Validating Protein-Ligand Binding with Molecular Dynamics: A Comprehensive Guide from Theory to Practice
This article provides a comprehensive framework for researchers and drug development professionals to validate protein-ligand binding using Molecular Dynamics (MD) simulations.
Validating Protein-Ligand Binding with Molecular Dynamics: A Comprehensive Guide from Theory to Practice
Abstract
This article provides a comprehensive framework for researchers and drug development professionals to validate protein-ligand binding using Molecular Dynamics (MD) simulations. It covers foundational principles, from understanding force fields and sampling limitations to advanced methodological applications like absolute binding free energy calculations and enhanced sampling. The guide details practical troubleshooting and optimization strategies for simulation stability and performance and establishes rigorous protocols for validating results against experimental data and benchmarking across methods. By integrating these four core intents, this resource aims to enhance the reliability and predictive power of MD simulations in drug discovery pipelines.
Understanding the Foundations: Why MD is a Powerful Tool for Binding Validation
The Critical Role of Binding Free Energy in Drug Discovery
In rational drug design, the binding free energy (ΔG) quantifies the strength of the interaction between a potential drug molecule (ligand) and its biological target (receptor) [1]. Accurate prediction of ΔG is paramount as it directly influences a drug's potency and efficacy, guiding researchers toward promising compounds and away from likely failures [2] [3]. Computational methods for predicting binding free energy have thus become indispensable tools, capable of significantly compressing the traditional 10-12 year drug discovery timeline and reducing the immense costs, which can exceed $2.6 billion per new drug [1]. This guide provides a comparative analysis of the primary computational methods used to predict binding free energy, detailing their protocols, accuracy, and application within the broader context of validating protein-ligand interactions through molecular dynamics research.
Comparative Analysis of Binding Free Energy Methods
The following table summarizes the key computational techniques used for binding free energy prediction, comparing their theoretical basis, applications, and performance.
Table 1: Comparison of Key Computational Methods for Binding Free Energy Prediction
| Method | Theoretical Basis | Primary Application | Reported Accuracy | Computational Cost |
|---|---|---|---|---|
| MM/PBSA & MM/GBSA | End-state analysis using molecular mechanics and implicit solvation [1] | Virtual screening, binding affinity ranking [1] | Lower accuracy; improves docking results but limited precision [1] | Moderate |
| Free Energy Perturbation (FEP) | Alchemical transformation through intermediate λ states [4] [5] [6] | Relative binding affinity for congeneric series [3] [6] | ~1-2 kcal/mol RMSE; approaching experimental reproducibility [3] [6] | High |
| Thermodynamic Integration (TI) | Alchemical transformation using ∂U/∂λ [1] [4] | Relative binding affinity calculations [1] | Comparable to FEP [1] [4] | High |
| Nonequilibrium Switching (NES) | Short, bidirectional out-of-equilibrium transformations [2] | High-throughput relative binding free energy [2] | Comparable to FEP/TI with 5-10X higher throughput [2] | Moderate-High |
| Potential of Mean Force (PMF) | Reversible work along a physical reaction coordinate [5] [3] | Absolute binding free energy, ligand pathways [5] | Computationally expensive; convergence challenges [5] | Very High |
| Accelerated MD (aMD) | Enhanced sampling with boost potential [7] | Ligand binding pathways and kinetics [7] | Captures binding events faster than conventional MD [7] | High |
The selection of an appropriate method depends on the specific drug discovery context. Relative binding free energy methods like FEP and TI are particularly valuable for lead optimization, where small, systematic modifications are made to a lead compound [3]. These alchemical methods calculate the free energy difference between two similar ligands by transforming one into another in both the bound and unbound states [3]. For projects requiring high throughput, NES offers a distinct advantage by replacing slow equilibrium simulations with many rapid, independent transitions that can be run concurrently, achieving 5-10 times higher throughput than traditional methods [2].
Table 2: Analysis of Method Capabilities and Limitations
| Method | Key Strengths | Key Limitations | Optimal Use Case |
|---|---|---|---|
| MM/PBSA/GBSA | Fast; good for post-docking refinement; less demanding than FEP [1] | Limited precision; accuracy depends on parameter tuning [1] | Initial screening and ranking of large compound libraries |
| FEP/TI | High accuracy for small modifications; widely validated [3] [6] | Challenging for large conformational changes or dissimilar ligands [3] | Lead optimization of congeneric series |
| NES | High throughput; highly parallelizable; fault-tolerant [2] | Relatively newer method with less established track record [2] | Large-scale compound evaluation in cloud environments |
| Enhanced Sampling (aMD) | Captures binding kinetics and pathways [7] | Specialized analysis required; not purely for affinity prediction [7] | Understanding binding mechanisms and metastable states |
Experimental Protocols and Validation Frameworks
Alchemical Free Energy Calculation Protocol
The following diagram illustrates the generalized workflow for alchemical free energy calculations, common to FEP and TI approaches:
Figure 1: Generalized workflow for alchemical binding free energy calculations.
