene-Asphaltene) | Very Slow | The definitive indicator for full system equilibrium. |
| Morse Parameter | Symbol | Typical Range | Description / Source |
|---|---|---|---|
| Dissociation Energy | (D_{ij}) | System-dependent | Bond dissociation energy; from experiment or quantum mechanics (CCSD(T), MP2). |
| Equilibrium Bond Length | (r_{0,ij}) | System-dependent | Same as in the original harmonic potential. |
| Width Parameter | (\alpha_{ij}) | ~2.1 ± 0.3 Å⁻¹ | Fits the Morse curve to the harmonic curve near the resting state; can be refined via IR/Raman spectroscopy. |
Experimental Protocols and Workflows
Protocol 1: Validating System Convergence via RDF Analysis
Objective: To determine if an MD simulation has reached true thermodynamic equilibrium beyond energy and density stabilization. [20]
Methodology:
- System Preparation: Construct your initial model with a density much lower than the target final density to ensure a random molecular distribution. [20]
- Energy Minimization: Perform energy minimization to eliminate excessive repulsive forces and prevent unstable atomic velocities. [20]
- Equilibration: Run a relatively long equilibration simulation under the desired temperature and pressure (NPT ensemble). [20]
- Trajectory Analysis: Calculate the RDFs for all key molecular components in your system throughout the simulation trajectory.
- Convergence Check: Monitor the RDF curves over time. The system is considered equilibrated only when the RDF for the slowest-converging component (e.g., asphaltene-asphaltene) shows a stable, smooth profile with a distinct peak. [20]
Protocol 2: Implementing Reactivity with Morse Potentials
Objective: To simulate bond dissociation and failure in materials using a reactive force field. [21]
Methodology:
- Parameter Identification: Identify the specific bond types (atom pairs ij) you wish to make reactive.
- Parameter Acquisition: For each reactive bond type, obtain:
- The equilibrium bond length ((r{0,ij})) from your existing harmonic force field.
- The bond dissociation energy ((D{ij})) from experimental data or high-level quantum mechanical calculations.
- The width parameter ((\alpha_{ij})), initially set to ~2.1 Å⁻¹ and refined against experimental vibrational spectra. [21]
- Force Field Modification: In your simulation input, replace the harmonic bond potential for the specified atom types with a Morse potential term: (E{\text{bond}} = D{ij} [1 - e^{-\alpha{ij}(r{ij} - r_{0,ij})}]^2).
- Simulation and Validation: Run the simulation with the modified parameters. Validate that the bulk and interfacial properties of the non-reactive parts of the system remain accurate. [21]
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for MD Troubleshooting
| Tool / Resource | Function | Application Context |
|---|---|---|
| GROMACS [22] [23] | A high-performance MD simulation package. | Used for running simulations, energy minimization, and equilibration in various fields, including drug solubility studies. [22] |
| CHARMM, AMBER, OPLS-AA [21] [23] | Biomolecular force fields. | Provide the foundational parameters for simulating proteins, nucleic acids, and lipids. Can be extended with reactive potentials. [21] |
| IFF-R (Reactive INTERFACE FF) [21] | A reactive force field using Morse potentials. | Enables bond breaking in complex materials and biomolecular systems; compatible with several standard force fields. [21] |
| ReaxFF [21] | A reactive bond-order force field. | An alternative method for simulating chemical reactions; generally more computationally expensive than Morse potential approaches. [21] |
| AlphaFold [24] | Protein structure prediction tool. | Provides accurate 3D structural models of protein targets when experimental structures are unavailable, crucial for setting up valid simulations. [24] |
| Machine Learning Models [22] | Predictive analysis of MD properties. | Uses descriptors like SASA and DGSolv to predict properties like solubility, helping to diagnose and anticipate simulation outcomes. [22] |
Force Field Limitations and Their Impact on Minimization Success
Energy minimization is a foundational step in molecular dynamics (MD) simulations, essential for relieving steric clashes and preparing a stable system for production runs. The success of this process is intrinsically linked to the choice and parameterization of the molecular mechanics force field. Force fields provide the mathematical framework and parameters that describe the potential energy of a system as a function of its atomic coordinates. Limitations in these force fields—whether due to incomplete parameter sets, inaccurate functional forms, or improper application—can directly lead to minimization failures, including non-convergence, unphysical structures, or catastrophic simulation instability. This guide addresses these critical failure points within the context of academic research, providing troubleshooting methodologies for researchers and drug development professionals.
FAQs: Addressing Common Force Field and Minimization Issues
Q1: My energy minimization fails with a message that "the forces have not converged" and shows a very high maximum force. What force field-related issues could cause this?
This common error indicates the minimizer cannot find a geometry where the maximum force is below the specified threshold. Force field-related causes often include [25] [2]:
- Missing or Incorrect Parameters: The topology may be missing bond, angle, or dihedral parameters for certain residues or ligands, leading to uncontrolled movement and high forces.
- Steric Clashes from Poor Initial Structure: Incorrect initial structures, often stemming from earlier force field application issues in building (e.g., with
pdb2gmx), can create severe atomic overlaps that the minimizer cannot resolve. - Incompatible Force Fields: Mixing parameters from different force fields (e.g., using a protein from CHARMM and a ligand from AMBER without proper cross-term parameterization) creates internal contradictions in the energy landscape, preventing convergence [25].
- Incorrect Treatment of Terminal Residues: As highlighted in GROMACS documentation, improper naming of N- or C-terminal residues (e.g., using "ALA" instead of "NALA" for the N-terminus in AMBER force fields) leads to missing atoms or incorrect bonding patterns, causing large forces [25].
Q2: During system setup, I get an error that a residue is not found in the residue topology database. How should I proceed?
This pdb2gmx error means the force field you selected does not contain a definition for the specified residue (e.g., a non-standard amino acid or a novel drug molecule) [25]. Your options are:
- Rename the Residue: If the residue is standard but named differently in the force field's database, rename it in your coordinate file [25].
- Use a Different Force Field: Switch to a force field that includes parameters for your residue.
- Manually Provide a Topology: You cannot use
pdb2gmxfor arbitrary molecules. You must create a topology for the residue manually or using other tools (e.g.,x2topor external programs likeACPYPEorPRODRG), and then include it in your system's top file [25]. - Parameterize the Residue: As a last resort, you may need to derive new parameters for the residue yourself, a complex process that requires significant expertise [25].
Q3: My simulation fails with a "segmentation fault" or "non-numeric atom coords - simulation unstable" error during or after minimization. Could this be force field-related?
Yes. While segmentation faults can have computational causes (faulty hardware, compiler issues), they often follow minimization and indicate profound instability, frequently tied to force field problems [2] [26].
- Severe Parameter Inconsistency: The force field parameters may be physically unrealistic for the system, leading to "blowing up" as atoms are propelled with enormous velocities. This is a known risk in reactive force fields like ReaxFF if parameters are inconsistent [27] [26].
- Topology Errors: Incorrect atom ordering, charge assignments, or bonded terms in the topology can generate impossible forces.
- Long-Range Electrostatics Mismanagement: Improper treatment of long-range electrostatic interactions can cause artifacts that destabilize the simulation.
Q4: What are the fundamental limitations of traditional force fields that can affect simulation accuracy?
Traditional, widely-used biomolecular force fields (AMBER, CHARMM, OPLS-AA, GROMOS) have inherent limitations that can impact minimization and subsequent dynamics [28] [29]:
- Fixed Atomic Charges: They use atom-centered point charges that do not account for polarization effects—the change in electron distribution due to the chemical environment. This can lead to inaccurate descriptions of interactions in heterogeneous systems like protein-ligand complexes [28].
- Non-Reactive Potentials: They use harmonic potentials for bonds, which cannot describe bond breaking and formation. This limits their application in studying chemical reactions or mechanical failure [29].
- Functional Form: The relatively simple functional form of the potential energy may not fully capture the complexity of quantum mechanical potential energy surfaces, sometimes requiring empirical corrections to compensate [28].
- Training Set Bias: Each force field is derived from a specific training set of molecules and properties, which can introduce a bias and reduce transferability to novel molecular systems [28].
Troubleshooting Guide: A Systematic Workflow
Follow this logical workflow to diagnose and resolve force field-related minimization problems. The diagram below outlines the key decision points.
Step-by-Step Diagnostic and Resolution Procedures
Step 1: Analyze the Error Message and Log File Carefully read the minimization output and log file. Specific warnings before the failure are crucial. Note the value and location of the maximum force, as this points to the problematic atom group [2].
Step 2: Check Topology and Parameter Files
- Diagnostic: Use tools like
gmx pdb2gmx -horgmx grompp -vto check for warnings about missing parameters or duplicate directives. The error "Found a second defaults directive" indicates multiple[defaults]sections in your topology, often from incorrectly includeditpfiles [25]. - Resolution: Ensure the
[defaults]directive appears only once in your main topology file. Comment out extra[defaults]sections in includeditpfiles. For missing parameters, consult the force field's literature or use parameter derivation tools [25].
Step 3: Validate the Initial Structure
- Diagnostic: Look for warnings about "long bonds," "missing atoms," or "atom ... not found in rtp entry" during
pdb2gmxexecution [25]. - Resolution: For missing hydrogens, consider using the
-ignhflag to letpdb2gmxadd them correctly. For missing heavy atoms, you must model them using external software before proceeding [25].
Step 4: Review the Minimization Protocol (mdp Parameters)
- Diagnostic: The minimizer stops but reports an unacceptably high maximum force [2].
- Resolution: The Steepest Descent algorithm, while robust for initial steps, can have slow convergence. As a hybrid approach, start with Steepest Descent to remove severe clashes, then switch to the Conjugate Gradient algorithm for finer convergence [30]. Ensure your
emtol(force tolerance) is realistic (e.g., 1000 kJ/mol/nm for initial minimization, 10-100 for finer minimization). Temporarily increasing theemstep(step size) can also help escape high-energy states.
Step 5: Test with a Simpler System If the complex system fails, isolate the problem. Run minimization on individual components: the protein alone, the ligand alone, and the solvent box alone. This identifies which component's topology or parameters are causing the instability.
Step 6: Verify Force Field Consistency Ensure all components of your system (protein, DNA, ligand, ions, water) are parameterized with the same force field family. Mixing force fields (e.g., AMBER for protein and CHARMM for water) without validated cross-compatibility is a common source of instability [25] [29].
Advanced Topic: Selecting and Evaluating Force Fields
Performance Comparison of Modern DNA Force Fields
Recent assessments of the AMBER force field family for DNA simulations reveal performance variations. The following table summarizes key findings from a 2023 study comparing force fields when paired with different water models [31].
Table: Performance Assessment of Recent AMBER DNA Force Fields (2023)
| Force Field | Recommended Water Model | Performance with B-DNA | Performance with Z-DNA | Key Characteristics |
|---|---|---|---|---|
| OL21 | OPC | Excellent, currently recommended | Significantly better than Tumuc1 | Incremental improvement from OL15; refined α/γ dihedrals [31]. |
| Tumuc1 | OPC or TIP3P | Similar to OL21 | Discrepancies observed | Comprehensive reparameterization of bonded terms using QM [31]. |
| bsc1 | TIP3P or SPC/E | Good | Adequate | Includes bsc0 modifications plus refinements to χ, ε, ζ dihedrals [31]. |
| OL15 | TIP3P or SPC/E | Good | Adequate | Predecessor to OL21; includes improvements to β dihedral [31]. |
Experimental Protocol: Force Field Assessment for DNA Systems
The quantitative data in the previous table was generated using the following detailed methodology, which can serve as a template for evaluating force fields in your own research context [31]:
- System Preparation: Start with experimental structures (NMR or high-resolution X-ray crystal structures). Remove crystallographic water and counterions to ensure a clean starting point.
- Solvation and Ion Addition: Solvate the DNA in a truncated octahedron box with a 10 Å buffer using the chosen water model (e.g., TIP3P, OPC). Add net-neutralizing counterions (Na⁺, Cl⁻) using the Joung and Cheatham ion parameters, plus excess salt to achieve a physiological concentration of ~200 mM.
- Equilibration: Perform a multi-step equilibration:
- Hold the solute under strong positional restraints (5 kcal mol⁻¹ Å⁻²) for 1 ns.
- Gradually reduce the restraints (0.5, then 0.1 kcal mol⁻¹ Å⁻²) in successive 1 ns simulations.
- Conclude with a short (1 ns) unrestrained equilibration. Use a 1 fs integration time step during equilibration.
- Production Simulation: Run multiple independent replicas (e.g., 5 per system) for an extended time (microsecond scale) in the NPT ensemble at 300 K. Use Langevin dynamics for temperature control and a barostat for pressure regulation. An integration time step of 4 fs can be enabled by employing hydrogen mass repartitioning.
- Data Analysis: Analyze the aggregated simulation time from all replicas, discarding the initial segment (e.g., first 1000 ns) as additional equilibration. Compare properties like helical parameters, backbone dihedrals, and root-mean-square deviation (RMSD) to experimental data.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table: Key Resources for Force Field Parameterization and Validation
| Reagent / Resource | Function in Research | Application Context |
|---|---|---|
| Residue Topology Database (RTP) | Defines atom types, connectivity, and bonded interactions for molecular building blocks (e.g., amino acids, nucleotides) [25]. | Essential for pdb2gmx to generate topologies for standard residues. |
| Restrained Electrostatic Potential (RESP) | A method for deriving atomic partial charges by fitting to the quantum mechanically calculated electrostatic potential [28]. | Used in AMBER force fields to assign charges for new molecules. |
| Joung-Cheatham Ion Parameters | Optimized parameters for monovalent ions (Na⁺, Cl⁻, K⁺) for use with specific water models like TIP3P and SPC/E [31]. | Critical for achieving physiological ionic conditions in simulations. |
| Hydrogen Mass Repartitioning (HMR) | A technique that allows for a larger MD integration time step (4 fs) by redistributing mass from hydrogens to the bonded heavy atoms [31]. | Used to accelerate production simulations after equilibration. |
| Reactive INTERFACE Force Field (IFF-R) | A reactive force field that replaces harmonic bonds with Morse potentials to simulate bond breaking and formation [29]. | Used for simulating chemical reactions, polymer failure, and other reactive processes. |
Force field limitations present significant but surmountable challenges in molecular dynamics minimization. Success hinges on a systematic approach: a thorough understanding of the force field's scope and parameters, careful system construction, and methodical troubleshooting. The field continues to advance with more accurate, polarizable, and reactive force fields. By applying the diagnostics and solutions outlined in this guide, researchers can effectively overcome minimization failures and build a solid foundation for reliable and meaningful molecular dynamics simulations.
Practical Implementation: Algorithm Selection and Parameter Optimization
Algorithm Comparison at a Glance
The table below summarizes the core characteristics of the Steepest Descent and L-BFGS algorithms to help you make an informed choice.
| Feature | Steepest Descent | L-BFGS |
|---|---|---|
| Core Principle | Follows the direction of the negative gradient (force) [32]. | Uses gradient and position history to approximate the Hessian (curvature) for a smarter search direction [33]. |
| Convergence Speed | Slow, linear convergence rate [34]. | Fast, superlinear convergence rate near the minimum [35]. |
| Computational Cost per Step | Very low; requires only force calculations [32]. | Low; requires force calculations and vector-vector operations [36] [33]. |
| Memory Usage | Very low (O(1)) [32]. |
Low (O(m*N), where m is memory size) [33]. |
| Robustness | High; guaranteed progress at every step [32] [35]. | Moderate; can fail on very ill-conditioned or non-convex problems without safeguards [35] [37]. |
| Typical Use Case in MD | Initial minimization from poor starting structures, systems with clashes [32] [2]. | Final minimization stages, geometry optimization of stable structures [36] [32]. |
Understanding the Algorithms
Steepest Descent
This is one of the simplest optimization algorithms. It calculates the force (the negative gradient of the potential energy) on each atom and moves the atoms in the direction of the force to lower the energy [32]. The step size is adaptive: it is increased by 20% after a successful step that lowers the energy and drastically reduced by 80% after a step that increases the energy [32]. While this makes it very robust and stable, even from poor starting points, its convergence is slow because it does not use information about the curvature of the energy landscape.
L-BFGS (Limited-Memory Broyden–Fletcher–Goldfarb–Shanno)
L-BFGS is a quasi-Newton method. It significantly improves upon Steepest Descent by using information from the last few steps to build an approximation of the energy landscape's curvature (the inverse Hessian matrix) [33]. This allows it to find better search directions and converge much faster once it's near a minimum. Its "Limited-Memory" nature means it does not store a dense matrix, making it suitable for large systems with thousands of atoms [36] [33].
Troubleshooting Common Scenarios
My energy minimization stops abruptly without force convergence.
This is a common issue, often encountered with Steepest Descent. The simulation stops because the algorithm can no longer find a step that lowers the energy, even though the forces are still high.
- Problem Analysis: This typically indicates your system is stuck in a very flat region of the energy landscape or on a "ledge" where tiny steps no longer yield progress [2]. The algorithm's adaptive step size becomes so small that it cannot make meaningful progress.
- Solution:
- Switch Algorithms: As suggested in forum discussions, use the output from the failed Steepest Descent run as the input for a more efficient algorithm like L-BFGS or Conjugate Gradient [2]. L-BFGS, with its curvature information, is often better at escaping these flat regions.