The standard protocol involves several key stages [4]:
System Preparation: The process begins with a high-quality receptor structure, often from X-ray crystallography or cryo-EM. Critical preparatory steps include:
- Adding missing loops or flexible regions
- Assigning correct protonation states of binding site residues at physiological pH
- Determining ligand tautomeric states
- Adding hydrogens, solvation, and counterions to neutralize the system
Thermodynamic Cycle Definition: Alchemical methods employ a non-physical pathway that connects the two end states of interest through a series of intermediate states characterized by a coupling parameter λ [4]. This parameter progressively transforms the Hamiltonian of the system from describing one ligand to describing the other.
Intermediate λ State Selection: The transformation is divided into discrete windows (typically 10-20), with careful attention to ensuring sufficient phase space overlap between adjacent windows for convergence [4] [5]. For example, a transformation might use λ values of 0, 0.2, 0.4, 0.6, 0.8, 0.9, and 1.0 [5]. Electrostatic and van der Waals transformations are often separated or use soft-core potentials to avoid singularities [4].
Equilibration and Production Sampling: Each λ window undergoes energy minimization, equilibration, and finally production molecular dynamics simulation. The conformational sampling must be adequate to represent the Boltzmann distribution for each state.
Free Energy Analysis: Using methods such as the Bennett Acceptance Ratio (BAR) [5] or Multistate BAR (MBAR) [4], the free energy differences between adjacent states are calculated and summed to give the total free energy difference.
Enhanced Sampling for Binding Pathways
While alchemical methods predict affinity, enhanced sampling techniques like accelerated Molecular Dynamics (aMD) provide insights into binding kinetics and pathways [7]. In aMD, a non-negative boost potential is added to the system's potential energy when it drops below a predefined threshold, effectively reducing energy barriers and accelerating transitions between low-energy states [7]. This approach has successfully captured millisecond-timescale events, such as the binding of agonists and antagonists to G protein-coupled receptors (GPCRs), in significantly shorter simulation times [7].
Validation Against Experimental Data
The gold standard for validating computational predictions is comparison with experimentally measured binding affinities, typically reported as dissociation constants (Kd), inhibition constants (Ki), or half-maximal inhibitory concentrations (IC50) [6]. The accuracy of experimental measurements themselves sets the upper limit for achievable predictive accuracy. Recent analyses suggest:
- The reproducibility of relative binding affinity measurements between different experimental assays has a root-mean-square difference of approximately 0.56-0.69 pKi units (0.77-0.95 kcal/mol) [6].
- Well-executed FEP calculations can achieve accuracy comparable to this experimental reproducibility, with errors in the range of 1-2 kcal/mol for many systems [3] [6].
- For congeneric series with careful system preparation, FEP has demonstrated the ability to achieve errors approaching 1 kcal/mol [6].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for Binding Free Energy Calculations
| Tool/Category | Specific Examples | Function/Purpose |
|---|---|---|
| Force Fields | CHARMM, AMBER, OPLS | Provide parameters for potential energy calculations; critical accuracy determinant [7] [6] |
| Solvation Models | TIP3P, GBSA, PBSA | Represent solvent effects; implicit vs. explicit trade-offs [1] [5] |
| Software Platforms | GROMACS, AMBER, NAMD, DESMOND, FEP+ | MD simulation engines with free energy capabilities [4] [5] |
| Enhanced Sampling | GaMD, MetaD, aMD | Accelerate rare events sampling; study binding pathways [7] [1] |
| System Preparation | GAAMP, CGenFF, tleap | Generate ligand parameters; prepare simulation systems [7] |
| Analysis Tools | alchemical-analysis.py, fastDRH | Analyze free energy calculations; automate workflows [1] [4] |
The following diagram illustrates how these tools integrate within a typical research workflow for binding free energy validation:
Figure 2: Tool integration in a binding free energy research workflow.
Binding free energy calculations have matured into powerful tools that provide critical insights for drug discovery. The current state of the art enables researchers to predict relative binding affinities with accuracy approaching experimental reproducibility for many systems. Method selection involves important trade-offs: MM/PB(GB)SA offers speed for initial screening, while FEP/TI provides higher accuracy for lead optimization at greater computational cost. Emerging methods like NES promise enhanced throughput through massive parallelism.
Future advancements will likely focus on improving force field accuracy, expanding the domain of applicability to more challenging targets like membrane proteins, and integrating machine learning approaches to accelerate sampling. As these methods continue to evolve, their role in rational drug design will expand, potentially transforming the efficiency and success rate of pharmaceutical development. For now, rigorous validation against experimental data and careful system preparation remain essential for obtaining reliable predictions that can genuinely guide drug discovery efforts.
The fundamental challenge in simulating protein-ligand binding with molecular dynamics (MD) is the vast disparity between computational and biological timescales. While functional conformational changes and binding events occur from microseconds to seconds in nature, all-atom MD simulations are typically limited to microsecond scales, creating a critical sampling bottleneck [8] [9]. This "sampling problem" arises because biomolecules navigate rough energy landscapes with multiple local minima separated by high energy barriers, causing simulations to become trapped in non-functional states [10]. For drug discovery professionals, this limitation is particularly acute as accurate binding affinity prediction requires sufficient sampling of all relevant conformational substates [10] [11].