- Check System Integrity: Verify your initial structure for unphysical clashes, incorrect bond lengths, or missing parameters that might create insurmountable energy barriers [2].
L-BFGS fails to start or converges poorly after many steps.
- Problem Analysis: L-BFGS relies on the history of steps and gradients. If the initial geometry is pathological or the energy landscape is highly non-convex, the history can contain inconsistent information, leading to a poor search direction [35] [37].
- Solution:
- Use Steepest Descent First: Always use a few dozen steps of Steepest Descent to first relax the worst steric clashes and bring the system to a smoother region of the energy landscape. Then, switch to L-BFGS for efficient convergence [32] [2].
- Ensure Proper Conditioning: The performance of L-BFGS, like other gradient-based methods, can be sensitive to the scaling of different variables. Poor conditioning can lead to slow convergence [34].
Essential Research Reagent Solutions
The table below lists key computational "reagents" for your energy minimization experiments.
| Item | Function |
|---|---|
| Steepest Descent Algorithm | Provides a robust, fall-back minimizer for initial stabilization of poorly configured systems and for overcoming initial steric clashes [32]. |
| L-BFGS Algorithm | The workhorse for efficient, high-precision energy minimization in most production scenarios, offering a good balance of speed and accuracy [36] [32]. |
| Position Restraints | Allows for the selective relaxation of parts of a system (e.g., solvent) while holding critical regions (e.g., protein backbone) fixed, which is a common equilibration protocol [2]. |
| Line Search (e.g., BFGSLineSearch) | An advanced component that determines the optimal step size along a search direction, stabilizing optimization and preventing oscillation [36]. |
Experimental Protocol: A Standard Minimization Workflow
For a typical molecular system (e.g., a protein-DNA complex in solvent), follow this two-stage protocol to ensure robust and efficient minimization [32] [2]:
Initial Robust Minimization with Steepest Descent
- Objective: Remove any severe steric clashes and bring the system to a stable, albeit not fully minimized, state.
- Integrator:
steep - Force Tolerance (
emtol):1000.0kJ/mol/nm (or a similarly loose tolerance) - Maximum Steps (
nsteps):500 - 1000 - This stage is expected to converge to machine precision but not to the desired force tolerance.
High-Precision Minimization with L-BFGS
- Objective: Achieve the desired force convergence for a stable starting configuration for molecular dynamics.
- Integrator:
l-bfgs - Force Tolerance (
emtol):10.0 - 500.0kJ/mol/nm (dependent on your production run requirements) - Maximum Steps (
nsteps):1000 - 5000 - Use the final structure from the Steepest Descent run as the input for this stage.
Decision Workflow for Energy Minimization
The following diagram outlines the logical process for selecting and troubleshooting energy minimization algorithms in your research.
Frequently Asked Questions
1. What are the most critical parameters for energy minimization in molecular dynamics?
The most critical parameters are the force tolerance (emtol), the maximum number of iterations (nsteps), and for the steepest descent algorithm, the initial step size (emstep). Proper tuning of these parameters is essential for achieving a stable, minimized system as a prerequisite to molecular dynamics production runs. [5]
2. My minimization fails to converge. What should I check first?
First, verify that your force tolerance (emtol) is set to an appropriate value, typically 100-1000 kJ mol⁻¹ nm⁻¹, and that the maximum number of iterations (nsteps) is high enough to allow the convergence to occur. Secondly, for the steepest descent integrator, ensure the initial step size (emstep) is not too large, as this can cause instability. A good starting point is 0.01 nm. [5]
3. How do I know if my energy minimization has been successful?
A successful minimization will terminate when the maximum force in the system falls below the value specified by emtol. The log file will typically state "converged to within the requested tolerance" or similar. You should also observe a steady, significant decrease in the potential energy throughout the process. [5]
4. Should I use steepest descent or conjugate gradient for minimization?
Steepest descent (integrator = steep) is more robust for initially relieving severe steric clashes in a structure, as it is less likely to fail on the first steps. The conjugate gradient algorithm (integrator = cg) is more efficient for later stages of minimization and for achieving a more precise minimum but can be less stable with poorly starting structures. [5]
5. What is the function of the nstcgsteep parameter?
The nstcgsteep parameter specifies how often a steepest descent step is performed when using the conjugate gradient algorithm. Periodically using steepest descent can improve the efficiency and stability of the conjugate gradient minimization. The default in GROMACS is to do this every 1000 steps. [5]
Troubleshooting Guides
Problem: Energy Minimization Fails to Converge
- Symptoms: The minimization hits the maximum number of iterations (
nsteps) without the maximum force dropping below the specified tolerance (emtol). The log file may show that the energy or force is no longer decreasing. - Diagnosis and Solutions:
- Force tolerance is too strict: A very low
emtol(e.g., 1 kJ mol⁻¹ nm⁻¹) requires an extremely precise minimum and can take a prohibitively long time to achieve.- Solution: Increase
emtolto a more reasonable value. For a preliminary minimization before a molecular dynamics run, a value of 100-1000 is often sufficient. [5]
- Solution: Increase
- Maximum iterations is too low: The system simply needs more steps to find the minimum.
- Solution: Increase the
nstepsparameter. For large or highly distorted systems, several thousand steps may be necessary.
- Solution: Increase the
- Underlying structural problems: Severe atomic clashes, missing atoms, or incorrect bonds can create forces that cannot be minimized effectively. [12]
- Solution: Inspect your initial structure for anomalies. Check the output of
pdb2gmxfor warnings about missing atoms or long bonds, and correct your input structure accordingly. [12]
- Solution: Inspect your initial structure for anomalies. Check the output of
- Force tolerance is too strict: A very low
Problem: Minimization Becomes Unstable (Crash or Blowing Up)
- Symptoms: The simulation crashes, or the potential energy becomes astronomically high (e.g.,
1e+NaN). - Diagnosis and Solutions:
- Step size is too large: This is a common cause with the steepest descent algorithm. A large initial step can cause atoms to be moved too far, leading to even larger forces and instabilities. [5]
- Solution: Drastically reduce the
emstepparameter. Tryemstep = 0.001nm. Theemstepvalue is also used as the initial step for the first step of conjugate gradient minimization. [5]
- Solution: Drastically reduce the
- Incorrect constraints: Using
constraints = all-bondswith a simple minimization integrator can sometimes cause issues.- Solution: For initial minimization, try using
constraints = noneor use the LINCS constraint algorithm (constraints = h-bondsorconstraints = h-angles). [38]
- Solution: For initial minimization, try using
- Step size is too large: This is a common cause with the steepest descent algorithm. A large initial step can cause atoms to be moved too far, leading to even larger forces and instabilities. [5]
Problem: Minimization is Inefficient (Extremely Slow)
- Symptoms: The energy is decreasing consistently but at a very slow rate, making it impractical to reach the desired tolerance.
- Diagnosis and Solutions:
- Inefficient algorithm for the stage of minimization:
- Solution: Use a hybrid approach. Start with the robust steepest descent algorithm (
integrator = steep) for 50-100 steps to relieve the worst clashes, then switch to the more efficient conjugate gradient (integrator = cg) to refine the structure to the desired tolerance. [5]
- Solution: Use a hybrid approach. Start with the robust steepest descent algorithm (
- Conjugate gradient is stuck:
- Solution: The
nstcgsteepparameter forces a steepest descent step periodically during conjugate gradient minimization, which can help the algorithm escape from shallow regions. Ensure this is enabled (default is every 1000 steps). [5]
- Solution: The
- Inefficient algorithm for the stage of minimization:
Parameter Tables for Energy Minimization
Table 1: Core Energy Minimization Parameters in GROMACS
| Parameter | MDP Option | Description | Typical Values / Range |
|---|---|---|---|
| Integrator | integrator |
Algorithm used for minimization. [5] | steep (steepest descent), cg (conjugate gradient) [5] |
| Force Tolerance | emtol |
Convergence criterion; minimization stops when the maximum force is below this value. [5] | 10.0 - 1000.0 [kJ mol⁻¹ nm⁻¹] [5] |
| Max Iterations | nsteps |
Maximum number of minimization steps allowed. [5] | 100 - 50000 (system dependent) |
| Initial Step Size | emstep |
Initial step size (in nm) for the steepest descent algorithm. [5] | 0.001 - 0.01 [nm] [5] |
| CG Steepest Desc. Freq. | nstcgsteep |
Frequency of steepest descent steps during conjugate gradient minimization. [5] | 1000 [steps] (default) [5] |
Table 2: Troubleshooting Guide Summary Table
| Symptom | Likely Cause | Suggested Action |
|---|---|---|
| Fails to converge | emtol too low or nsteps too small. [5] |
Increase emtol; increase nsteps. |
| Crashes immediately | Severe steric clashes; emstep too large. [12] [5] |
Check structure for clashes; reduce emstep to 0.001. |
| Energy becomes NaN | Instability from large forces/step size. [5] | Reduce emstep; try a more robust integrator (steep). |
| Extremely slow progress | Inefficient algorithm for the current state. | Use steep first, then switch to cg; check nstcgsteep. |
Experimental Protocols
Protocol 1: Standard Two-Stage Energy Minimization for a Solvated Protein-Ligand System
This protocol is designed to efficiently minimize a typical system while avoiding instability.
- System Preparation: The system (e.g., protein, ligand, solvated in a water box with ions) is prepared using the standard
gromppcommand to generate a run input file (.tpr). A sample.mdpfile for the first stage is provided below. - Stage 1 - Steepest Descent: Run the first minimization stage using a robust steepest descent algorithm to relieve major atomic clashes.
- MDP Parameters:
integrator = steepemtol = 1000.0nsteps = 500emstep = 0.01
- Execution:
gmx grompp -f stage1.mdp -c system.gro -p topol.top -o min1.tprgmx mdrun -v -deffnm min1
- MDP Parameters:
- Stage 2 - Conjugate Gradient: Use the output from Stage 1 as input for a more precise minimization using the conjugate gradient algorithm.
- MDP Parameters:
integrator = cgemtol = 10.0nsteps = 5000nstcgsteep = 1000
- Execution:
gmx grompp -f stage2.mdp -c min1.gro -p topol.top -o min2.tprgmx mdrun -v -deffnm min2
- MDP Parameters:
- Validation: Check the output of
gmx energyfor the potential energy of themin2run to ensure a steady decrease and final convergence.
Protocol 2: Troubleshooting Severe Steric Clashes
If a system fails the standard protocol with instabilities, this aggressive protocol can be used.
- Ultra-Conservative Steepest Descent:
- MDP Parameters:
integrator = steepemtol = 1000.0nsteps = 100emstep = 0.001# Very small step size for stabilityconstraints = none# Remove constraints to simplify initial minimization
- Run this step and visually inspect the resulting structure. If stable, use its output as input for Protocol 1.
- MDP Parameters:
The Scientist's Toolkit
Table 3: Essential Software and Analysis Tools
| Item | Function | Source / Command |
|---|---|---|
| GROMACS | Molecular dynamics simulation package used for energy minimization and production runs. [22] | https://www.gromacs.org |
| pdb2gmx | GROMACS tool to generate topology and coordinates from a PDB file. Critical for preparing the initial system. [12] | gmx pdb2gmx -f protein.pdb -p topol.top -ff forcefield |
| grompp | GROMACS preprocessor. Assembles the .tpr run input file from the .mdp parameters, topology, and coordinates. [12] |
gmx grompp -f min.mdp -c system.gro -p topol.top -o min.tpr |
| mdrun | GROMACS MD engine. Executes the minimization or simulation. [22] | gmx mdrun -v -deffnm min |
| energy | GROMACS analysis tool to extract energy terms (like Potential Energy) from the output file for plotting and validation. | gmx energy -f min.edr -o potential.xvg |
Workflow and Relationship Diagrams
Energy Minimization Troubleshooting Workflow
Relationship Between Key Minimization Parameters
FAQs: Core Parameter Definitions and Selection
Q1: What are the precise definitions of emtol and emstep, and what are their typical units and value ranges?
emtol and emstep are critical parameters that control the convergence and step size in GROMACS energy minimization.
Table 1: Core Energy Minimization Parameters
| Parameter | Definition | Units | Typical Value Range | Function |
|---|---|---|---|---|
emtol |
The convergence tolerance; minimization stops when the maximum force (Fmax) on any atom falls below this value. |
kJ mol⁻¹ nm⁻¹ | 10 - 1000 (MD prep) [39] [40] [41] | Sets the quality of the minimized structure. |
emstep |
The initial step size for the steepest descent algorithm. | nm | 0.001 - 0.01 [39] [40] | Controls how far atoms move in each step. |
Q2: How do I choose the correct integrator for energy minimization?
The integrator mdp option specifies the algorithm used for minimization. For energy minimization, the primary choices are steep (steepest descent) and cg (conjugate gradient) [5].
steep: A robust steepest descent algorithm. It is less efficient than conjugate gradient but is more stable for poorly structured starting configurations, making it ideal for the initial stages of minimization [5].cg: A conjugate gradient algorithm. It is more efficient than steepest descent once the initial large forces have been reduced, but it may fail if the initial forces are very high [5] [40]. The parameternstcgsteepdetermines how often a steepest descent step is performed during a conjugate gradient minimization to improve stability [5].
Q3: My minimization fails with "forces have not converged to the requested precision." What are the primary causes?
This is a common error indicating that the minimization stopped before the forces reached the desired emtol. The two most frequent underlying causes are:
- Extremely high initial forces: The algorithm cannot reduce the forces to the target
emtolbecause the starting configuration is too unstable [39] [41]. This can be due to atomic overlaps, incorrect topology, or misplaced molecules. - Machine precision limit: The minimization has converged to the lowest energy possible for the given configuration and precision setting, but this is still above the requested tolerance [39] [40]. The message often states it "converged to machine precision."
Troubleshooting Guide: Energy Minimization Failures
Error: Incomplete Convergence and High Forces
Problem Description
The energy minimization run terminates with a warning that the forces have not converged to the requested Fmax < [emtol], despite the Potential Energy (Epot) becoming more negative. The maximum force (Fmax) remains orders of magnitude too high, for example, 1.91991e+05 versus a target of 10 [39].
Diagnosis Methodology This error requires a systematic diagnosis to isolate the root cause. The following workflow outlines the recommended investigative process, from initial checks to advanced solutions.
Diagram 1: Troubleshooting workflow for minimization convergence failures.
Experimental Protocols for Resolution
Protocol A: Two-Stage Minimization with Optimized Parameters This protocol is effective when dealing with severe atomic overlaps or a poor initial structure [42].
- Stage 1 - Coarse Minimization:
- Objective: Quickly relieve severe clashes and high-energy contacts.
- Parameters: Use the
steepintegrator with a relaxedemtolof 100-1000 and a conservativeemstepof 0.001. A higheremtolprevents the algorithm from getting stuck on machine precision for a very bad configuration [39] [40]. - Sample MDP Snippet:
- Stage 2 - Fine Minimization:
Protocol B: Topology and Charge Validation Incorrect system topology or net charge is a major source of high, irresolvable forces [42].
- Validate Residue Topologies: Ensure all molecules (protein, ligand, ions) have correct residue topology (
rtp) entries in the force field database. The errorResidue not found in residue topology databaseindicates a missing entry [43]. - Check Total System Charge: Use
gmx gromppto report the total charge of the system. A large net charge in a periodic system creates infinite electrostatic energy, preventing minimization. Neutralize the system by adding appropriate counter-ions usinggmx genion[42].
Protocol C: Constraint Handling Overly rigid constraints can prevent the system from relaxing properly.
- Action: In the minimization mdp file, set
constraints = none. This allows all atoms, including bond vibrations, to move freely during minimization, which can help resolve clashes [39] [41]. Constraints should be re-enabled for the subsequent equilibration and production runs.
Error: Minimization Stalls with No Energy Change
Problem Description
The minimization stops after very few steps (e.g., 15 steps) with a message that there was "no change in the energy since last step" or that the step size was too small, even though Fmax is still very high [39] [41].
Diagnosis and Solution This typically indicates that the steepest descent algorithm can no longer find a downhill direction given its current step size and the local energy landscape.
Table 2: Advanced MDP Parameters for Stalled Minimization
| Parameter | Function | Recommended Adjustment |
|---|---|---|
emstep |
Initial step size for steepest descent. | Increase cautiously (e.g., to 0.01 or 0.02) to allow the algorithm to escape shallow local minima [40]. |
nstcgsteep |
Frequency of steepest descent steps during CG minimization. | For integrator=cg, setting this (e.g., nstcgsteep = 1000) periodically resets the search direction, improving stability [5] [40]. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Components for Energy Minimization Setup
| Reagent / Component | Function | Technical Specification |
|---|---|---|
| Force Field (.itp files) | Defines the potential energy function and parameters for all molecules. | Examples: AMBER ff99SB-ILDN, CHARMM36. Must be consistent for all components [44]. |
| Molecular Topology (.top file) | Describes the system composition: atoms, bonds, angles, dihedrals, and non-bonded interactions. | Generated via pdb2gmx or manually assembled. Must be error-free [43] [42]. |
| Coordinate File (.gro/.pdb) | Contains the initial 3D atomic coordinates of the system. | Must be structurally sound, with no severe atomic overlaps or missing atoms [43]. |
| Water Model | Solvates the system and provides a realistic environment. | Examples: SPC/E, TIP3P, TIP4P. Chosen for compatibility with the force field [44]. |
| Ions | Neutralizes the system's net charge and mimics physiological conditions. | Added via gmx genion. Critical for managing electrostatic interactions in Periodic Boundary Conditions [42]. |
FAQs
Q1: Why does my simulation become unstable immediately after adding solvent?
This is often caused by atomic overlaps between the solute and solvent molecules during the insertion process. These overlaps create extremely high potential energy points that the energy minimization cannot resolve, leading to a simulation "crash." To prevent this, ensure you use the correct commands and parameters for solvent addition, such as gmx solvate, which includes algorithms to minimize these overlaps. Always check the final system density after solvation to ensure it is realistic [42].
Q2: How can I tell if my system is properly neutralized, and why is it important?
An improperly neutralized system, particularly one with a net charge under Periodic Boundary Conditions (PBC), can lead to severe artifacts in electrostatic calculations, causing unrealistic forces and simulation instability. You can check the total charge of the system in your topology file (.top). Use tools like gmx genion to replace solvent molecules with ions to achieve a net zero charge. Incorrect charge assignments in the topology are a common source of this problem [42].
Q3: My energy minimization won't converge. What should I check first?
Convergence issues in energy minimization frequently stem from a poor starting configuration. This includes atomic overlaps, extremely high initial energy, or incorrect constraints. First, try using a two-stage minimization approach: initially using the steepest descent algorithm for its robustness in handling bad structures, followed by a conjugate gradient method for finer convergence. Also, verify that all constraints and restraints in your molecular dynamics parameter (.mdp) file are necessary and correctly defined [42].
Q4: When should I use constraints on my system? Constraints are essential for freezing the fastest degrees of freedom in the system, such as bond vibrations involving hydrogen atoms. This allows you to use a larger, more computationally efficient integration time step (e.g., 2 fs instead of 1 fs). They are commonly applied to all bonds, making water molecules rigid. However, for the energy minimization stage, consider using a flexible water model and avoiding bond constraints to allow the system to relax more freely and resolve conflicts [45] [7].
Troubleshooting Guides
Common Errors and Solutions
| Error Symptom | Potential Cause | Recommended Solution |
|---|---|---|
| Unstable pressure/temperature | Incorrect barostat/thermostat coupling parameters; insufficient equilibration time. | Use the c-rescale barostat; extend equilibration until properties stabilize; refine coupling constants [45] [42]. |
| "Blow-up" during simulation | Atomic overlaps from poor solvation/ion placement; incorrect constraint handling; time step too large. | Re-run solvation/ion placement; minimize without constraints; reduce time step; use position restraints during initial equilibration [45] [42]. |
| Energy minimization non-convergence | Poor starting structure with atomic clashes; inadequate minimization steps or parameters. | Use a two-stage minimization (steepest descent then conjugate gradient); increase the number of steps (nsteps); ensure a reasonable initial structure [45] [42]. |
| Ion clustering/distribution errors | Random ion placement too close to solute or each other. | Use gmx genion for controlled placement; verify ion distribution visually; ensure appropriate ion concentration [42]. |
| Artifacts from box boundary | Solute too close to the box edge; insufficient padding. | Ensure a minimum of 1.0 nm between the solute and the box edge after solvation [42]. |
Workflow for Robust System Preparation
Step-by-Step Experimental Protocol
1. Topology and Box Definition
- Generate System Topology: Use a tool like
gmx pdb2gmxto generate the molecular topology for your protein or solute, carefully selecting an appropriate force field [45] [46]. - Define Simulation Box: Use
gmx editconfto place the solute in a simulation box (e.g., cubic, dodecahedron). The box size must provide sufficient padding—typically at least 1.0 nm—between the solute and the box edges to prevent artificial self-interaction across periodic boundaries [42].
2. Solvation and Ion Placement
- Add Solvent: Use
gmx solvateto fill the box with solvent molecules (e.g., SPC, TIP3P water models). Inspect the output structure visually to check for gross overlaps [45] [42]. - Neutralize System: Use
gmx genionto replace solvent molecules with ions (e.g., Na⁺, Cl⁻) to achieve a net zero charge for the system. It is good practice to first create a run input file withgmx gromppfor this step [45].
3. Energy Minimization
- Run Minimization: Execute energy minimization using
gmx mdrun. The goal is to find the nearest local energy minimum and relieve any atomic clashes introduced during setup. - Convergence Criteria: Monitor the minimization log file. Convergence is typically achieved when the maximum force (as indicated in the log) falls below a chosen threshold (e.g., 100.0 kJ/mol/nm). The potential energy should also stop decreasing significantly [45] [42].
4. System Equilibration
- NVT Equilibration: Perform equilibration in the NVT ensemble (constant Number of particles, Volume, and Temperature) for 50-100 ps. Apply position restraints to the solute heavy atoms to allow the solvent to relax around it. Use a thermostat like Nosé-Hoover [45] [42].
- NPT Equilibration: Perform equilibration in the NPT ensemble (constant Number of particles, Pressure, and Temperature) for 100-200 ps or until density stabilizes. Continue with solute restraints. Use a barostat like C-rescale (Berendsen) to maintain constant pressure [45] [42].
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function |
|---|---|
gmx pdb2gmx |
Processes a protein coordinate file (.pdb) and generates a topology, choosing a specific force field and water model [45]. |
gmx editconf |
Modifies the simulation box dimensions and type, crucial for creating adequate padding around the solute [45]. |
gmx solvate |
Solvates a molecular system by filling the simulation box with solvent molecules, a key step for simulating biological conditions [45] [42]. |
gmx genion |
Replaces water molecules with ions to neutralize the system's charge or achieve a specific ion concentration [45] [42]. |
gmx grompp |
The GROMACS preprocessor; reads the topology, coordinates, and simulation parameters (.mdp file) to produce a run input file (.tpr) [45]. |
gmx mdrun |
The main GROMACS MD engine; performs the energy minimization, equilibration, and production simulation [45]. |
.mdp file |
A plain-text file containing all the parameters that control the simulation protocol, from integrator settings to temperature and pressure coupling [42]. |
Energy minimization is a fundamental step in Molecular Dynamics (MD) simulations, serving to remove unfavorable atomic clashes and steric conflicts within a system prior to production runs. For protein-ligand complexes, which are often constructed from diverse structural sources, this process is particularly crucial. A properly minimized system establishes a stable foundation for subsequent dynamics, while failures at this stage propagate through the entire simulation pipeline, leading to inaccurate results or complete algorithmic failure. This guide addresses the diagnosis and resolution of common minimization failures encountered during protein-ligand simulation studies, providing a structured troubleshooting framework for researchers.
Diagnosing Minimization Failures: A Troubleshooting Guide
Energy minimization failures manifest through specific error messages and numerical indicators. The table below categorizes common symptoms, their underlying causes, and immediate diagnostic actions.
Table 1: Common Minimization Failure Symptoms and Initial Diagnosis
| Observed Symptom/Error | Potential Underlying Cause | Immediate Diagnostic Action |
|---|---|---|
| Force Non-Convergence: "Energy minimization has stopped, but the forces have not converged to the requested precision Fmax < 1000" [10]. | Severe steric clashes, incorrect topology, or inappropriate position restraints [10]. | Check system for atomic clashes; verify restraint definitions in .mdp file. |
| Excessively High Potential Energy (e.g., 1.3e+32) [10]. | Catastrophic structural breakdown, often from a single bad contact (e.g., a misplaced water molecule or ligand) [10]. | Identify the atom with the Maximum Force (from log file) and visualize its local environment. |
pdb2gmx Error: "Residue ‘XXX’ not found in residue topology database" [47]. |
The force field does not contain parameters for a specific residue or molecule in your input file [47]. | Verify residue naming; prepare a ligand topology file separately if needed. |
pdb2gmx Error: "Atom X in residue YYY not found in rtp entry" [47]. |
Atom naming in the coordinate file does not match the expected names in the force field's residue topology [47]. | Rename atoms in your structure to match force field conventions. |
grompp Error: "Atom index n in position_restraints out of bounds" [47]. |
Position restraint files are included in the wrong order relative to their corresponding molecule topology [47]. | Ensure #include order for position restraints immediately follows their molecule's topology. |
grompp Error: "Found a second defaults directive" [47]. |
The [defaults] directive appears more than once in the topology, often from incorrectly mixed force fields [47]. |
Check all .itp files and comment out extra [defaults] sections. |
Workflow for Systematic Diagnosis
When a minimization failure occurs, follow this logical pathway to identify the root cause.
Resolving Common Issues: Protocols and Methodologies
Protocol A: Remediating Severe Steric Clashes and Force Non-Convergence
This protocol addresses the most common minimization failure, characterized by high potential energy and force non-convergence [10].
Detailed Methodology:
- Identify the Epicenter: From the minimization log file, locate the line stating "Maximum force" and note the atom number. For example:
Maximum force = 1.3916486e+11 on atom 5791[10]. - Visualize the Conflict: Use a molecular visualization tool (e.g., VMD [48], PyMOL [48]) to display the system and highlight the problematic atom. This often reveals a misplaced ligand, water molecule, or ion causing a catastrophic clash.
- Intervention and Re-minimization:
- Manual Adjustment: If the clash is isolated, manually delete or reposition the offending molecule in the coordinate file.
- Two-Stage Minimization: Implement a gentler, multi-stage approach:
- Stage 1: Run minimization with steepest descent for 50-100 steps with a soft-core potential or a very weak force constant (e.g., 10-50 kJ/mol/nm²) on the protein and ligand backbone. This allows the system to "untangle" without exploding.
- Stage 2: Proceed with standard minimization using the desired parameters (e.g., conjugate gradient) to achieve final convergence.
- Check Position Restraints: Ensure that the
define = -DPOSRESstatement in your.mdpfile is not accidentally restraining atoms in conflicting positions. Temporarily remove this line to test if it resolves the issue [10].
Protocol B: Correcting Topology and Parameter Errors
Errors during pdb2gmx or grompp indicate problems with the molecular description of the system [47].
Detailed Methodology:
- Ligand Parameterization: For non-standard residues (e.g., drug-like ligands),
pdb2gmxwill fail. Use specialized tools to generate the ligand topology:- CGenFF/CHARMM-GUI: For the CHARMM force field, use the CGenFF web server to obtain topology and parameters.
- ACPYPE/GAUSSIAN: For the AMBER force field, use tools like ACPYPE which can interface with quantum chemistry software for parameter derivation.
- PRODRG & SwissParam: Online servers that provide quick initial parameters for testing.
- Systematic Topology Assembly: Manually assemble your system's topol` file, ensuring correct order.
- Force Field Consistency: Never mix topologies from different force fields (e.g., CHARMM and AMBER) without careful conversion. This causes "second defaults directive" errors [47]. Use parameters and topologies derived for a single, consistent force field.
The Scientist's Toolkit: Essential Research Reagents and Software
Successful minimization and MD require a suite of specialized software and resources.
Table 2: Essential Research Reagent Solutions for Protein-Ligand MD
| Tool / Resource Name | Type | Primary Function in Minimization/MD Setup |
|---|---|---|
| GROMACS [47] | MD Suite | The core engine for running energy minimization and molecular dynamics simulations. |
| pdb2gmx [47] | Tool | Generates protein topology and coordinates in a format compatible with the chosen force field. |
| CHARMM-GUI [48] | Web Server | Provides a user-friendly interface for building complex systems (e.g., membrane-protein-ligand) and generating all necessary input files. |
| VMD [48] | Visualization | Critical for visualizing initial structures, diagnosing clashes, and analyzing simulation trajectories. |
| PyMOL [48] | Visualization | Used for structure manipulation, editing, and high-quality rendering of molecular systems. |
| SwissParam [47] | Web Server | Provides topologies and parameters for small molecules compatible with the CHARMM and GAFF force fields. |
| CGenFF [47] | Web Server | The official server for generating CHARMM-compatible parameters and topologies for small organic molecules. |
| RCSB PDB [48] | Database | Source for initial experimental protein-ligand complex structures used as simulation starting points. |
Frequently Asked Questions (FAQs)
Q1: My minimization fails with "forces have not converged," but the system looks fine visually. What could be wrong?
A1: Subtle issues can cause this. First, check your .mdp parameter emtol (force tolerance); it might be set too stringently. Second, ensure all molecules, especially water and ions, have correct topology definitions. A single misplaced ion with incorrect charge can prevent convergence. Third, try using the -double precision option in grompp for higher numerical accuracy, as the standard message suggests [10].
Q2: How do I handle a ligand or non-standard residue that pdb2gmx does not recognize?
A2: The pdb2gmx tool is only for standard amino acids and nucleic acids. You must generate the ligand topology separately using tools like CGenFF (for CHARMM) or Acpype (for AMBER/GAFF). Once you have the ligand's .itp file, you manually include it in your main topology (.top) file and add the ligand name to the [ molecules ] section [47].
Q3: What is the recommended minimization algorithm (emtype) for a typical protein-ligand system in water?
A3: For initial stabilization of a system with potential clashes, the steepest descent algorithm is robust and efficient for the first 50-500 steps. It is not easily trapped by local gradients. After the steepest descent converges or the energy drop plateaus, switch to the conjugate gradient algorithm to achieve more precise convergence to a local energy minimum.
Q4: Are position restraints necessary during minimization, and why would I use them? A4: Position restraints are not always necessary but are highly recommended. They allow you to relax the solvent and ions around the protein-ligand complex without distorting the experimentally determined structure of the core complex. This is typically done by applying restraints to the heavy atoms of the protein and ligand backbone, which are then gradually released in subsequent equilibration phases [47].
A successful minimization protocol is the cornerstone of a stable MD simulation. By systematically diagnosing error messages, visualizing structural problems, and applying the corrective protocols outlined in this guide, researchers can overcome the most common hurdles. Adherence to a methodical workflow for system building, topology generation, and parameter selection ensures that protein-ligand simulations begin from a stable, physically realistic state, thereby enhancing the reliability and interpretability of all subsequent dynamical analysis.
Diagnosing and Resolving Minimization Failures: A Systematic Approach
Convergence failure is a frequent and formidable challenge in computational chemistry and molecular dynamics (MD) simulations, particularly during energy minimization and self-consistent field (SCF) procedures. When calculations oscillate or stall, researchers lose valuable time and computational resources, potentially jeopardizing project timelines in drug development and materials science. This guide identifies the root causes of these failures—including problematic initial configurations, inadequate algorithmic settings, and insufficient sampling—and provides systematic troubleshooting protocols to overcome them. Understanding these convergence dynamics is essential for producing reliable, reproducible simulation data that forms the foundation of scientific conclusions in molecular research.
Frequently Asked Questions
Q1: Why does my energy minimization fail with "forces have not converged" errors?
This common error occurs when the maximum force on any atom exceeds the requested convergence tolerance within the allowed step limit. In one documented case, a protein-ligand complex minimization failed with Fmax = 1.9×10⁵ kJ/mol/nm even after 15 steps of steepest descent algorithm [39]. The solution involves both algorithmic adjustments and system preparation:
- Increase iteration limits: Raise nsteps from 5000 to 50000-100000
- Loosen initial tolerance: Begin with fmax=100-1000, then gradually tighten
- Remove constraints: Set
constraints = noneto eliminate artificial force restrictions - Verify topology: Check for missing atoms, incorrect bonds, or steric clashes in initial structure
Q2: What causes oscillatory behavior in SCF calculations, and how can it be resolved?
SCF oscillations occur when electron density updates create cyclic patterns instead of converging toward a stable solution. This is particularly problematic in metallic systems, slabs, and molecules with degenerate states [49]. Multiple strategies exist to break these cycles:
- Conservative mixing: Decrease SCF%Mixing to 0.05 (from default 0.1-0.2)
- DIIS adjustments: Reduce DIIS%Dimix to 0.1 and set Adaptable=false
- Alternative algorithms: Switch to MultiSecant method (
SCF Method MultiSecant) or LISTi method (Diis Variant LISTi) - Finite temperature: Apply electronic temperature (kT=0.01-0.001 Hartree) to smear occupations
Q3: How can I distinguish between true convergence and premature stalling?
True convergence shows stable properties with fluctuations smaller than the required tolerance, while premature stalling displays plateaus with periodic small variations. Assessment methods include [50] [51]:
- Multiple metrics: Monitor energy, RMSD, RMSF, and secondary structure evolution simultaneously
- Property correlation: Check if energy and force convergence correspond
- Extended sampling: Continue calculations beyond the apparent plateau to confirm stability
- Statistical analysis: Use Markov chain Monte Carlo (MCMC) approaches to assess sampling adequacy
Table 1: Troubleshooting SCF Convergence Failures
| Problem Symptom | Possible Causes | Solution Strategies | Key Parameters to Adjust |
|---|---|---|---|
| Violent oscillations | Overly aggressive mixing | Conservative mixing, DIIS dimix reduction | SCF%Mixing=0.05, DIIS%Dimix=0.1 [49] |
| Slow convergence, many HALFWAY iterations | Poor numerical precision, insufficient k-points | Increase integration grid quality, add k-points | NumericalQuality Good, KSpace%Quality [49] |
| Progressive then stuck | Inadequate basis set, frozen core | Start with SZ basis, remove frozen core | Basisfunction type, FrozenCore None [49] |
| Cyclic pattern | DIIS interpolation issues | Alternative convergence algorithms | SCF%Method=MultiSecant [49] |
Advanced Diagnostic Protocols
Energy Landscape Analysis
Convergence failures often reflect complex energy landscapes. During protein stretching simulations, energy minima flatten and disappear while new minima emerge, creating barriers to convergence [52]. The inherent structure formalism separates vibrational motion from structural transitions, providing clearer insight into true convergence problems versus temporary trapping.