In the post-AlphaFold era, where static protein structures have become readily available, the major frontier has shifted to understanding dynamic conformational transitions between functionally important states [12] [8]. This review comprehensively compares current enhanced sampling methodologies, evaluating their performance in overcoming temporal barriers for protein-ligand binding validation, with specific emphasis on computational efficiency, predictive accuracy, and practical implementation requirements.
Enhanced Sampling Methodologies: A Comparative Analysis
Collective Variable-Based Enhanced Sampling
Collective variable (CV)-based methods enhance sampling by applying bias potentials along carefully selected reaction coordinates that describe the slowest degrees of freedom during conformational transitions [13].
- Metadynamics discourages revisiting previously sampled states by "filling free energy wells with computational sand" through the addition of repulsive Gaussian potentials, enabling exploration of the entire free energy landscape for applications including protein folding and conformational changes [10].
- Replica-Exchange Molecular Dynamics (REMD) employs parallel simulations at different temperatures, with periodic exchange of configurations between replicas based on Metropolis criteria, allowing efficient barrier crossing in higher-temperature replicas while maintaining proper Boltzmann sampling at the target temperature [10].
- True Reaction Coordinate (tRC) Methods represent a recent breakthrough where bias is applied specifically along the few essential coordinates that determine the committor probability. These tRCs control both conformational changes and energy relaxation, enabling dramatic acceleration of processes like HIV-1 protease flap opening and ligand unbinding by 10⁵ to 10¹⁵-fold compared to standard MD [8].
Advanced Sampling Without Predefined Coordinates
For systems where suitable CVs are unknown a priori, several methods enable enhanced sampling without requiring predefined reaction coordinates.
- FRESEAN Mode Analysis identifies anharmonic low-frequency vibrations from short equilibrium trajectories, providing highly reproducible collective variables that naturally drive conformational transitions. This approach has successfully sampled conformational changes in proteins including lysozyme, HIV-1 protease, and KRAS within a single day on standard HPC hardware [9].
- Generalized Simulated Annealing (GSA) employs an artificial temperature that decreases during simulation, making it particularly suitable for characterizing very flexible systems and large macromolecular complexes at relatively low computational cost [10].
Free Energy Calculation Methods
Accurate binding affinity prediction requires calculating free energy differences between bound and unbound states.
- MM/PBSA and MM/GBSA methods compute binding free energies by combining molecular mechanics energy with Poisson-Boltzmann or Generalized Born solvation terms, and surface area-based nonpolar contributions. Recent advancements enable application to membrane protein-ligand systems through multitrajectory approaches and automated membrane parameter calculation [14].
- Free Energy Perturbation (FEP) and Thermodynamic Integration (TI) provide high accuracy but require extensive sampling of intermediate states, making them computationally prohibitive for large-scale screening [15] [11].
- Fragmentation Approaches like the Generalized Many-Body Expansion for Density Matrices (GMBE-DM) partition large biomolecules into smaller fragments for quantum mechanical calculations, achieving strong correlation with experimental binding free energies (R² = 0.84) in under 5 minutes per complex [11].
Table 1: Performance Comparison of Enhanced Sampling and Binding Affinity Methods
| Method | Acceleration/Speed | Accuracy (Correlation/Success) | System Size Limitations | Key Applications |
|---|---|---|---|---|
| True Reaction Coordinates [8] | 10⁵ to 10¹⁵-fold acceleration | Follows natural transition pathways; enables NRT generation | Requires energy relaxation simulations | HIV-1 protease flap opening, PDZ allostery |
| FRESEAN Mode Analysis [9] | ~1 day computation time | Reproducible low-frequency modes (correlation >0.9) | Tested on proteins up to multi-domain systems | Lysozyme, HIV-1 Pr, MCL-1, RBP, KRAS dynamics |
| GMBE-DM [11] | <5 minutes per complex | R² = 0.84 with experimental ΔG | 200-400 atom binding pockets | CDK2 and JAK1 ligand ranking |
| D3-ML [11] | <1 second per complex | R² = 0.87 with experimental ΔG | Scalable to large systems | High-throughput virtual screening |
| MMPBSA with Ensemble [14] | Medium throughput | Significant improvement over single-trajectory | Adapted for membrane proteins | P2Y12R conformational changes |
| Metadynamics [10] [13] | Dependent on CV quality | Accurate FES with optimal CVs | Low-dimensional CVs essential | Protein folding, molecular docking |
| REMD [10] | More efficient than MD with positive activation energy | Broad conformational sampling | Many replicas needed for large systems | Peptide and protein folding |
Experimental Protocols and Methodologies
True Reaction Coordinate Identification Protocol
The identification and application of true reaction coordinates involves a rigorous multi-step process [8]:
- Potential Energy Flow Calculation: Compute energy flows through individual coordinates using the equation (d{W}{i}=-\frac{\partial U\left({{\bf{q}}}\right)}{\partial {q}{i}}d{q}_{i}), which represents the energy cost of motion for each coordinate.
- Generalized Work Functional Analysis: Generate an orthonormal coordinate system (singular coordinates) that disentangles true reaction coordinates from non-essential coordinates by maximizing potential energy flows.
- tRC Validation: Verify that identified coordinates control both conformational changes and energy relaxation processes.
- Enhanced Sampling: Apply bias potentials directly to the identified tRCs to achieve dramatic acceleration of conformational transitions while maintaining physical pathways.