Diagnostic Workflow:
- Identify inherent structures through quenched dynamics (energy minimizations of snapshots)
- Track basin transitions instead of instantaneous energy values
- Map disconnectivity graphs to visualize the relationship between minima
- Calculate barrier heights between frequently visited states
Force Field and Solvation Model Validation
For intrinsically disordered proteins (IDPs) and flexible systems, convergence problems may stem from force field limitations rather than algorithmic issues [51]. The CHARMM36m force field corrects oversampling of left-handed α-helices, while AMBER ff14IDPs improves φ/ψ distributions for disorder-promoting residues. Solvation models significantly impact convergence:
Table 2: Solvation Model Effects on Convergence
| Solvation Model | Advantages | Convergence Challenges | Recommended Applications |
|---|---|---|---|
| TIP3P (explicit) | Standard for folded proteins | May over-stabilize structures | Globular proteins, stable complexes [51] |
| TIP4P-D | Improved for IDPs, reduces compaction | Slower due to enhanced sampling | Disordered regions, flexible linkers [51] |
| Continuum implicit | Faster sampling, no viscosity effects | May lack specific interactions | Initial screening, large systems [51] |
Quantitative Convergence Criteria
Establishing appropriate convergence thresholds is essential for meaningful simulations. Overtightening criteria wastes computational resources, while loose criteria produce unreliable results [53]. For fragmentation methods, the convergence error is typically much smaller than the fragmentation error (~1 kcal/mol), allowing for looser SCF criteria without sacrificing accuracy.
Table 3: Recommended Convergence Thresholds
| Calculation Type | Energy Threshold | Force Threshold | Application Notes |
|---|---|---|---|
| Geometry optimization | ΔE < 1×10⁻⁵ Ha/atom | Fmax < 0.01 Ha/Bohr | Standard molecules [54] |
| MD equilibration | Energy fluctuation < 2% | -- | Production run prerequisites [50] |
| Fragmentation methods | ΔE < 1×10⁻⁴ Ha/fragment | -- | Can loosen without significant error propagation [53] |
| Protein-ligand complex | ΔE < 2×10⁻⁵ Ha/atom | Fmax < 0.05 Ha/Bohr | Initial optimization [39] |
The Scientist's Toolkit: Essential Research Reagents
Successful convergence requires appropriate computational "reagents" – the algorithms, parameters, and models that drive simulations toward physical solutions.
Table 4: Research Reagent Solutions for Convergence Problems
| Tool Category | Specific Solutions | Function | Key References |
|---|---|---|---|
| Optimization algorithms | BFGS, LBFGS, FIRE, MDMin | Locate energy minima through Hessian updates or dynamics | [54] |
| SCF convergence accelerators | DIIS, MultiSecant, LISTi | Extrapolate density matrices to accelerate SCF convergence | [49] [53] |
| Enhanced sampling | Finite electronic temperature, simulated annealing | Escape local minima while progressing toward global minimum | [49] [50] |
| Advanced solvation | TIP4P-D, COSMO, PCM | Balance computational cost with accurate solvation effects | [51] |
Workflow Diagrams for Troubleshooting
Diagram 1: Convergence troubleshooting workflow.
Diagram 2: Energy landscape changes that cause convergence failures.
Implementation Protocols
Automated Convergence Management
For challenging systems like proteins with flexible regions, implement adaptive convergence criteria that evolve during optimization [49]:
This protocol begins with loose criteria (electronic temperature kT=0.01 Hartree) when gradients are large, then tightens them (kT=0.001 Hartree) as the system approaches the minimum.
Fragmentation Method Optimization
For large systems, fragmentation methods can accelerate convergence while maintaining accuracy [53]. The key insight is that convergence errors propagate less significantly than fragmentation approximation errors:
- Looser SCF criteria: 10⁻⁴ to 10⁻⁶ a.u. for fragments vs. 10⁻⁶ to 10⁻⁸ for full systems
- Electrostatic embedding: Point charges from environment stabilize fragment calculations
- Systematic fragmentation: Overlapping fragments with conjugate caps prevent boundary artifacts
Convergence failures in molecular simulations stem from diverse sources including algorithmic limitations, problematic energy landscapes, and inadequate sampling. By applying the diagnostic framework and solution strategies presented here—including SCF parameter adjustment, optimization algorithm selection, and energy landscape analysis—researchers can systematically overcome oscillations and stalling. The provided workflows, tolerance guidelines, and implementation protocols offer a comprehensive reference for achieving robust convergence across diverse molecular systems, from small drug candidates to complex biomolecular assemblies. Through methodical application of these principles, computational scientists can enhance the reliability and efficiency of their simulation workflows, accelerating drug discovery and materials design.
FAQs: Understanding and Identifying Steric Clashes
What is a steric clash and why is it problematic in molecular simulations?
A steric clash is a structural artifact where two non-bonding atoms in a molecule are positioned impossibly close to each other, resulting in an unphysical overlap of their Van der Waals radii [55] [56]. This is problematic because:
- High Energy Penalties: Severe clashes create high Van der Waals repulsion energy, making the structure physically unrealistic and unstable [55].
- Simulation Failures: Energy minimization and molecular dynamics (MD) simulations often fail to converge or crash when severe clashes are present, as the initial forces are excessively large [55] [57].
- Poor Model Quality: A high number of clashes calls the reliability of a structural model into question, which is critical for applications like drug design [56].
How can I quantitatively measure clashes in my protein structure?
You can use the clashscore, a metric defined as the number of clashes per 1,000 atoms (including hydrogens), where a clash is counted when Van der Waals radii overlap by ≥ 0.4 Å [56]. Alternatively, a more granular metric defines clashscore as the total Van der Waals repulsion energy (in kcal/mol) divided by the number of atomic contacts, providing an energy-based assessment [55]. For high-resolution crystal structures, an acceptable clashscore is typically ≤ 0.02 kcal.mol⁻¹.contact⁻¹ [55].
Table 1: Clashscore Examples from the PDB
| PDB ID | Resolution | Total Atoms | Number of Clashes | Clashscore | Percentile Rank |
|---|---|---|---|---|---|
| 6zx4 | 0.96 Å | 5,850 | 6 | 1.0 | ~90th |
| 6ef8 | 3.7 Å | 43,806 | 558 | 13 | ~35th |
Data sourced from Proteopedia analysis [56].
What are the common error messages indicating severe steric clashes during GROMACS energy minimization?
During energy minimization in GROMACS, the following messages often indicate unresolved steric clashes:
- Force Non-Convergence:
"Energy minimization has stopped, but the forces have not converged to the requested precision Fmax < 1000"[2] [58]. This means the algorithm cannot reduce the maximum force below your threshold, often due to severe atomic overlaps. - Excessively High Forces: The output shows a
Maximum forcevalue that remains very high (e.g., 7.0e+04 kJ/mol/nm or more) on a specific atom [2]. - Segmentation Faults: In severe cases, the minimization or subsequent equilibration may crash with a
Segmentation faulterror, which can be caused by extreme forces from bad contacts [2].
Troubleshooting Guides: Resolving Minimization Failures
How do I resolve force non-convergence and segmentation faults caused by severe clashes?
Follow this systematic protocol to resolve severe clashes in poor initial structures.
Initial Assessment and Pre-processing:
- Validate Your Structure: Use validation servers like MolProbity to identify and visualize clashes before simulation [56]. This helps confirm clashes are the primary issue.
- Check Topology and Atom Names: Ensure your topology is correct. Use
pdb2gmxwith-ignhto let GROMACS add correct hydrogens if the nomenclature in your input file is inconsistent [12].
Specialized Minimization Protocol: If standard minimization fails, a multi-stage approach is needed.
Table 2: Step-by-Step Minimization Protocol for Clash-Ridden Structures
| Step | Goal | Recommended Method & Parameters | Rationale |
|---|---|---|---|
| 1. Pre-Relaxation | Remove the worst overlaps | Use Steepest Descent with a very small step size (emstep = 0.0001 or smaller) and increased emtol (e.g., 5000-10000). Run for a few hundred steps. |
Gently "nudge" atoms apart without causing instability. A small step size prevents drastic, destabilizing moves [42]. |
| 2. Robust Minimization | Further reduce clashes | Switch to Conjugate Gradient algorithm. It is more efficient than Steepest Descent for navigating complex energy landscapes after the initial pre-relaxation [42]. | |
| 3. Advanced Refinement | Resolve persistent, severe clashes | Use a dedicated clash-resolution tool like Chiron [55] or Rosetta fast relax [55]. These protocols use specialized sampling (e.g., Discrete Molecular Dynamics) to efficiently resolve clashes with minimal backbone perturbation. | Standard molecular mechanics force fields can get trapped; these methods are designed specifically for this task [55]. |
| 4. Final EM in GROMACS | Prepare for MD | Return to GROMACS for final minimization with standard parameters (emtol = 1000, integrator = steep or cg) to ensure compatibility with subsequent simulation steps. |
GROMACS-Specific Solutions:
- Remove Constraints: Temporarily set
constraints = nonein your.mdpfile during the initial minimization stages to allow maximum flexibility for clash resolution [2] [58]. - Version Check: Be aware that minimization behavior can differ between GROMACS versions. If convergence fails in a newer version, check for reported bugs and consider using a patched version (e.g., 2022.3+ for a known GPU minimization issue) [58].
- Double Precision: Compiling and running GROMACS in double precision can provide higher accuracy for difficult minimization problems [2].
How can I avoid steric clashes when building initial structures?
Prevention is the best strategy. Key practices include:
- Use High-Quality Templates: For homology modeling, start with high-resolution template structures to minimize introduced clashes [55].
- Proper Protonation: Use tools that correctly add and place hydrogen atoms, as incorrect protonation is a common source of clashes [12] [56].
- Refine Loops and Side Chains: Pay special attention to model regions like loops and side chains. Use refinement tools that include backbone and side-chain repacking to maintain favorable packing while removing clashes [55].
Experimental Protocols & Workflows
Protocol: Automated Clash Resolution with Chiron
Chiron is an automated protocol designed to resolve severe steric clashes efficiently using Discrete Molecular Dynamics (DMD) simulations [55].
Methodology:
- Clash Identification: The input structure is analyzed for atomic overlaps. A clash is quantitatively defined as any non-bonded atomic overlap resulting in Van der Waals repulsion energy > 0.3 kcal/mol, with exceptions for bonded atoms, disulfide bonds, and hydrogen-bonded atoms [55].
- DMD Simulation: The system undergoes all-atom DMD simulations.
- Sampling and Selection: Multiple independent simulations are run. The resulting structure with the lowest clashscore is selected as the output [55].
Benchmarking: Benchmark studies indicate Chiron is robust and efficient at resolving severe clashes in large structures and homology models where traditional methods like simple molecular mechanics minimization may fail [55].
Workflow: Integrated Clash Management for Molecular Dynamics
The following diagram illustrates a logical workflow for diagnosing and resolving steric clashes as part of MD setup.
Table 3: Key Software Tools for Clash Detection and Resolution
| Tool Name | Type | Primary Function | Application Context |
|---|---|---|---|
| MolProbity | Validation Server | All-atom structure validation; calculates clashscore and visualizes clashes [56]. | Quality control for any protein structure before simulation. |
| Chiron | Automated Refinement Server | Rapidly resolves severe steric clashes using DMD simulations with minimal backbone perturbation [55] [59]. | Refining low-resolution structures, homology models, and other clash-ridden models. |
| Rosetta | Modeling Suite | Knowledge-based protein design and refinement; fast relax protocol can resolve clashes [55]. |
Protein structure refinement and de novo design (best for proteins <250 residues) [55]. |
| GROMACS | Simulation Suite | Molecular dynamics and energy minimization; first-line tool for removing moderate clashes [2] [57]. | Standard energy minimization and MD after initial clash refinement. |
| WHAT_CHECK | Validation Tool | Qualitatively identifies steric clashes based on distance cutoffs [55]. | Part of the WHAT IF software suite for structure checking. |
| wwPDB Validation Service | Validation Server | Provides comprehensive validation reports for PDB entries, including clash analysis [56] [60]. | Assessing the quality of existing PDB structures or prior to deposition. |
Frequently Asked Questions (FAQs)
FAQ 1: What does the error "Residue 'XXX' not found in residue topology database" mean and how can I resolve it?
This error occurs when pdb2gmx cannot find a topology for the residue "XXX" in the selected force field's database. This is typically because the residue is not a standard amino acid or nucleic acid, or its name does not match the database entry [12].
- Solution: Your options are to:
- Rename the residue in your coordinate file if a suitable entry exists under a different name [12].
- Find a topology file for the molecule, convert it to an
.itpfile, and include it in your main topology [12]. - Parameterize the residue yourself, though this is complex and time-consuming [12].
- Use a different force field that already includes parameters for this residue [12].
FAQ 2: Why does my energy minimization fail to converge, reporting a "Stepsize too small" error or a segmentation fault?
This indicates that the minimizer cannot reduce the forces further, often due to a physically unrealistic starting structure. In severe cases, this can lead to a segmentation fault during subsequent molecular dynamics (MD) runs [2] [61].
- Solution:
- Check Atomistic clashes: Inspect your initial configuration for atoms that are excessively close, which can generate infinite forces [61].
- Review Topology Parameters: Ensure all bonds, angles, and dihedrals have correct parameters. Missing or incorrect parameters can cause instabilities [62].
- Re-run Minimization: Sometimes, using the output of one minimization as the input for another round with a different algorithm (e.g., conjugate gradients after steepest descent) can help [2].
FAQ 3: What does the warning "Invalid order for directive [defaults]" during grompp mean?
This error signifies that the directives in your topology (.top) or include (.itp) files appear in an incorrect sequence. The [defaults] section, for instance, must be the very first directive in the topology [12].
- Solution: Reorder your topology file according to the GROMACS reference manual. Typically, the structure should be:
[defaults]>[atomtypes]>[moleculetype]>[ molecules ]. Ensure that[*types]directives for any new molecules are placed before the[moleculetype]directive [12].
FAQ 4: How do I handle missing torsion parameters for specific chemical patterns, such as ">C-N-N=N-"?
This is a common issue when simulating non-standard molecules or ligands. The force field lacks a predefined torsion potential for that specific combination of atom types [63].
- Solution: You will need to derive the missing dihedral parameters. This is typically done by performing a torsion scan around the rotatable bond using quantum mechanical (QM) calculations and then fitting the molecular mechanics (MM) parameters to match the QM potential energy surface [64].
Troubleshooting Guides
Guide 1: Diagnosing and Correcting Missing Parameters
Missing parameters for bonds, angles, and dihedrals are a primary source of energy minimization failure and simulation instability [62].
- Symptoms: Warnings about unassigned parameters, empty
c0columns in your topology file, or fatal errors duringgrompp[62] [63]. - Diagnosis: Carefully inspect the
grompplog output and your topology file for lines indicating missing interactions. - Resolution Protocol:
- Identify the missing term: Note the specific atom types involved in the interaction [63].
- Check for known fixes: Search literature or force field repositories for published parameters for your molecule or similar chemical moieties.
- Parameterize the term: If no parameters exist, you must derive them. The standard workflow involves:
The diagram below illustrates the logical workflow for resolving missing parameters.
Guide 2: Addressing Systematic Force Field Errors
Systematic errors are inherent in the force field parameters themselves and can lead to consistent deviations from experimental observations, such as miscalculated hydration free energies (HFEs) [65].
- Identification: Systematic errors can be identified by benchmarking simulation results (e.g., HFEs, densities) against experimental data for a set of small molecules. Statistical analysis of the errors often reveals biases associated with specific elements [65].
- Correction: Empirical corrections can be applied post-simulation. For example, a study on GAFF identified systematic errors for molecules containing Cl, Br, I, and P, and applied an Element Count Correction (ECC) to significantly improve HFE predictions [65].
Table 1: Example Systematic Error Identification and Correction for Hydration Free Energies (HFEs) [65]
| Element | Systematic Error in HFE (GAFF) | Correction Method | Improvement in RMSE (kcal/mol) |
|---|---|---|---|
| Cl | Over-estimation | Element Count Correction (ECC) | ~1.44 (overall) |
| Br | Over-estimation | Element Count Correction (ECC) | ~1.44 (overall) |
| I | Over-estimation | Element Count Correction (ECC) | ~1.44 (overall) |
| P | Over-estimation | Element Count Correction (ECC) | ~1.44 (overall) |
Experimental Protocols
Protocol: On-the-Fly Force Field Optimization for Novel Molecules
This protocol outlines a modern, efficient method for deriving missing intramolecular parameters, particularly dihedrals, using machine learning-accelerated quantum calculations [64].