This protocol successfully accelerated the ligand dissociation process in HIV-1 protease, which has an experimental lifetime of 8.9×10⁵ seconds, to just 200 picoseconds in simulation [8].
FRESEAN Mode Analysis Workflow
The FRESEAN (Frequency-Selective Anharmonic) mode analysis protocol enables rapid identification of collective variables without prior knowledge of conformational changes [9]:
- Equilibrium Simulation: Run five independent 20-ns all-atom MD trajectories with randomized initial velocities.
- Coarse-Graining: Convert all-atom trajectories to a simplified representation (two beads per residue) preserving collective low-frequency vibrations.
- Mode Calculation: Apply FRESEAN analysis to isolate anharmonic low-frequency vibrations contributing most to the zero-frequency vibrational density of states.
- CV Selection: Select modes 7-9 (excluding translational and rotational modes 1-6) as collective variables for enhanced sampling.
- Biased Sampling: Perform enhanced sampling simulations using the identified low-frequency modes as CVs to explore conformational transitions.
This workflow consistently reproduces low-frequency vibrational modes across independent replicas (with 2D subspace correlations >0.9) and has successfully captured known conformational transitions in test proteins including lysozyme, HIV-1 protease, and KRAS [9].
MMPBSA for Membrane Proteins Protocol
Advanced MMPBSA calculations for membrane protein-ligand systems require specific modifications to address the membrane environment [14]:
- System Preparation: Model missing loops using tools like Modeller, select appropriate membrane lipid composition, and embed the protein in a lipid bilayer using CHARMM-GUI Membrane Builder.
- Multi-Trajectory Approach: Assign distinct protein conformations (pre- and post-ligand binding) as receptors and complexes instead of using a single trajectory.
- Ensemble Simulations: Run multiple replicas to improve conformational sampling depth.
- Membrane Parameter Automation: Use enhanced tools in Amber to automatically determine membrane thickness and location rather than relying on default values.
- Entropy Corrections: Apply truncated normal mode analysis for more efficient entropy calculations.
- Continuum Dielectric Treatment: Ensure consistent dielectric treatment in electrostatic energy calculations between GB and PB calculations.
This protocol demonstrated significant improvement in accuracy for simulating P2Y12R conformational changes upon ligand binding [14].
Diagram 1: Method Selection Framework for Enhanced Sampling. This roadmap guides researchers in selecting appropriate sampling strategies based on their system characteristics and available prior knowledge.
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 2: Essential Computational Tools for Enhanced Sampling Studies
| Tool/Resource | Function | Key Features | Accessibility |
|---|---|---|---|
| PySAGES [13] | Advanced sampling library | GPU acceleration, multiple backend support, ML framework integration | Open-source, Python-based |
| AMBER [14] | MD simulation package | Enhanced MMPBSA for membrane proteins, automated membrane parameters | Commercial with academic licensing |
| FRESEAN Analysis [9] | Low-frequency mode identification | Anharmonic vibrations, coarse-grained representation, high reproducibility | Custom implementation |
| LABind [16] | Binding site prediction | Graph transformer, cross-attention mechanism, unseen ligand capability | Open-source |
| GMBE-DM [11] | Quantum fragmentation | Density matrix reconstruction, linear-scaling, high accuracy | Research implementation |
| True RC Method [8] | Reaction coordinate identification | Energy relaxation analysis, natural pathway preservation | Research implementation |
| GPCRmd [12] | Specialized database | MD trajectories for GPCR proteins, standardized analysis | Public database |
Comparative Performance Analysis
Acceleration Efficiency and Binding Affinity Accuracy
Recent methodological advances demonstrate dramatic improvements in both sampling efficiency and binding affinity prediction accuracy.
Sampling Acceleration: True reaction coordinate methods have achieved unprecedented acceleration factors of 10⁵ to 10¹⁵ for protein conformational changes, reducing processes that would require years of standard MD simulation to tractable timescales [8]. FRESEAN mode analysis enables complete characterization of protein conformational ensembles within approximately one day on standard HPC hardware [9].
Binding Affinity Prediction: Quantum fragmentation methods like GMBE-DM achieve strong correlation with experimental binding free energies (R² = 0.84) while requiring less than 5 minutes per complex without extensive parallelization [11]. The machine learning-corrected dispersion potential D3-ML demonstrates even stronger ranking performance (R² = 0.87) with sub-second runtime per complex, making it ideal for high-throughput virtual screening [11].
Membrane Protein Advancements: Enhanced MMPBSA protocols for membrane proteins significantly improve accuracy for systems like P2Y12R, addressing critical challenges in simulating membrane protein-ligand interactions [14].