Objective: To obtain optimized dihedral parameters for a rotatable bond in a novel molecule not fully covered by existing force fields.
Required Tools and Reagents:
- Software: A quantum chemistry package (e.g., Gaussian, ORCA), molecular dynamics software (e.g., GROMACS, OpenMM), and RDKit for cheminformatics operations.
- Computational Model: A fine-tuned neural network potential (NNP) like DPA-2-TB, which uses delta-learning to correct semi-empirical methods (GFN2-xTB) for high accuracy and speed [64].
Procedure:
- Molecule Fragmentation:
- Use an automated fragmentation tool to break the target molecule into smaller fragments, each containing the rotatable bond of interest.
- Cap the broken bonds with methyl groups or hydrogen atoms to maintain valency [64].
Flexible Torsion Scan:
- For the generated fragment, systematically rotate the dihedral angle of the target bond in increments (e.g., 15°).
- At each step, allow all other structural degrees of freedom (bond lengths, other angles) to relax. This generates a relaxed potential energy surface.
- This scan is performed on a high-accuracy potential surface. Using the fine-tuned DPA-2-TB model can reduce computation time from days to minutes compared to standard QM methods [64].
Parameter Optimization:
- Using the relaxed geometries and energies from the torsion scan, optimize the MM dihedral force constants (
kχ), multiplicities (n), and phases (δ) to minimize the difference between the MM and high-accuracy potential energy surfaces [64].
- Using the relaxed geometries and energies from the torsion scan, optimize the MM dihedral force constants (
Parameter Matching and Application:
- A node-embedding-based similarity metric is used to create a unique "fingerprint" for the fragment. This fingerprint allows the newly optimized parameters to be automatically matched to other molecules with the same chemical environment in the future [64].
The workflow for this protocol is visualized below.
The Scientist's Toolkit: Key Research Reagents and Solutions
Table 2: Essential Resources for Force Field Troubleshooting and Development
| Resource Name | Type | Primary Function | Reference |
|---|---|---|---|
| FreeSolv Database | Database | A benchmark database of experimental and calculated hydration free energies for small molecules, used for validating and identifying force field errors. | [65] |
| DPA-2-TB Model | Computational Model | A fine-tuned neural network potential that accelerates quantum mechanical torsion scans by using delta-learning, drastically reducing computation time. | [64] |
| CHARMM General Force Field (CGenFF) & General AMBER Force Field (GAFF) | Force Field | Widely used additive force fields for small molecules and drug-like compounds. Often the starting point for parameterization. | [66] |
| 3D-RISM | Implicit Solvent Model | An integral equation theory-based solvation model used to rapidly calculate solvation properties and identify systematic force field errors. | [65] |
| Element Count Correction (ECC) | Correction Method | An empirical post-calculation correction that improves hydration free energy predictions by accounting for systematic errors per element type. | [65] |
FAQs on Energy Minimization Failures
Q1: My energy minimization fails with "convergence to machine precision achieved, but forces are not small enough." What should I do? This indicates that the algorithm has stalled. The recommended action is to switch from conjugate gradients to the Steepest Descent algorithm. Steepest Descent is more robust for initial minimization, especially when starting from structures with bad contacts or high initial forces, as it is less likely to get trapped [32].
Q2: How do I choose the right minimizer for my protein-ligand system? The choice depends on your system and the presence of constraints. Use L-BFGS for the fastest convergence if your system does not require parallelization and has no constraints. Use Conjugate Gradients for minimization prior to normal-mode analysis, as it cannot be used with constraints like the SETTLE algorithm for water. Use Steepest Descent for its robustness in the initial stages of minimization or when other algorithms fail [32].
Q3: What is a reasonable force tolerance (emtol) to set for a protein in water?
A reasonable value for the force tolerance (emtol) is between 10.0 and 100.0 kJ mol⁻¹ nm⁻¹. This can be estimated from the root mean square force of a harmonic oscillator. For a weak oscillator at 1 K, this force can be 7.7 kJ mol⁻¹ nm⁻¹, so a value between 1 and 10 is acceptable, but a slightly larger value is often used in practice to avoid endless iterations due to numerical noise [32].
Q4: Why is multi-stage minimization recommended for membrane protein systems? Multi-stage protocols are crucial for complex systems like membrane proteins. A recommended approach is to first minimize the energy of the solute (e.g., the protein and any ligands) with position restraints applied to all atoms, allowing the solvent and ions to relax around a fixed structure. This is followed by a full, unrestrained minimization of the entire system. This step-wise relaxation prevents large, destabilizing movements in the initial stages [67].
Troubleshooting Guides
Problem: Minimization fails due to atomic clashes in the initial structure.
- Symptoms: The job crashes immediately or within a few steps with errors related to force or energy.
- Solution:
- Apply Restraints: Use a multi-stage protocol. Begin with energy minimization while applying strong position restraints on the protein and ligand heavy atoms. This allows the solvent to relax and fill any voids without the protein collapsing.
- Use a Robust Algorithm: For this first stage, use the Steepest Descent algorithm with a conservative step size and a low number of steps (e.g.,
integrator = steepest,nsteps = 50). - Full Minimization: Once the solvent is relaxed, perform a second stage of minimization without any restraints, using a more efficient algorithm like L-BFGS to achieve the final convergence [67].
Problem: Energy minimization is prohibitively slow for a large system.
- Symptoms: The minimization takes an exceptionally long time to meet the force tolerance criterion.
- Solution:
- Switch Algorithms: If you are using Conjugate Gradients, switch to L-BFGS, which has been found to converge faster in practice [32].
- Adjust Parameters: For Steepest Descent, ensure the initial step size (
emstep) is not too small. The software will dynamically adjust it, but a reasonable starting value (e.g., 0.01 nm) is important. - Review Tolerance: Check if your force tolerance (
emtol) is set too stringently. For initial equilibration, a tolerance of 100-1000 kJ mol⁻¹ nm⁻¹ may be sufficient before moving to molecular dynamics.
Problem: The system does not reach a true equilibrium state after minimization and equilibration.
- Symptoms: Properties like pressure continue to drift significantly during subsequent equilibration runs, or Radial Distribution Function (RDF) curves remain noisy and non-convergent.
- Solution:
- Extend Minimization: Ensure energy minimization is complete. Inadequate minimization can prevent the system from reaching equilibrium later.
- Verify Convergence: Do not rely solely on energy and density. Monitor pressure and RDFs, particularly for key interactions (e.g., asphaltene-asphaltene in asphalt systems), as these can take much longer to converge [20].
- Increase Temperature: If applicable, a slightly higher temperature during equilibration can accelerate convergence [20].
Energy Minimization Algorithm Comparison
The table below summarizes the key algorithms available in GROMACS for energy minimization, aiding in the selection of the most appropriate one for a given problem [32].
| Algorithm | Key Principle | Strengths | Weaknesses | Typical Use Case |
|---|---|---|---|---|
| Steepest Descent | Moves atoms in the direction of the negative gradient of the potential energy. | Very robust; easy to implement; good for initial steps. | Inefficient close to the energy minimum. | Initial minimization of poorly structured systems with bad contacts. |
| Conjugate Gradient | Uses a sequence of conjugate (non-interfering) directions for search. | More efficient than Steepest Descent near the minimum. | Cannot be used with constraints (e.g., SETTLE water). | Minimization prior to normal-mode analysis where high accuracy is needed. |
| L-BFGS (Limited-memory Broyden–Fletcher–Goldfarb–Shanno) | Builds an approximation of the inverse Hessian matrix from a history of steps. | Fastest convergence; low memory requirements. | Not yet parallelized; performance can degrade with sharp cut-offs. | General-purpose minimization for most production systems where speed is desired. |
Multi-Stage Minimization Protocol for Robust Convergence
This protocol provides a detailed methodology for a two-stage energy minimization, which is critical for stabilizing complex molecular systems before molecular dynamics production runs [67].
A. Preparation
- Obtain and Prepare Structure: Start with a PDB file of your protein. Remove crystallographic water molecules and separate any ligand coordinates. The ligand topology must be defined separately.
- Generate Topology: Use the
pdb2gmxcommand to generate the molecular topology and GROMACS-compatible coordinate file (.gro). Select an appropriate force field (e.g.,ffG53A7is recommended in Gromacs v5.1 for explicit solvent). - Define the Simulation Box: Use
editconfto place the protein in the center of a periodic box (e.g., cubic, dodecahedron) with a minimum distance (e.g., 1.4 nm) from the box edge. - Solvate and Add Ions: Solvate the box using the
solvatecommand. Then, neutralize the system's charge by adding counterions (e.g., Na⁺, Cl⁻) using thegenioncommand.
B. Two-Stage Energy Minimization
- Stage 1: Solvent and Ion Relaxation (with position restraints on solute)
- Objective: To relax the solvent and ions around the fixed protein, relieving any bad contacts or voids.
- Method: Create a parameter file (
min_restrained.mdp) with the following key settings: - Execute:
- Stage 2: Full System Minimization
- Objective: To minimize the energy of the entire system without restraints.
- Method: Create a second parameter file (
min_final.mdp) with potentially more efficient settings: - Execute:
The Scientist's Toolkit: Essential Materials and Reagents
The table below lists key resources required for setting up and running molecular dynamics simulations, as per the standard protocol [67].
| Item | Function / Description | Example / Format |
|---|---|---|
| Protein Structure File | The initial atomic coordinates of the system. | PDB file from RCSB Protein Data Bank. |
| Force Field | A set of mathematical functions and parameters describing interatomic forces. | GROMACS-supported fields like ffG53A7, CHARMM, AMBER. |
| Molecular Topology File | Describes the molecular system: atoms, bonds, angles, dihedrals, and force field parameters. | .top file (generated by pdb2gmx). |
| Molecular Geometry File | Contains the coordinates and velocities of all atoms in the system. | .gro file (GROMACS format). |
| Parameter File | Contains all the settings for the simulation run (minimization, dynamics, output). | .mdp file. |
| GROMACS Suite | The molecular dynamics simulation software used to perform all calculations. | Open-source MD package (e.g., GROMACS 2025.3). |
| Molecular Visualization Tool | Software for visual inspection and rendering of molecular structures. | RasMol, VMD, PyMOL. |
Energy Minimization Decision Workflow
The following diagram illustrates the logical decision process for selecting and applying energy minimization algorithms and protocols.
Machine Learning Force Fields (MLFFs) aim to narrow the gap between the high accuracy of quantum mechanical (ab initio) methods and the computational efficiency of classical force fields [68]. By learning the relationship between atomic structure and potential energy from reference data, MLFFs can achieve quantum-chemical accuracy for broad chemical spaces, enabling more reliable molecular dynamics (MD) simulations and energy minimization tasks [69] [68]. However, their integration into research workflows introduces new challenges. This guide provides targeted troubleshooting and FAQs to help researchers overcome common obstacles when using MLFFs.
Frequently Asked Questions (FAQs)
1. My MLFF-produced energies are accurate on the test set, but my molecular dynamics simulation becomes unstable. Why?
This is a common pitfall where good performance on a static test set does not guarantee stability during MD. Instability often arises from the model encountering configurations far outside its training data during simulation. The 2023 TEA Challenge highlighted that MLFFs can produce seemingly reasonable yet incorrect results when they extrapolate beyond their learned chemical space [69]. To diagnose, check if atomic positions or forces exhibit unphysical values shortly before the simulation crashes.
2. How can I improve the transferability of my MLFF to different molecular systems?
Transferability remains a significant challenge [69] [70]. The solution is to improve the diversity and coverage of your training data. Relying solely on data from a single system or a narrow range of configurations is insufficient. For robust models, the training dataset should encompass a wide range of chemical environments, potential reaction pathways, and relevant states of the system you intend to simulate [68].
3. What are the major limitations of using Density Functional Theory (DFT) data for training MLFFs?
While DFT is a common source of training data, it has known inaccuracies and inconsistencies. Different DFT approximations (functionals) can yield varying results for the same system [70]. Furthermore, MLFFs trained solely on DFT data inherit these inaccuracies and are limited in their ability to surpass the quality of the reference method. For higher accuracy, research is moving towards using more precise, albeit computationally expensive, methods like Coupled Cluster (CC) theory for generating training data [70].
4. My simulation is computationally expensive even with an MLFF. How can I improve its efficiency?
The computational cost depends on the MLFF architecture and implementation. The TEA Challenge 2023 revealed large variations in the computational resources required by different models to produce the same amount of simulation data [69]. Consider trying a different, potentially lighter, MLFF architecture. Furthermore, ensure you are using optimized simulation software and taking advantage of hardware acceleration (e.g., GPUs) where supported [71].
Troubleshooting Guides
Guide 1: Diagnosing and Resolving Energy Minimization Failures
Energy minimization failures when using MLFFs often manifest as convergence failures, unphysical geometries, or "NaN" (Not a Number) values for energies or forces.
Step 1: Verify Training Data Quality and Coverage The root cause is often inadequate training data. Generate a visualization of your model's training data distribution (e.g., using Principal Component Analysis) and plot the configurations where minimization fails on the same graph. If the failure points lie outside the dense regions of your training data, your model is extrapolating unreliably [69].
- Solution: Augment your training dataset with quantum-mechanical calculations (e.g., DFT) that cover the configuration space where failures occur. This includes regions near the intended minimized structure.
Step 2: Analyze Force and Energy Predictions Before starting a minimization, evaluate the MLFF on a set of known structures (e.g., from your training or test set).
- Action: Calculate the Root Mean Square Error (RMSE) in forces and energies against the quantum-mechanical reference. Compare these errors to established benchmarks, such as those from the TEA Challenge (see Table 1). High errors indicate a poorly trained or insufficient model [69].
- Solution: Retrain the model, potentially adjusting hyperparameters or increasing model size, to achieve lower test errors.
Step 3: Check for Numerical Instabilities Examine the output logs for extremely large force values or NaN/Infinity in the energy.
- Action: This can be caused by the model receiving an atomic configuration with interatomic distances much shorter or longer than any seen during training, leading to a pathological prediction [69].
- Solution: Implement safeguards in your simulation code to halt the simulation and log the atomic configuration if forces exceed a reasonable threshold. Use this configuration to retrain or improve the model.
Guide 2: Addressing Poor Transferability and Generalization
This issue occurs when an MLFF performs well on its original training system but fails on a related but distinct system (e.g., a different isomer, a molecule with the same functional groups but different backbone).
Step 1: Identify the Chemical Space Gap Perform a comparative analysis of the chemical environments in the original training set versus the target system for simulation.
- Action: Use descriptors like the Smooth Overlap of Atomic Positions (SOAP) to characterize and compare the atomic environments in both datasets [69]. A significant mismatch indicates a coverage problem.
- Solution: The most reliable fix is to expand the training set to be more chemically diverse, explicitly including data from the type of system where the model is failing.
Step 2: Evaluate the Reference Method Consider the limitations of the reference method used to generate your training data.
- Action: If your model was trained on DFT data, be aware that DFT itself may not accurately describe the new system's properties (e.g., dispersion interactions, reaction barriers) [70].
- Solution: For critical applications, validate your MLFF's predictions on small representative fragments of the new system using a higher-level quantum chemistry method like CCSD(T) [70].
Step 3: Employ a More Universal Architecture Some MLFF architectures are designed for broader applicability.
- Solution: Consider using models that explicitly incorporate long-range electrostatics and charge equilibration, as these are crucial for modeling ionic systems, interfaces, and response properties across a wide chemical space [71].
Key Experimental Data & Protocols
Quantitative Performance of MLFF Architectures
The TEA Challenge 2023 provides a recent, quantitative comparison of several MLFF architectures across diverse systems. The table below summarizes error metrics and resource usage, which are critical for selecting an appropriate model.
Table 1: MLFF Performance Metrics from TEA Challenge 2023 Analysis [69]
| MLFF Architecture | Reported Force RMSE (kcal mol⁻¹ Å⁻¹) | Param. Count | Relative Comp. Cost (1M MD steps) |
|---|---|---|---|
| MACE | ~0.1 - 1.0 (system-dependent) | 2,983,184 | Benchmark-dependent |
| SO3krates | ~0.1 - 1.0 (system-dependent) | 487,613 | Lower than MACE |
| sGDML | ~0.1 - 1.0 (system-dependent) | 123,000 (kernel model) | Lowest |
| FCHL19* | ~0.1 - 1.0 (system-dependent) | Kernel-based | Moderate |
Note: The specific force error for each architecture varies significantly based on the system (molecules, materials, interfaces) and the quality of its training. The parameter count and computational cost are key differentiators [69].
Core Protocol: Benchmarking an MLFF for Stability
Before relying on an MLFF for production simulations, it is essential to benchmark its stability.
- Model Training: Train your chosen MLFF architecture on a provided quantum-mechanical dataset.
- Initial Validation: Evaluate the model on a held-out test set of structures to confirm low RMSE for energy and forces.
- Molecular Dynamics Stress Test: Run a relatively short (e.g., 10-100 ps) MD simulation in the NVE (microcanonical) ensemble starting from a stable configuration.
- Stability Analysis: Monitor the total energy of the system. In a stable NVE simulation, the total energy should be conserved. A significant drift or sudden divergence in energy indicates an unstable model that is likely to fail during longer simulations or minimization [69].