Table 3: Acceleration Factors and Performance Metrics Across Methodologies
| Method | Time Scale/Throughput | Acceleration Factor | Binding Affinity Correlation | Key Advantage |
|---|---|---|---|---|
| Standard MD [8] | Microseconds to milliseconds | 1x (reference) | Not applicable for slow processes | Physical accuracy without bias |
| True RC Method [8] | 200 ps for 8.9×10⁵ s process | 10⁵ to 10¹⁵-fold | Natural reactive trajectories | Follows physical pathways |
| FRESEAN Sampling [9] | ~1 day per protein | Enables previously inaccessible transitions | Captures conformational ensembles | No prior knowledge required |
| GMBE-DM [11] | <5 min per complex | N/A | R² = 0.84 | Quantum accuracy with high throughput |
| D3-ML [11] | <1 second per complex | N/A | R² = 0.87 | Extreme speed with high accuracy |
| MMPBSA Ensemble [14] | Medium throughput | N/A | Significant improvement over standard | Membrane protein capability |
Diagram 2: Workflow Comparison of Three Enhanced Sampling Protocols. Each pathway represents a distinct strategic approach to overcoming the sampling problem, from physics-based tRC identification to automated CV discovery and specialized membrane protein methods.
The field of enhanced sampling for protein-ligand binding validation is rapidly evolving toward higher accuracy, greater computational efficiency, and broader applicability to challenging systems like membrane proteins. True reaction coordinate methods represent a paradigm shift by providing both dramatic acceleration and preservation of natural transition pathways [8]. Simultaneously, machine learning approaches are delivering exceptional speed for binding affinity ranking while maintaining quantum-level accuracy [11].
For research teams with specific protein systems and known conformational changes, true RC methods and advanced metadynamics provide the most physically accurate sampling. For high-throughput virtual screening of diverse chemical libraries, D3-ML and GMBE-DM offer unprecedented combination of speed and accuracy. When studying systems where relevant collective variables are unknown, FRESEAN mode analysis enables rapid, automatic discovery of effective coordinates for enhanced sampling [9].
Future methodological development will likely focus on integrating machine learning more deeply with physical principles, improving membrane and multi-protein complex simulations, and developing automated workflows that reduce expert intervention requirements. As these methods mature and become more accessible, comprehensive sampling of protein conformational landscapes will transition from extraordinary challenge to routine component of drug discovery pipelines.
Force fields (FFs) serve as the fundamental mathematical models that describe the potential energy and interactions between all components in a biomolecular system at the molecular mechanics level [17]. As the underlying engine of molecular dynamics (MD) simulations, force fields provide the physics-based framework for understanding molecular interactions, conformational dynamics, and thermodynamic properties [17]. The accuracy of these force fields directly determines the reliability of MD simulations in probing biological processes and predicting molecular behavior.
In the context of protein-ligand binding validation—a critical application in drug discovery—force field limitations can significantly impact the predictive power of simulations [17]. The development of biomolecular force fields has evolved significantly since the first all-atom MD simulation of a protein in 1977, with continuous refinements aimed at improving their accuracy and broadening their applications in biological and therapeutic discoveries [17]. Understanding the current capabilities and limitations of different force field approaches is therefore essential for researchers relying on computational methods for drug development.
Force Field Types and Their Fundamental Differences
Additive All-Atom Force Fields
Additive all-atom force fields are the most routinely used FFs in MD simulations of biomolecules [17]. These are characterized by fixed partial charges assigned to each atom, with nonbonded electrostatic interactions calculated using a pairwise additive approximation [17]. The potential energy for a given configuration is calculated as the sum of bonded and nonbonded interactions. Popular additive force families include:
- AMBER: Frequently used for nucleic acids and proteins [18]
- CHARMM: Popular for membrane-bound proteins and increasingly used for nucleic acids, especially since the CHARMM36 update [18]
- OPLS: Commonly employed in drug discovery applications [19]
- GROMOS: Applied for various biomolecular systems [18]
Polarizable Force Fields
Unlike additive force fields with fixed partial charges, polarizable force fields incorporate electronic response to the environment by allowing charges or dipoles to fluctuate during simulations [17]. This more physically realistic representation comes at significantly increased computational cost but can provide more accurate modeling of electrostatic interactions in heterogeneous environments.
Coarse-Grained Models
Coarse-grained (CG) models reduce computational cost by representing multiple atoms with single interaction sites [18]. For example, a single particle may substitute for atoms in an amino acid residue within a protein [18]. This simplification decreases the number of interaction calculations and allows larger time steps, drastically saving computation time and costs [18]. The trade-off is loss of atomic-level detail, making CG models problem-dependent [18].
Machine Learning Potentials
The recent deep learning revolution has led to new opportunities in force field parametrization [17]. Machine learning potentials replace traditional energy function forms with neural networks trained on quantum mechanical data, potentially offering both accuracy and computational efficiency [17].