- Comparison: The TEA Challenge recommends running the same simulation with different MLFF architectures. Consistent results across models increase confidence in the simulation's correctness [69].
Essential Research Reagents & Tools
Table 2: Key Software and Data Resources for MLFF Research
| Item Name | Function / Description | Relevance to MLFF Development |
|---|---|---|
| LAMMPS | A highly flexible MD simulator with support for many MLFFs [72]. | The primary engine for running large-scale MD simulations using trained MLFF potentials. |
| Quantum Chemistry Codes (VASP, etc.) | Software to perform DFT or higher-level ab initio calculations [72]. | Generates the reference energy and force data required for training and validating MLFFs. |
| MPNICE (Schrödinger) | An MLFF architecture with explicit charge equilibration, covering 89 elements [71]. | Useful for modeling systems requiring accurate electrostatics, such as ionic materials and interfaces. |
| Universal Models for Atoms (UMA) | A suite of pretrained universal models for atoms [71]. | Offers high-accuracy, broad-coverage models that can be a starting point for research. |
| TEA Challenge Datasets | Standardized datasets for molecules, materials, and interfaces [69]. | Provides benchmarks for fairly comparing the performance of different MLFF architectures. |
Workflow and System Diagrams
MLFF Development and Validation Workflow
The following diagram outlines the key stages in developing and validating a reliable Machine Learning Force Field.
MLIP Inference within a Molecular Dynamics Loop
This diagram illustrates how a trained Machine Learning Interatomic Potential is integrated into a Molecular Dynamics simulation to calculate forces, a process critical for both dynamics and energy minimization.
Ensuring Reliability: Validation Protocols and Performance Benchmarking
Frequently Asked Questions (FAQs)
Q1: My geometry optimization stopped, reporting that forces are converged to machine precision but did not reach the requested Fmax. Has my system found a true energy minimum?
This is a common sign of false convergence. The optimizer can no longer make progress, but the forces remain high. This is frequently caused by atom overlaps or other steric clashes in your initial structure that create extremely steep potential energy gradients [39] [1]. The calculation stops because the algorithm detects no further energy change, not because a stable minimum has been found.
Q2: In my molecular dynamics simulation, the system energy and density stabilize quickly. Can I be sure the system is fully equilibrated?
No. Rapid stabilization of energy and density is often necessary but not sufficient to prove true equilibrium. These properties can reach a plateau while other critical metrics, like pressure or Radial Distribution Function (RDF) curves, remain unconverged [20] [73]. True convergence, especially for studying nanoscale interactions, may require much longer simulation times until key RDF curves show smooth, stable peaks [20].
Q3: What does the error "Maximum force = inf on atom X" mean, and how can I resolve it?
An infinite force (inf) is a clear indicator of severe atom overlaps in your structure [1]. The repulsive forces between atoms occupying the same space become computationally infinite. To resolve this:
- Check topology-coordinate mismatch: Ensure all atoms in your coordinate file (.gro, .pdb) are correctly defined in your molecular topology file (.top) [1].
- Validate ligand construction: Incorrectly built or parameterized ligands are a common source of such clashes [1].
- Review initial geometry: Carefully check your starting structure for impossible bond lengths or atom placements.
Q4: The geometry optimization did not converge within the allowed number of steps. What should I do?
This is a direct "ERROR: GEOMETRY NOT CONVERGED" or "ERROR: More cycles needed" [74]. Recommended actions include:
- Increasing the maximum number of optimization steps.
- Loosening the convergence criteria (e.g., a larger
fmax) for an initial, rough optimization. - Providing a better initial guess geometry closer to the expected minimum.
- Switching to a different optimization algorithm that may be more suitable for your system's potential energy surface [54].
Diagnostic Tables & Protocols
Table 1: Interpreting Common Convergence Messages and Errors
| Error / Warning Message | Likely Cause | Recommended Troubleshooting Action |
|---|---|---|
| "Geometry NOT CONVERGED" [74] | Exceeded max steps, poor initial geometry. | Increase steps; improve initial structure; try different algorithm (e.g., LBFGS instead of Steepest Descent) [54]. |
| "Energy minimization has stopped, but the forces have not converged..." [39] | Atom overlaps; system stuck in a high-energy state. | Check for steric clashes; validate topology; consider using double precision calculation [1]. |
| "Maximum force = inf" [1] | Severe atom overlap creating infinite repulsive force. | Correct atom coordinates in input file; ensure ligand topology matches structure [1]. |
| "Fmax = 1.91991e+05" (Very High Force) [39] | Significant structural problems, bad contacts. | Run a slower, more robust initial minimization (e.g., Steepest Descent) before a more efficient algorithm. |
| SCF procedure did not converge [74] | Electronic structure calculation instability. | Disable KeepOrbitals; try different SCF versions; follow SCF troubleshooting guides [74]. |
| RDF curves are fluctuating/irregular [20] | System not equilibrated; simulation time too short. | Extend simulation time until RDF curves are smooth; use asphaltene-asphaltene RDF as a key convergence metric [20]. |
Table 2: Essential Research Reagent Solutions for Simulation Troubleshooting
| Reagent / Tool | Function in Troubleshooting |
|---|---|
| Double Precision Compilation | Provides higher numerical accuracy, which can sometimes resolve "infinite force" errors by more precisely handling steep gradients [1]. |
| Soft-Core Potentials | Used in free energy calculations to avoid infinite forces when atoms or molecules "disappear," preventing atom-overlap errors [1]. |
| Mixed-Resolution Models | Combines atomistic detail in the region of interest with coarse-grained models for the rest of the system. Drastically reduces computational cost for sampling conformational changes in large proteins [75]. |
| Machine Learning Potentials (MLPs) | Trained on ab initio data to enable nanosecond-scale simulations with quantum mechanics-level accuracy, helping to achieve proper equilibration of interface structures [76]. |
| Anisotropic Network Model (ANM) | A coarse-grained model that efficiently generates plausible protein conformers based on low-frequency normal modes, useful for exploring conformational diversity before costly atomistic simulation [75]. |
Experimental Protocols for Validation
Protocol 1: Systematic Workflow for Distinguishing True and False Minima
This protocol provides a step-by-step methodology to diagnose and resolve convergence issues, based on analysis of common failure modes [39] [1] [73].
Diagram 1: Convergence Diagnosis Workflow.
Step-by-Step Procedure:
- Inspect Forces: After a failed or suspicious convergence, first check the output log for the maximum force (
Fmax). Values that are extremely high (e.g., > 10^5 kJ/mol/nm) or reported asinfindicate severe structural issues [39] [1]. - Path A - High Forces: If high forces are found, suspect atom overlaps.
- Action: Visually inspect the structure using molecular graphics software (e.g., VMD, PyMol) focusing on the atom(s) reported in the error. Manually correct impossible contacts or adjust the initial configuration [1].
- Action: Meticulously check that the ligand and residue definitions in your topology file perfectly match the atom names and counts in your coordinate file [1].
- Path B - Low Forces but Suspected False Minimum: If forces are low but you suspect false convergence (e.g., unstable energy in subsequent MD).
- Action: Monitor convergence beyond just energy and density. Plot key metrics like system pressure and relevant Radial Distribution Functions (RDFs) over time [20] [73].
- Action: Significantly extend the simulation time. For complex systems, convergence of interaction networks (e.g., asphaltene-asphaltene RDFs) can require orders of magnitude more time than energy stabilization [20].
- Iterate: Repeat the validation steps until all diagnostic metrics confirm a stable, physically reasonable state.
Protocol 2: Advanced Validation Using Partial vs. Full Equilibrium
This protocol is based on research showing that systems can reach partial equilibrium where some properties stabilize, while others, crucial for the biological or chemical question, have not [73].
Methodology:
- Define Multiple Relevant Properties (A~i~): Select a set of properties to monitor. These should include both global metrics and specific, functionally relevant ones [73].
- Global Metrics: Total potential energy, system density, RMSD of the backbone.
- Specific Metrics: Key hydrogen bond distances, dihedral angles of active site residues, radius of gyration, or RDFs between specific molecular components [20].
- Calculate Running Averages: For each property A~i~, calculate its running average ⟨A~i~⟩(t) from simulation time 0 to t.
- Analyze for Plateauing: Plot ⟨A~i~⟩(t) for all properties over the course of a long simulation (e.g., multi-microsecond for biomolecules) [73].
- Validate Convergence: A property is considered "converged" when its running average plateaus and fluctuations around the final average value become small for a significant portion of the trajectory. The system is only fully equilibrated when all key properties of interest meet this criterion [73].
Diagram 2: Partial vs Full Equilibrium Validation.
Frequently Asked Questions
Q: Why does my molecular dynamics simulation fail during energy minimization after I modify a ligand's structure? A: Energy minimization failures often occur due to steric clashes or unrealistic bond geometries introduced during structure modification. The force field calculates extremely high energies from these clashes, causing the minimizer to fail. Before minimization, always perform structural reasonableness checks to identify atoms too close together (steric clashes), incorrect bond lengths, or anomalous bond angles [77].
Q: How can I programmatically check new molecular structures for severe steric incompatibilities? A: Implement a two-step validation protocol. First, use a rule-based approach to flag atoms within a defined van der Waals radius threshold (e.g., less than 70-80% of the sum of their radii). Second, use the UFF (Universal Force Field) or similar to perform a single-point energy calculation; an abnormally high potential energy indicates significant steric strain or clashes that need resolution [77].
Q: What are the consequences of applying bond-length constraints during a simulation, and how do I account for them? A: Applying constraints allows for longer simulation time steps by freezing the fastest vibrational motions. However, this restricts the system's phase space and can introduce free energy errors. To correct for this, you can calculate the free energy cost of applying or removing constraints during post-processing using methods that consider enthalpy, vibrational entropy, and Jacobian free energy terms. This can reduce free energy errors by about 90% [78].
Q: My automated topology generation for a novel macrocycle failed. What structural checks should I perform? A: Complex systems like macrocycles require robust checks. Ensure your topology generation tool can handle cyclic and branched molecules. Key checks include:
- Ring Closure: Verify that all bonds in the ring can form with reasonable geometry.
- Chirality: Confirm the stereochemistry of chiral centers is correctly defined.
- Bond Order: Validate bond orders (single, double, aromatic) are correctly assigned, as this affects bond length and angle parameters. Frameworks like Vermouth are designed to automate these checks for complex, non-linear molecules [79].
Experimental Protocols for Structural Validation
Protocol 1: Pre-minimization Steric and Geometric Screening
- Input Structure: Obtain your initial 3D molecular structure (e.g., from a PDB file or a file in SDF format) [77] [79].
- Steric Clash Check:
- Calculate all interatomic distances.
- Flag any pair of non-bonded atoms where the distance is less than the sum of their van der Waals radii multiplied by a factor of 0.7-0.8.
- Manually or algorithmically adjust the conformation to resolve severe clashes.
- Bond and Angle Validation:
- Using a library of standard bond lengths and angles (e.g., from the force field you intend to use), compare your structure's parameters.
- Flag any bond length deviation greater than 0.2 Å or angle deviation greater than 20° from the standard value for further inspection.
- Topology Assignment:
- Use a reliable topology generator (e.g., Martinize2 for coarse-grained systems) to assign parameters [79].
- Carefully review any warning or error messages generated during this process, as they often point to structural ambiguities or missing parameters.
Protocol 2: Constraint Correction in Free Energy Calculations
This methodology corrects for the free energy cost of imposing constraints, improving accuracy [78].
- Run Constrained Simulation: Perform your free energy simulation (e.g., using BAR, MBAR) with the necessary bond or angle constraints applied.
- Post-Processing Analysis: For each sampled frame in your trajectory, calculate the free energy difference (
ΔG_cons) of releasing the constraint. This involves:- Find the Energy Minimum: Optimize the constrained coordinate(s) to find the local energy minimum given the current soft degrees of freedom (e.g., dihedral angles).
- Calculate Hessian: Compute the Hessian matrix (matrix of second derivatives) for the constrained system at this minimum.
- Compute Free Energy Terms: Calculate the enthalpy (
ΔH_cons), vibrational entropy (-TΔS_cons), and Jacobian term (ΔG_jac) associated with the constraint.
- Apply Correction: The total constraint correction is
ΔG_correction = -ΔG_cons. Apply this correction to your final free energy estimate.
Data Presentation: Standard Geometric Parameters
Table 1: Typical Bond Length and Angle Ranges for Common Atom Pairs in Biomolecules
| Atom Pair | Bond Type | Typical Length (Å) | Notes |
|---|---|---|---|
| C-C | Single | 1.50 - 1.54 | Alkane chains |
| C=C | Double | 1.31 - 1.35 | Alkene, aromatic |
| C-N | Single | 1.43 - 1.50 | Peptide backbone |
| C=O | Double | 1.20 - 1.23 | Carbonyl group |
| C-H | Single | 1.08 - 1.10 | |
| N-H | Single | 1.00 - 1.02 | |
| O-H | Single | 0.96 - 0.98 |
| Angle Type | Typical Value (Degrees) | Example |
|---|---|---|
| C-C-C | 109.5 - 114.0 | Alkane chain |
| C-C-H | ~109.5 | |
| C-N-C | ~120.0 | |
| Tetrahedral | 109.5 | SP3 carbon |
| Trigonal Planar | 120.0 | SP2 carbon |
Table 2: Summary of Common Structural Issues and Corrective Actions
| Issue Type | Diagnostic Sign | Corrective Action |
|---|---|---|
| Steric Clash | High initial potential energy; non-bonded atoms too close. | Perform a short, steepest descent minimization; adjust conformation manually. |
| Incorrect Bond Length | Large force field energy term for bonds; deviation from standard values. | Rebuild the fragment; check for correct bond order assignment. |
| Anomalous Angle | Large force field energy term for angles. | Manually adjust the angle or use a conformer search algorithm. |
| Topology Error | Simulation crashes immediately; parameter not found. | Verify atom types and bonding in the topology generator. |
The Scientist's Toolkit: Essential Research Reagents & Software
Table 3: Key Software Tools for Structural Checks and Topology Generation
| Tool Name | Primary Function | Application Context |
|---|---|---|
| Vermouth/Martinize2 | A unified Python library and program for automated topology generation [79]. | Converting all-atom structures to coarse-grained representations (e.g., for Martini force field); handles proteins, macrocycles, and polymers. |
| Chem3DLLM | A multimodal large language model for generating and optimizing 3D molecular structures [77]. | Structure-based drug design; generating 3D ligand conformations conditioned on protein pockets. |
| Reinforcement Learning with Scientific Feedback (RLSF) | A training paradigm that uses physical/chemical priors as rewards to guide 3D structure generation [77]. | Optimizing generated molecular conformations for stability, low energy, and high binding affinity. |
| SHAKE/LINCS | Algorithms to constrain bond lengths and angles during molecular dynamics simulations [78]. | Allowing for longer integration time steps by freezing the fastest vibrational motions. |
Workflow Visualization
Workflow for troubleshooting structural issues in molecular dynamics setups.
Methodology for calculating free energy corrections in constrained simulations.
Frequently Asked Questions (FAQs)
FAQ 1: What are the most common causes of energy minimization failures in molecular dynamics simulations? Energy minimization often fails due to structural anomalies in the initial configuration. The most frequent causes include atom overlaps leading to infinite forces, incorrect topology parameters that create unrealistic bonds or angles, missing atoms in the coordinate file, and force field parameters that are inconsistent with the system's atom types [47] [1] [61]. These issues manifest as errors like non-finite forces or failure to converge to the requested force tolerance.
FAQ 2: My minimization reports "infinite force" on an atom. How do I resolve this? An "infinite force" error indicates severe atom overlaps [1] [61]. To resolve this:
- Inspect and Correct Initial Coordinates: Visually check the structure around the problematic atom (e.g., atom 1251 as in one case [1]) for steric clashes.
- Verify Topology-Configuration Consistency: Ensure every atom in your coordinate file (like
.groor.pdb) has a matching, correctly defined atom type and parameters in your topology file (.top) [1]. Mismatches here are a common source of error. - Use Soft-Core Potentials: For free energy calculations, soft-core potentials can help avoid infinite forces during minimization [61].
FAQ 3: The minimization converged to machine precision but did not achieve the requested Fmax. Is this acceptable?
This is a common and often acceptable outcome, particularly for systems with high water content [61]. It means the algorithm has found the nearest local minimum given the initial structure. If the potential energy (Epot) is reasonable for your system (e.g., around -10^5 to -10^6 for solvated systems), it is generally safe to proceed to the equilibration phase. The key is to check if the Fmax value is low enough for your subsequent simulation needs [61].
FAQ 4: How does hardware selection impact the speed and scale of my MD simulations? Hardware is critical for performance. GPUs dramatically accelerate computation, with high-end models like the NVIDIA RTX 6000 Ada (48 GB VRAM) being ideal for large systems, while the RTX 4090 offers a strong balance of price and performance for smaller simulations [80]. For CPUs, prioritize clock speed over core count for most MD workloads [80]. Specialized hardware like the Molecular Dynamics Processing Unit (MDPU) has been shown to reduce time and power consumption by up to 10^3 to 10^9 times compared to CPU/GPU setups while maintaining ab initio accuracy [81].
FAQ 5: What is the benefit of using cloud platforms like Fovus for OpenMM simulations? Cloud HPC platforms can offer significant cost efficiency and scalability. Benchmarking results for OpenMM on Fovus show simulation costs can be as low as $6.59 per microsecond for a small protein system (2,489 atoms) and simulation speeds can reach 3359 nanoseconds per day when optimized for time [82]. These platforms automate hardware selection and leverage spot instances, allowing researchers to run large-scale simulations without managing physical infrastructure [82].
Performance Benchmarking Data
| System (Number of Atoms) | Descriptive Name | Min. Cost ($/µs) | Max. Speed (ns/day) | Best Cost Efficiency (ns/$) |
|---|---|---|---|---|
| System 1: gbsa (2,489) | Dihydrofolate Reductase (DHFR) – Implicit | $6.59 | 3358.7 | 151.6 |
| System 2: pme (23,558) | Dihydrofolate Reductase (DHFR) – Explicit-PME | $14.62 | 2048.8 | 68.4 |
| System 3: amoebapme (23,558) | AMOEBA DHFR | $875.87 | 40.6 | 1.1 |
| System 4: apoa1rf (92,224) | Apolipoprotein A1 – RF | $46.19 | 1012.3 | 22.6 |
| System 5: apoa1pme (92,224) | Apolipoprotein A1 – PME | $66.06 | 760.0 | 15.1 |
Table 2: Performance Comparison of MD Hardware and Architectures
| Processing Unit / Architecture | Key Feature | Reported Performance Gain | Ideal Use Case |
|---|---|---|---|
| MDPU (Molecular Dynamics Processing Unit) [81] | Specialized, computing-in-memory (CIM) | ~10^3 (vs. MLMD) to ~10^9 (vs. AIMD) lower ηt and ηp | Large-scale, long-duration AIMD-accuracy simulations |
| Universal MLIPs (uMLIPs) trained on OMat24 [83] | Trained on non-equilibrium configurations | Mean absolute percentage errors <6% for cleavage energy | Predicting surface stability and interfacial properties |
| NVIDIA RTX 6000 Ada [80] | 48 GB GDDR6 VRAM | Suitable for largest and most complex simulations | Memory-intensive simulations with large particle counts |
Experimental Protocols and Methodologies
Protocol 1: Standard Workflow for System Setup and Energy Minimization in GROMACS
This protocol outlines the steps to prepare a molecular system for simulation, a critical phase where many initialization errors occur [47] [1].
1. Generate Topology
- Command:
gmx pdb2gmx -f protein.pdb -o protein_processed.gro -p topol.top -water spc -ff [forcefield] - Troubleshooting:
- "Residue not found in database": The residue name in your PDB file may not match the force field's database. Rename the residue or choose a different force field that contains it [47].
- "Atom not found in rtp entry" or "Missing atoms": The atom names in your coordinate file may not match the force field's expectations. Use the
-ignhflag to ignore hydrogens and letpdb2gmxadd them correctly, or rename the atoms in your input file [47].
2. Define the Simulation Box
- Command:
gmx editconf -f protein_processed.gro -o protein_box.gro -c -d 1.0 -bt cubic
3. Solvate the System
- Command:
gmx solvate -cp protein_box.gro -cs spc216.gro -o protein_solvated.gro -p topol.top - Troubleshooting: Ensure the box size is appropriate. A box that is too large will waste computational resources; one that is too small can cause artifacts. Confusion between Ångström and nanometers can lead to a box 10^3 times larger than intended [47].
4. Add Ions
- Command:
gmx grompp -f ions.mdp -c protein_solvated.gro -p topol.top -o ions.tprfollowed bygmx genion -s ions.tpr -o protein_solvated_ions.gro -p topol.top -pname NA -nname CL -neutral
5. Energy Minimization
- Command:
gmx grompp -f em.mdp -c protein_solvated_ions.gro -p topol.top -o em.tprfollowed bygmx mdrun -v -deffnm em - Troubleshooting:
This protocol enables scalable, AI-driven molecular dynamics by integrating a machine-learned interatomic potential (MLIP) with the LAMMPS MD package.
1. Environment Setup
- Ensure you have LAMMPS (September 2025 release or later) built with Kokkos, MPI, ML-IAP, and Python support.
- A working Python environment with PyTorch and your trained MLIP model is required.
2. Develop the ML-IAP-Kokkos Interface
- Create a Python class that inherits from
MLIAPUnified(fromlammps.mliap.mliap_unified_abc). - Implement the
__init__function to specify element types and cutoff radius. - Implement the
compute_forcesfunction to perform inference using your PyTorch model. The function should calculate energies and forces using the atomic data (positions, types, neighbor lists) passed from LAMMPS.
3. Serialize and Save the Model
- Save your model object using
torch.save(mymodel, "my_model.pt").
4. Run LAMMPS with the Custom Model
- In your LAMMPS input script, load the potential with:
- Execute LAMMPS with Kokkos support on a GPU:
lmp -k on g 1 -sf kk -pk kokkos newton on neigh half -in sample.in
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for MD Simulations
| Item | Function / Explanation | Example Use Case |
|---|---|---|
| Force Fields | Parameterized sets of equations describing potential energy as a function of atomic coordinates [84]. | CHARMM, AMBER, GROMOS for biomolecules; ReaxFF for reactive systems [84] [27]. |
| Specialized MD Hardware | Application-specific hardware designed to overcome von Neumann architecture bottlenecks [81]. | MDPU for AIMD-accurate, large-scale simulations with massive reductions in time and power consumption [81]. |
| Cloud HPC Platforms | AI-optimized, serverless high-performance computing platforms that automate resource management [82]. | Fovus for running high-throughput OpenMM simulations with optimized cost and speed [82]. |
| MLIP Interfaces | Software bridges that connect machine learning models to traditional MD engines [85]. | ML-IAP-Kokkos interface for integrating PyTorch-based models into LAMMPS for AI-driven MD [85]. |
Troubleshooting Guides
Guide 1: Resolving "Infinite Force" and Atom Overlap Errors
Logical Flow for Troubleshooting Infinite Force Errors
Guide 2: Addressing LINCS/SETTLE/SHAKE Warnings
These warnings indicate that constraint algorithms are failing, often signaling deeper system instability [61].
- Diagnose the Root Cause: LINCS warnings are often symptoms, not the primary problem. The underlying issue is usually that forces in the system are becoming too large.
- Revisit Equilibration Protocol:
- Ensure the system was properly energy minimized before dynamics.
- Use a gradual equilibration strategy: first run in the NVT ensemble to stabilize temperature, then switch to NPT for pressure equilibration.
- Check Topology Parameters:
- Verify all bonded parameters (bonds, angles, dihedrals) are physically reasonable and consistent with the force field.
- For ligands or non-standard residues, ensure the generated topology is correct.
- Adjust Simulation Parameters:
- If the system is stable but produces occasional LINCS warnings, you can increase the
lincs_iterandlincs_orderparameters in your.mdpfile for greater constraint accuracy.
- If the system is stable but produces occasional LINCS warnings, you can increase the
Guide 3: Optimizing for System Size and Scalability
Decision Flow for Hardware and Architecture Selection Based on System Size
Welcome to the Technical Support Center
This resource provides targeted troubleshooting guides and FAQs for researchers grappling with simulation failures, framed within a broader thesis on molecular dynamics energy minimization. The content is structured to help you diagnose and resolve specific issues based on the computational approach you are using.
The table below summarizes the core characteristics, common failure points, and strengths of traditional force fields versus machine learning interatomic potentials (ML-IAPs).
| Feature | Traditional Force Fields | ML-Accelerated Approaches (ML-IAPs) |
|---|---|---|
| Functional Form | Pre-defined, physics-based (e.g., Lennard-Jones, bond-order) [86]. | Data-driven, flexible model learned from quantum mechanical data [86]. |
| Computational Cost | Relatively low, efficient for large systems [86]. | Higher than traditional MD, but much lower than ab initio methods [86]. |
| Accuracy & Transferability | Limited transferability; accuracy can vary significantly for chemistries outside parameterization [86]. | Near ab initio accuracy for systems within the training data distribution [86]. |
| Data Dependence | Relies on expert parameterization for specific molecules/conditions [86]. | Requires extensive, high-quality quantum mechanical training datasets [27]. |
| Common Failure Modes | Topology errors, inadequate energy minimization [12] [2]. | Data fidelity issues, model generalizability, force discontinuities [86] [27]. |
| Typical Error Messages | "Residue not found in database", "Forces have not converged" [12] [2]. | Warnings about inconsistent parameters or polarization catastrophes [27]. |
Troubleshooting Guides & FAQs
Traditional Force Fields
Q: My energy minimization stops abruptly, reporting that "forces have not converged." What should I do?
A: This is a common issue where the minimizer cannot reduce the maximum force below the specified threshold (emtol). The system may be stuck in a local high-energy state [2].
Recommended Protocol:
- Verify Topology and Structure: Ensure your initial structure is physically realistic. Check for:
- Missing atoms: Look for warnings about missing atoms in
pdb2gmxoutput [12]. - Long bonds: These indicate structural problems that prevent stable minimization [12].
- Incorrect residue naming: Terminal residues in proteins may require specific prefixes (e.g.,
NALAfor N-terminal alanine in AMBER force fields) [12].
- Missing atoms: Look for warnings about missing atoms in
- Adjust Minimization Parameters: In your
.mdpfile, try the following:- Increase the maximum force tolerance (
emtol): Start with a looser tolerance (e.g., 1000) and progressively tighten it in subsequent runs [2]. - Change the algorithm: Switch from steepest descent (
integrator = steep) to conjugate gradient (integrator = cg), or vice-versa [2]. - Increase the number of steps (
nsteps): Allow the minimizer more time to converge. - Reduce the constraint accuracy: If using constraints (e.g., on bonds with hydrogen), try setting
constraints = noneto see if the system can minimize without them [2].
- Increase the maximum force tolerance (
Q: I get a "Residue 'XXX' not found in residue topology database" error frompdb2gmx. How can I fix this?
A: This error means the force field you selected does not contain a definition for the molecule or residue "XXX" [12].
Recommended Protocol:
- Check Residue Name: Verify the residue name in your coordinate file matches the expected name in the force field's database.
- Find a Topology: Search the literature or force field repositories for a pre-existing topology (
.itpfile) for your molecule. You can include this file directly in your system topology (.topfile) [12]. - Parameterize the Molecule: If no topology exists, you will need to parameterize the molecule yourself, which is a complex process often requiring specialized tools and expertise [12].
ML-Accelerated Approaches (ML-IAPs)
Q: The geometry optimization with my ML-IAP does not converge and seems unstable. Why?
A: This is often caused by discontinuities in the force calculation, a known challenge for some ML-IAPs like ReaxFF. These occur when bond orders cross a cutoff threshold, leading to sudden changes in the computed forces [27].
Recommended Protocol:
- Modify the Bond Order Cutoff: Decrease the
BondOrderCutoffparameter (e.g., from the default 0.001). This includes more angles in the calculation, making the energy surface smoother and improving optimization stability [27]. - Use Tapered Bond Orders: Enable the
TaperBOoption, which applies a smoothing function to bond orders, effectively reducing the sharpness of discontinuities [27]. - Enable 2013 Torsion Angles: Set the
Torsionsparameter to2013to use a newer formula that behaves more smoothly at lower bond orders [27].
Q: I receive a warning about "Suspicious force-field EEM parameters." What does this mean?
A: This warning indicates a potential for a "polarization catastrophe" in the charge equilibration method. It happens when the EEM parameters (eta and gamma) for an atom type do not satisfy the relation eta > 7.2*gamma. This can lead to unphysically large charge transfers at short interatomic distances [27].
- Solution: Review and correct the electronegativity equalization method (EEM) parameters in your force field file to meet the stability condition [27].
Experimental Protocols & Workflows
Protocol 1: Troubleshooting a Failed Traditional Force Field Minimization
This protocol is designed to systematically address the common error where energy minimization fails to converge.
1. Initial System Setup (Pre-processing):
- Tool:
pdb2gmx - Action: Generate system topology.
- Check: Carefully review the screen output for any warnings about missing atoms, long bonds, or unknown residues [12]. Address these issues before proceeding.
2. Energy Minimization Configuration:
- Tool:
grompp(to prepare the binary input) - Action: Create a
.tprfile using a conservative.mdpparameter set. - Sample
minim.mdpparameters:
3. Execute and Diagnose:
- Tool:
mdrun - Action: Run the minimization and inspect the log file.
- Diagnosis: If forces do not converge, check the "Maximum force" value and the atom it acts upon. A very high force on a specific atom often indicates a local structural problem [2].
4. Iterative Refinement:
- If the first run fails, use the output structure as a new starting point.
- Gradually tighten
emtolor switch minimization algorithms. As a last resort, consider temporarily turning off constraints (constraints = none) [2].
Protocol 2: Training a Robust Machine Learning Interatomic Potential
This protocol outlines the key steps for generating a reliable ML-IAP, highlighting stages where errors can propagate.
1. Data Generation:
- Method: Perform ab initio molecular dynamics (AIMD) simulations using Density Functional Theory (DFT).
- Goal: Create a diverse set of atomic configurations that span the relevant chemical space.
- Critical Consideration (Data Fidelity): The accuracy of the ML-IAP is fundamentally limited by the quality of this training data. Using higher-level DFT functionals (e.g., meta-GGA) can improve generalizability [86].
2. Material Representation & Feature Encoding:
- Method: Use geometrically equivariant Graph Neural Networks (GNNs).
- Goal: Convert atomic coordinates into a representation that preserves physical symmetries (rotation, translation).
- How it works: The network learns local atomic environments through message-passing, ensuring that scalar outputs (e.g., energy) are invariant, and vector outputs (e.g., forces) are equivariant under these transformations [86].
3. Model Training:
- Architecture: Employ state-of-the-art frameworks like NequIP or DeePMD.
- Targets: Train the model to predict total energy and atomic forces that match the ab initio reference data.
- Success Metrics: Aim for low Mean Absolute Error (MAE) on energies (e.g., < 1 meV/atom) and forces (e.g., < 20 meV/Å) on a held-out test set [86].
4. Validation and Deployment:
- Action: Run MD simulations with the trained ML-IAP and compare properties (e.g., radial distribution functions, diffusion coefficients) against AIMD or experimental data to validate its transferability beyond the training set.
Workflow Visualization
Traditional vs. ML-Accelerated MD Workflow
This diagram contrasts the troubleshooting pathways for simulations using traditional force fields versus ML-IAPs, aligning with the thesis context on energy minimization failures.
ML-IAP Training and Validation Cycle
This diagram details the iterative workflow for creating and validating a Machine Learning Interatomic Potential, highlighting the critical role of data.
The Scientist's Toolkit: Essential Research Reagents & Software
| Tool / Reagent | Function / Description | Commonly Associated Issues |
|---|---|---|
| GROMACS | A software package for performing molecular dynamics simulations; primarily used with traditional force fields. | Topology errors, energy minimization failures, segmentation faults [12] [2]. |
| AMBER Tools | A suite of programs for molecular dynamics simulation, particularly with the AMBER force field. | Requires careful handling of terminal residues and parameter conversion when transferring to other MD engines [2]. |
| DeePMD-kit | An open-source package for building and running ML-IAPs based on the Deep Potential model. | Accuracy is dependent on the quality and breadth of the input ab initio data [86]. |
| NequIP | A framework for developing E(3)-equivariant ML-IAPs, known for high data efficiency and accuracy. | Enforcing strict equivariance can increase computational cost [86]. |
| ReaxFF | A reactive force field that allows for bond formation and breaking; can be combined with ML approaches. | Geometry optimization failures due to discontinuities in the force calculation [27]. |
| QM9/MD17/MD22 Datasets | Public benchmark datasets of quantum mechanical calculations for small organic molecules [86]. | May not be sufficient for training universal potentials for complex, extended materials systems [86]. |
Troubleshooting Guide: Energy Minimization
Energy Minimization Fails to Converge
Problem Description The energy minimization process stops before forces reach the requested precision (Fmax). The error message indicates: "Energy minimization has stopped, but the forces have not converged to the requested precision Fmax < 10" [39].
Example Error Output
Solution Steps
- Check your starting configuration for atomic overlaps or extremely high initial energy values, which can prevent minimization convergence [42].
- Verify constraints and restraints: Overly stringent constraints can impede proper energy minimization. Try relaxing constraints or setting
constraints = nonein your mdp file [39]. - Adjust minimization parameters: Increase the number of steps (
nsteps) or try different algorithms (e.g., conjugate gradient after initial steepest descent) [42]. - Review system topology: Ensure no missing bonds, angles, or incorrect charge assignments that could cause structural instabilities [42].
Unstable Temperature and Pressure Control During Equilibration
Problem Description Observable properties like temperature or pressure fail to reach a stable average, showing abrupt spikes or drifts during equilibration [42].
Solution Steps
- Extend equilibration time: Depending on system size and complexity, significantly increase the equilibration duration until properties stabilize [87].
- Adjust coupling parameters: Refine thermostat and barostat time constants based on preliminary trials. For Nose-Hoover thermostats, increase the thermostat chain length if experiencing strong, persistent oscillations [87].