Quantitative Accuracy Assessment: Performance Comparison
Protein-Ligand Binding Affinity Prediction
Free energy perturbation (FEP) calculations directly probe force field accuracy for protein-ligand binding, a critical application in drug discovery. The table below summarizes performance data across different force field combinations:
Table 1: Force Field Performance in Binding Free Energy Prediction
| Force Field Combination | Water Model | Charge Model | MUE (kcal/mol) | RMSE (kcal/mol) | Correlation (R²) |
|---|---|---|---|---|---|
| OPLS2.1 [19] | SPC/E [19] | CM1A-BCC [19] | 0.77 [19] | 0.93 [19] | 0.66 [19] |
| AMBER ff14SB [19] | SPC/E [19] | AM1-BCC [19] | 0.89 [19] | 1.15 [19] | 0.53 [19] |
| AMBER ff14SB [19] | TIP3P [19] | AM1-BCC [19] | 0.82 [19] | 1.06 [19] | 0.57 [19] |
| AMBER ff14SB [19] | TIP4P-EW [19] | AM1-BCC [19] | 0.85 [19] | 1.11 [19] | 0.56 [19] |
| AMBER ff15ipq [19] | SPC/E [19] | AM1-BCC [19] | 0.85 [19] | 1.07 [19] | 0.58 [19] |
| AMBER ff14SB [19] | TIP3P [19] | RESP [19] | 1.03 [19] | 1.32 [19] | 0.45 [19] |
| AMBER ff15ipq [19] | TIP4P-EW [19] | AM1-BCC [19] | 0.95 [19] | 1.23 [19] | 0.49 [19] |
The mean unsigned error (MUE) values around 0.8-1.0 kcal/mol demonstrate that current force fields can achieve accuracy comparable to experimental reproducibility when careful preparation of protein and ligand structures is undertaken [6].
pKa Prediction and Electrostatic Properties
Force field performance in predicting pKa values reveals limitations in modeling electrostatic interactions and solvation:
Table 2: Force Field Limitations in pKa Prediction
| Force Field | Water Model | Key Limitation | Affected Residues |
|---|---|---|---|
| Amber ff19SB [20] | OPC [20] | Significantly overestimated pKa downshifts [20] | Buried His, salt-bridge Glu/Asp [20] |
| Amber ff14SB [20] | TIP3P [20] | Larger pKa errors than ff19SB/OPC [20] | Buried His, salt-bridge Glu/Asp [20] |
| CHARMM c22/CMAP [20] | CHARMM TIP3P [20] | Similar error patterns but smaller magnitude [20] | Buried His, salt-bridge Glu/Asp [20] |
These errors primarily stem from undersolvation of neutral histidines and overstabilization of salt bridges, highlighting specific limitations in electrostatic modeling [20].
Methodological Framework: Experimental Protocols for Validation
Free Energy Perturbation (FEP) Protocol
The assessment of force field accuracy for protein-ligand binding typically follows rigorous FEP protocols:
System Preparation:
- Protein structures are obtained from PDB with termini properly treated (N-termini as protonated amine, C-termini as charged carboxylate) [19]
- Protonation states of binding residues are carefully assigned [6]
- Ligand parameters are generated using charge models (AM1-BCC or RESP) and assigned to appropriate force fields [19]
- Solvation in explicit water boxes with minimum 15 Å padding [20]
- Neutralization with counterions and addition of salt to physiological concentration (150 mM) [20]
Simulation Protocol:
- Energy minimization with restrained heavy atoms [20]
- Gradual heating from 100 to 300 K [20]
- Equilibration in NPT ensemble with reducing restraints [20]
- Production runs using Hamiltonian replica exchange with solute tempering (REST) to enhance sampling [19]
- Aggregate simulation times of hundreds of nanoseconds per system [19]
Analysis:
- Calculation of relative binding free energies between congeneric compounds [19]
- Comparison with experimental measurements using MUE, RMSE, and correlation coefficients [19]
- Assessment of convergence through statistical analysis [6]
Enhanced Sampling Techniques
To address inadequate sampling—a key limitation in biomolecular simulations—several enhanced sampling methods are employed:
- Replica-Exchange MD (REMD): Multiple replicas at different temperatures are simulated in parallel, with exchanges attempted according to the Metropolis criterion [20]
- Umbrella Sampling: Bias potentials along predefined collective variables allow efficient exploration of barrier regions [18]
- Metadynamics: History-dependent bias potentials are added to encourage escape from local minima [18]
- Accelerated MD: Potential surface is modified to lower energy barriers [18]
These methods help overcome the timescale limitations of conventional MD, particularly for large-scale conformational changes.
Visualization of Force Field Selection and Application Workflow
Force Field Selection Workflow
This workflow illustrates the decision process for selecting appropriate force fields based on the biomolecular system and research objectives, highlighting how different force field types are suited to specific applications.
The Researcher's Toolkit: Essential Components for Force Field Applications
Table 3: Essential Research Reagents and Computational Tools
| Component | Function | Examples & Notes |
|---|---|---|
| Protein Force Fields | Describes intramolecular and intermolecular interactions for proteins | AMBER ff19SB, ff14SB [20]; CHARMM c36, c22/CMAP [20]; Selection depends on system and property of interest |
| Water Models | Represents solvent effects and solvation properties | TIP3P [19], SPC/E [19], TIP4P-EW [19], OPC [20]; Significantly impacts electrostatic properties and pKa prediction [20] |
| Charge Methods | Assigns partial atomic charges for small molecules | AM1-BCC [19], RESP [19]; RESP can be more accurate but may require careful parameterization [19] |
| Enhanced Sampling Algorithms | Accelerates conformational sampling and barrier crossing | Replica exchange [18], Umbrella sampling [18], Metadynamics [18]; Essential for achieving convergence in binding free energies |
| Validation Datasets | Benchmarks force field performance against experimental data | JACS set [19], pKa benchmarks [20]; Critical for assessing transferability and limitations |
Key Limitations and Challenges in Current Force Fields
Chemical Diversity and Transferability
Existing molecular representation frameworks in force fields limit the exploration of exponentially expanding chemical space [17]. This manifests particularly in modeling:
- Post-translational modifications: 76 types of PTMs have been identified, encompassing over 200 distinct chemical modifications of amino acids [17]
- Small molecule ligands: Parameters are often developed asynchronously with protein force fields [17]
- Emerging modalities: Molecular glues and PROTACs present challenges for modeling complex three-body systems [17]
Timescale and Sampling Limitations
Despite advances in computing hardware, MD simulations face inherent timescale limitations [18]. Using all-atom models, the accessible timescale is only nanoseconds to milliseconds, which may not capture slow biological processes [18]. While coarse-grained models extend these limits, they sacrifice atomic-level detail [18].