- Choose appropriate algorithms:
System Topology and Solvation Errors
Problem Description Structural instabilities occur due to incorrect system topology or solvation issues, leading to simulation failures [42].
Solution Steps
- Generate reliable topology files: Use
gmx pdb2gmxfor proper topology generation and always check total system charge [42]. - Verify solvent insertion: Use
gmx solvatewith appropriate parameters, check system density after solvation, and ensure sufficient padding between solute and box edge [42]. - Ensure proper ion placement: Use
gmx genionfor ion placement, select appropriate concentration, and check initial ion distribution to prevent clustering [42].
Frequently Asked Questions (FAQs)
What are the best practices for validating minimized structures against experimental data?
Answer After successful minimization, correlate computational observables with experimental data:
- Compare structural properties: Calculate radius of gyration (using
gmx gyrate) and compare with experimental light scattering data [88]. - Analyze coordination numbers: For solvated systems, compare with neutron diffraction data.
- Validate dynamics: Calculate diffusion coefficients and compare with experimental values.
- Use multiple validation techniques: Combine computational and experimental approaches like Molecular Dynamics with Multi-Angle Light Scattering (MALS) for comprehensive conjugate analysis [88].
How do I choose the right time step for molecular dynamics simulations?
Answer The time step is crucial for accuracy and efficiency [87]:
| System Type | Recommended Time Step | Rationale |
|---|---|---|
| Systems with hydrogen atoms | ≤1 fs | To resolve high vibrational frequencies of light atoms [87] |
| Systems with only heavy atoms | 1-2 fs | Heavier atoms have lower vibration frequencies [87] |
| High-temperature simulations | Smaller time step | Atoms experience larger forces [87] |
| Systems far from equilibrium | Smaller time step | Large forces act on particles [87] |
Protocol: Start with 1 fs for most systems, then assess conservation of total energy in NVE simulation to determine if larger values are possible [87].
How long should equilibration continue before starting production runs?
Answer Equilibration should continue until:
- Observable properties stabilize: Temperature, pressure, and energy reach stable averages with reasonable fluctuations [42].
- System density matches experimental values (for condensed phase systems).
- Potential energy plateaus with only minor fluctuations.
Equilibration times can vary from picoseconds to hundreds of nanoseconds depending on system size and the longest relaxation time present [87]. Always verify equilibration by monitoring time evolution of key observables before beginning production simulations.
What should I do when encountering memory allocation problems during production runs?
Answer For memory allocation errors with large systems or long timescales [42]:
- Scale hardware appropriately: Ensure computational resources match simulation demands.
- Optimize simulation settings: Use memory-efficient algorithms and minimize unnecessary computation.
- Adjust trajectory output frequency: Balance detail and file size by optimizing how often states are saved.
- Monitor disk usage: Ensure adequate storage space and use compression if needed.
Experimental Validation Protocols
Table 1: Quantitative Metrics for Structural Validation
| Validation Metric | Computational Command | Experimental Correlative Technique | Acceptance Criteria |
|---|---|---|---|
| Radius of Gyration | gmx gyrate |
Multi-Angle Light Scattering (MALS) [88] | ≤5% deviation from experimental value |
| Root Mean Square Deviation (RMSD) | gmx rms |
X-ray Crystallography | ≤2Å for backbone atoms |
| Solvent Accessible Surface Area | gmx sasa |
NMR Chemical Shift Perturbation | Consistent with experimental trends |
| Coordination Numbers | gmx rdf |
Neutron Diffraction | First peak position within 0.1Å |
Table 2: Research Reagent Solutions for Validation Studies
| Reagent/Standard | Function/Purpose | Application Context |
|---|---|---|
| BSA (Bovine Serum Albumin) | Validation standard for conjugate analysis [88] | Protein conjugate characterization |
| Thyroglobulin | Practice standard for analysis validation [88] | Method development and training |
| Appropriate Molecular Standards | Instrument validation and system constants determination [88] | Photodetector normalization, inter-detector alignment |
Methodologies and Workflows
Protein Conjugate Analysis Protocol
This ASTRA method enables characterization of conjugates like copolymers and PEGylated proteins using three signals (UV, LS, RI) to calculate mass fraction and molar mass of two conjugate components [88].
Step-by-Step Methodology:
- Sample Preparation: Prepare conjugate samples in compatible solvents following manufacturer specifications.
- Instrument Calibration: Validate instrument using BSA molecular standards [88].
- Data Collection: Simultaneously collect UV, light scattering (LS), and refractive index (RI) signals.
- Data Analysis: Calculate mass fraction and molar mass using the three-signal combination method.
- Validation: Practice and validate analysis using Thyroglobulin as specified in technical note TN1006 [88].
Moments and Averages Calculation
For macromolecules with continuous distributions or discrete populations, molar mass, radius and viscosity moments are calculated in ASTRA software [88].
Key Methodology Points:
- Molar Mass Moments: Determine different average types (number, weight, z-average).
- Radius Moments: Calculate size distribution parameters.
- Viscosity Moments: Characterize hydrodynamic properties.
- Relevance Assessment: Identify which moments are most relevant for online versus batch light scattering techniques [88].
Workflow Visualization
Energy Minimization Troubleshooting Pathway
Experimental Validation Workflow
MD Simulation Setup and Equilibration
Conclusion
Successful molecular dynamics energy minimization requires a comprehensive understanding of both theoretical principles and practical implementation details. By systematically addressing common failure modes through proper algorithm selection, parameter optimization, and rigorous validation, researchers can significantly improve simulation reliability. The integration of machine learning force fields presents promising opportunities for enhancing both accuracy and efficiency in minimization protocols. Future directions should focus on developing adaptive minimization strategies, improving force field accuracy for complex biomolecular systems, and creating standardized benchmarking frameworks to facilitate reproducibility across computational studies in drug discovery and biomedical research.
References
- 1. Error in minimisation - User discussions - GROMACS forums [https://gromacs.bioexcel.eu/t/error-in-minimisation/9312]
- 2. Energy minimization stops abruptly without force convergence ... [https://gromacs.bioexcel.eu/t/energy-minimization-stops-abruptly-without-force-convergence-and-segmentation-fault/12899]
- 3. Minimize Structure [https://www.cgl.ucsf.edu/chimera/current/docs/ContributedSoftware/minimize/minimize.html]
- 4. Common errors when using GROMACS [https://manual.gromacs.org/current/user-guide/run-time-errors.html]
- 5. Molecular dynamics parameters (.mdp options) [https://manual.gromacs.org/current/user-guide/mdp-options.html]
- 6. Basics of numerical energy minimization techniques used ... [https://mattermodeling.stackexchange.com/questions/8760/basics-of-numerical-energy-minimization-techniques-used-in-molecular-dynamics]
- 7. Best Practices for Foundations in Molecular Simulations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6884151/]
- 8. Stationary Points on the Potential Energy Surface (PES) [https://calcus.cloud/learn/potential_energy_surface]
- 9. Potential energy surface [https://en.wikipedia.org/wiki/Potential_energy_surface]
- 10. Minimization issue - User discussions - GROMACS forums [https://gromacs.bioexcel.eu/t/minimization-issue/9769]
- 11. Representing crystal potential energy surfaces via a ... [https://www.sciencedirect.com/science/article/abs/pii/S1359645424007535]
- 12. Common errors when using GROMACS [https://manual.gromacs.org/2025.1/user-guide/run-time-errors.html]
- 13. Molecular Dynamics Simulation (MDSim) Module [https://bioinformatics.utep.edu/UPBiT/MDSim-UPBiT.php]
- 14. Conjugate gradient method [https://en.wikipedia.org/wiki/Conjugate_gradient_method]
- 15. Chapter 5 Conjugate Gradient Methods [https://indrag49.github.io/Numerical-Optimization/conjugate-gradient-methods-1.html]
- 16. L-BFGS Algorithm [https://medium.com/@ravisankarit/l-bfgs-algorithm-63bafb76cd89]
- 17. Numerical optimization based on the L-BFGS method [https://towardsdatascience.com/numerical-optimization-based-on-the-l-bfgs-method-f6582135b0ca/]
- 18. Unconstrained optimization: L-BFGS and CG - C++, C#, Java [https://www.alglib.net/optimization/lbfgsandcg.php]
- 19. Structure optimization — ASE documentation [https://wiki.fysik.dtu.dk/ase/ase/optimize.html?highlight=bfgs]
- 20. Investigation of equilibrium and convergence in MD ... [https://www.sciencedirect.com/science/article/abs/pii/S0950061825004659]
- 21. Implementing reactivity in molecular dynamics simulations ... [https://www.nature.com/articles/s41467-024-50793-0]
- 22. Machine learning analysis of molecular dynamics ... [https://www.nature.com/articles/s41598-025-11392-1]
- 23. Molecular Dynamics Simulations in Drug Discovery and ... [https://www.mdpi.com/2227-9717/9/1/71]
- 24. Computational-aided drug design strategies for drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12634332/]
- 25. Common errors when using GROMACS [https://manual.gromacs.org/2025.0/user-guide/run-time-errors.html]
- 26. Non-numeric atom coords - simulation unstable' error [https://matsci.org/t/error-on-proc-7-non-numeric-atom-coords-simulation-unstable-error/60538]
- 27. Troubleshooting and warnings — ReaxFF 2025.1 ... - SCM [https://www.scm.com/doc/ReaxFF/Troubleshooting.html]
- 28. Supplemental: Overview of the Common Force Fields [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/S01-Force_Fields_Overview/index.html]
- 29. Implementing reactivity in molecular simulations with... dynamics [https://www.nature.com/articles/s41467-024-50793-0?error=cookies_not_supported&code=fd775d70-06f0-4d3a-8ad0-46e2c120398b]
- 30. Mastering Steepest Descent for Energy Minimization [https://www.numberanalytics.com/blog/mastering-steepest-descent-energy-minimization]
- 31. Assessing the Current State of Amber Force Field ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10339676/]
- 32. Energy Minimization - GROMACS 2025.3 documentation [https://manual.gromacs.org/documentation/2025.3/reference-manual/algorithms/energy-minimization.html]
- 33. Limited-memory BFGS [https://en.wikipedia.org/wiki/Limited-memory_BFGS]
- 34. When does L-BFGS outperform GD? [https://scicomp.stackexchange.com/questions/36166/when-does-l-bfgs-outperform-gd]
- 35. A robust BFGS algorithm for unconstrained nonlinear ... [https://arxiv.org/html/1212.5929v2]
- 36. Optimization algorithm — Atomistic Simulation Tutorial [https://docs.matlantis.com/atomistic-simulation-tutorial/en/2_3_opt-algorithm.html]
- 37. Continuing with optimization using lbfgs/bfgs when ... [https://discourse.mc-stan.org/t/continuing-with-optimization-using-lbfgs-bfgs-when-optimization-is-terminated/34858]
- 38. What parameters should be changed in mdp file to use wth ... [https://gromacs.bioexcel.eu/t/what-parameters-should-be-changed-in-mdp-file-to-use-wth-charmm-force-field/6039]
- 39. Energy minimization error - GROMACS forums [https://gromacs.bioexcel.eu/t/energy-minimization-error/478]
- 40. MD minimization - User discussions - GROMACS forums [https://gromacs.bioexcel.eu/t/md-minimization/571]
- 41. Minimization error - User discussions - GROMACS forums [https://gromacs.bioexcel.eu/t/minimization-error/6463]
- 42. 12 GROMACS Errors: From Setup to Execution [https://parssilico.com/blogs/99-12-gromacs-errors-from-setup-to-execution]
- 43. Common errors when using GROMACS [https://manual.gromacs.org/2024.4/user-guide/run-time-errors.html]
- 44. Validating Molecular Dynamics Simulations Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420231/]
- 45. System preparation - GROMACS 2025.4 documentation [https://manual.gromacs.org/current/user-guide/system-preparation.html]
- 46. Preparation of the Inputs Files for Advanced Sampling ... [https://hackmd.io/@WeiTseHsu/mdtutorial-01]
- 47. Common errors when using GROMACS [https://manual.gromacs.org/documentation/2025.4/user-guide/run-time-errors.html]
- 48. Focus on α5-Containing GABA (A) Receptors - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12645288/]
- 49. Troubleshooting — BAND 2025.1 documentation - SCM [https://www.scm.com/doc/BAND/Troubleshooting/Trouble_Shooting.html]
- 50. Convergence and equilibrium in molecular dynamics ... [https://www.nature.com/articles/s42004-024-01114-5]
- 51. Optimization of Molecular Dynamics Simulations of c- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7400636/]
- 52. Energy Landscape Distortions and the Mechanical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1305495/]
- 53. Optimization of convergence criteria for fragmentation ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261417308187]
- 54. Structure optimization — ASE documentation [https://wiki.fysik.dtu.dk/ase/ase/optimize.html]
- 55. Automated Minimization of Steric Clashes in Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3058769/]
- 56. Clashes - Proteopedia, life in 3D [https://proteopedia.org/wiki/index.php/Clashes]
- 57. Introduction — GROMACS 2019-rc1 documentation [https://manual.gromacs.org/documentation/2019-rc1/reference-manual/introduction.html]
- 58. Minimization converges with gromacs 2021.5, but fails with ... [https://gromacs.bioexcel.eu/t/minimization-converges-with-gromacs-2021-5-but-fails-with-gromacs-2022-2/5287]
- 59. Automated minimization of steric clashes in protein structures [https://pubmed.ncbi.nlm.nih.gov/21058396/]
- 60. User guide to the wwPDB EM validation reports [http://www.wwpdb.org/validation/2017/EMValidationReportHelp]
- 61. Navigating mdrun Challenges in GROMACS Simulations [https://blog.bioinfoquant.com/navigating-mdrun-challenges-in-gromacs-simulations-part-i]
- 62. GROMOS54a7 ff leads to missing parameters in topology after ... [https://gromacs.bioexcel.eu/t/gromos54a7-ff-leads-to-missing-parameters-in-topology-after-s-s-bonds-between-cys-residues-were-app/6708]
- 63. Missing Torsion Parameter For >C-N-N=N- · Issue #336 [https://github.com/openforcefield/openforcefield/issues/336]
- 64. Efficient and Precise Force Field Optimization for ... [https://arxiv.org/html/2406.09817v1]
- 65. Identifying Systematic Force Field Errors Using a 3D-RISM ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9921782/]
- 66. Force fields for small molecules - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6733265/]
- 67. Protocol for Molecular Dynamics Simulations of Proteins [https://en.bio-protocol.org/en/bpdetail?id=2051&type=0]
- 68. Machine Learning Force Fields - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC8391964/]
- 69. Crash testing machine learning force fields for molecules ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc06529h]
- 70. Energy & Force Regression on DFT Trajectories is Not ... [https://arxiv.org/html/2502.03660v1]
- 71. Machine learning force fields for improved materials modeling [https://www.schrodinger.com/materials-science/learn/white-papers/machine-learning-force-fields-for-improved-materials-modeling/]
- 72. Research Status of Molecular Dynamics Simulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12565893/]
- 73. Convergence and equilibrium in molecular dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10850365/]
- 74. Error messages — ADF 2025.1 documentation - SCM [https://www.scm.com/doc/ADF/Appendices/Error_messages.html]
- 75. A Computationally Efficient Method to Generate Plausible ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12344705/]
- 76. An artificial intelligence accelerated ab initio molecular ... [https://www.nature.com/articles/s41597-025-05338-5]
- 77. Chem3DLLM: 3D Multimodal Large Language Models for ... [https://arxiv.org/html/2508.10696v1]
- 78. Correcting for the free energy costs of bond or angle ... [https://www.sciencedirect.com/science/article/abs/pii/S030441651400302X]
- 79. Martinize2 and Vermouth: Unified Framework for Topology ... [https://elifesciences.org/reviewed-preprints/90627]
- 80. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOopwmFpc2EbmE7x8W8Nm_hMUNjhqTI9EKXV_-WtxWrGtvTpsTG8m]
- 81. High-speed and low-power molecular dynamics processing unit (MDPU) with ab initio accuracy | npj Computational Materials [https://www.nature.com/articles/s41524-024-01422-3]
- 82. Fovus Delivers OpenMM Molecular Dynamics Simulations for as Low as $6.59/µs, as fast as 3359 ns/day - Fovus [https://fovus.co/2025/05/02/fovus-delivers-openmm-molecular-dynamics-simulations-for-as-low-as-6-59-%C2%B5s-as-fast-as-3359-ns-day/]
- 83. 1 Introduction [https://arxiv.org/html/2508.21663]
- 84. Energy Minimization | IntechOpen [https://www.intechopen.com/chapters/74105]
- 85. Enabling Scalable AI-Driven Molecular Dynamics ... [https://developer.nvidia.com/blog/enabling-scalable-ai-driven-molecular-dynamics-simulations/]
- 86. A critical review of machine learning interatomic potentials ... [https://www.oaepublish.com/articles/jmi.2025.17]
- 87. Molecular Dynamics - QuantumATK [https://docs.quantumatk.com/manual/technicalnotes/molecular_dynamics/molecular_dynamics.html]
- 88. Discover Tips and Tricks with our Technical Notes - Waters [https://www.wyatt.com/blogs/discover-tricks-and-tips-with-our-technical-notes.html]