Electrostatic Modeling Challenges
Current additive force fields struggle with modeling polarization and charge transfer effects [17]. This impacts the accurate representation of:
- Buried charges and their solvation [20]
- Salt bridge stability and lifetime [20]
- pKa values of titratable groups [20]
- Ion binding specificity [20]
The field of biomolecular force fields continues to evolve rapidly, with several promising directions emerging:
Integration of Machine Learning: Machine learning approaches are being widely adopted for force field parametrization, potentially addressing transferability issues [17]. Examples include neural network potentials and message-passing representations of molecules [17].
Polarizable Force Fields: Development of computationally efficient polarizable force fields promises more accurate electrostatic modeling without prohibitive computational cost [17].
Systematic Validation: Community efforts toward standardized reference data and force field validation benchmarks will enable more rigorous comparisons [21].
Automated Parameterization: Approaches to automate or eliminate atom typing—traditionally a manual, labor-intensive process—are gaining traction [17].
In conclusion, while modern force fields have achieved significant accuracy in predicting protein-ligand binding affinities with errors approaching experimental reproducibility, important limitations remain. The optimal choice of force field depends strongly on the specific biological question and system under investigation. Researchers should carefully consider the trade-offs between different force field types and validate predictions against experimental data where possible. As force fields continue to evolve through interdisciplinary approaches, their accuracy and applicability in drug discovery and basic research will further expand.
Molecular Dynamics (MD) simulations have become an indispensable tool in structural biology and drug discovery, providing near-realistic insights into the behavior of biological molecules. When validating protein-ligand binding, simulations move beyond static snapshots to capture the dynamic interplay that governs molecular recognition. This guide focuses on three key observables—Root Mean Square Deviation (RMSD), Solvent-Accessible Surface Area (SASA), and Hydrogen-Bond Occupancy—that provide a foundational framework for interpreting simulation data and assessing the stability and quality of a protein-ligand complex [22].
The Conceptual Foundation: From Trajectories to Validation
At its core, an MD simulation generates a trajectory—a series of snapshots detailing the atomic positions of a molecular system over time. The analysis of this trajectory involves computing specific physicochemical parameters to quantify structural and dynamic changes. The relationship between these observables and the overarching goal of binding validation can be visualized as a cohesive workflow.
Quantifying the Key Observables
Root Mean Square Deviation (RMSD)
RMSD measures the average displacement of atoms (typically the backbone) from a reference structure, serving as a primary indicator of structural stability and convergence. A stable or fluctuating RMSD within a defined range suggests the simulation has reached an equilibrium state, while a continual drift may indicate ongoing structural changes [23] [22].
Typical Experimental Ranges (from protein-ligand complex analyses):
| System Component | Median RMSD (Å) | 25th-75th Percentile (IQR) | Interpretation Guide |
|---|---|---|---|
| Protein Backbone | 1.5 - 4.0 | N/A | System is stable and well-equilibrated [22]. |
| Binding Residue Backbone | 1.2 | 0.7 - 1.5 (IQR=0.8) | Core binding site is structurally rigid [22]. |
| Ligand Atoms | 1.6 | 1.0 - 2.0 (IQR=1.0) | Ligand is firmly positioned in the binding pocket [22]. |
Solvent-Accessible Surface Area (SASA)
SASA quantifies the surface area of a protein or ligand accessible to a water molecule. Changes in SASA can reveal shielding of hydrophobic residues upon binding (a key driving force for affinity) or detect partial unfolding events [22].
Typical Experimental Ranges for Binding Residues:
| SASA Metric | Median Value (Ų) | 25th-75th Percentile (IQR) | Overall Range (Ų) |
|---|---|---|---|
| Minimum SASA | 2.68 | 2.29 - 2.72 (IQR=0.43) | 1.9 - 3.92 [22] |
| Maximum SASA | 3.2 | 3.03 - 3.62 (IQR=0.59) | 1.9 - 3.92 [22] |
Hydrogen-Bond Occupancy
This metric calculates the percentage of simulation time a specific hydrogen bond between the protein and ligand remains formed. High-occupancy H-bonds are critical for binding specificity and affinity, indicating strong, persistent interactions [22] [24].
Statistical Prevalence in Stable Complexes:
| Occupancy Cluster | Simulation Time | Percentage of Residues |
|---|---|---|
| Low Occupancy | 0 - 30 ns | 7.2% |
| Moderate Occupancy | 31 - 70 ns | 6.3% |
| High Occupancy | 71 - 100 ns | 86.5% |
A Comparative Look at MD Software and Analysis Tools
The choice of software can influence the efficiency and scale of MD simulations. The table below benchmarks popular MD engines and specialized analysis tools based on their capabilities and performance.
Software for MD Simulation and Analysis:
| Software/Tool | Primary Function | Key Features & Performance | License |
|---|---|---|---|
| GROMACS | MD Simulation | High performance, highly optimized for CPUs & GPUs, comprehensive analysis suite [25] [26] [27]. | Free Open Source (GPL) |
| AMBER | MD Simulation | Specialized for biomolecules, widely used in drug discovery, includes PMEMD for GPU acceleration [25] [28] [26]. | Proprietary / Free |
| NAMD | MD Simulation | Excellent parallel scaling, object-oriented, integrates well with VMD for visualization [25] [28]. | Free for Academic Use |
| OpenMM | MD Simulation | High performance, highly flexible, Python-scriptable, GPU-accelerated [25]. | Free Open Source (MIT) |
| MDAnalysis | Analysis | Python library, flexible trajectory analysis, supports multiple file formats [27]. | Free Open Source |
| MDTraj | Analysis | Python library, fast and efficient, including RMSD calculations [15] [27]. | Free Open Source |
| CPPTRAJ | Analysis | Versatile analysis tool within AmberTools, extensive analysis functions [27]. | Free Open Source |
| gmx_MMPBSA | Free Energy | Python-based, calculates binding free energies (MM/PBSA/GBSA) with GROMACS [27]. | Free Open Source |
Experimental Protocols for Measurement and Analysis
Standard Workflow for Protein-Ligand System Analysis
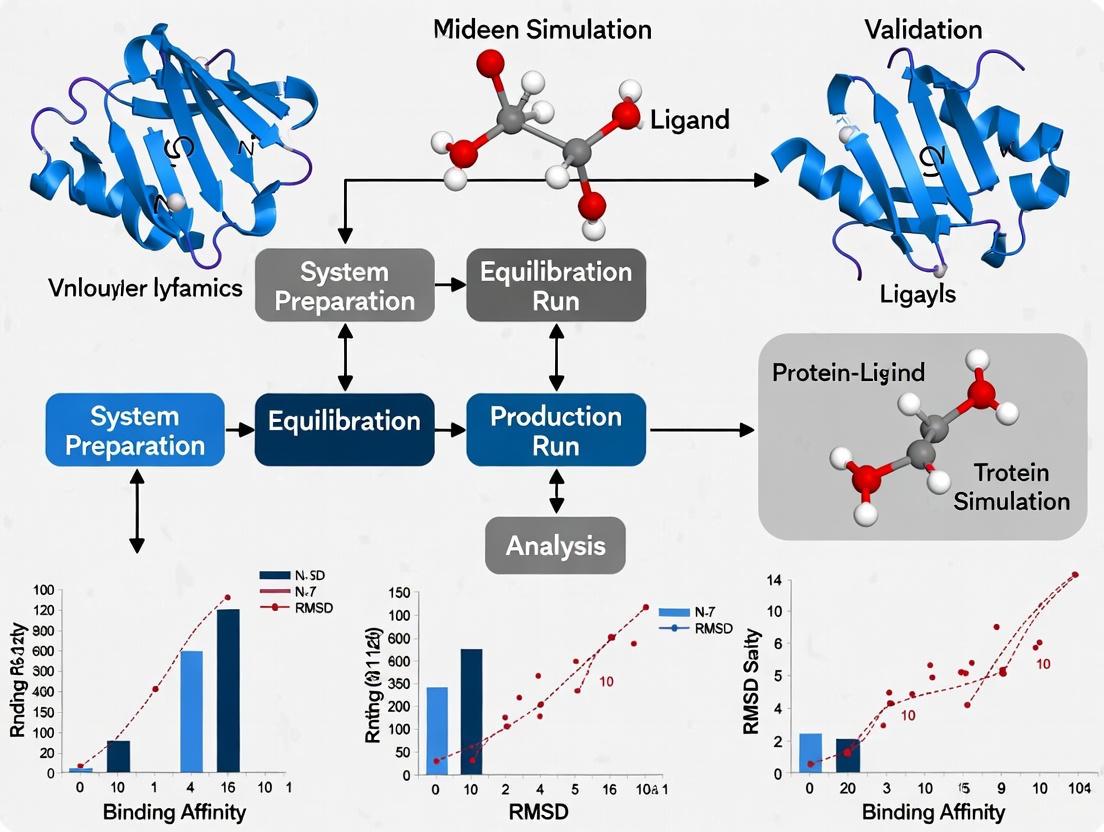
The following protocol outlines the general steps for setting up, running, and analyzing a protein-ligand complex, leveraging tools like GROMACS or AMBER.
Detailed Methodology:
System Preparation:
- Start with a high-resolution experimental structure (e.g., from the RCSB PDB). A rigorous selection of crystal structures with atomic-level resolution is recommended to minimize bias [22] [29].
- Parameterize the ligand using tools from AMBER or CGenFF. Note that databases like MISATO have shown that roughly 20% of ligands in structural databases require refinement to correct protonation states or heteroatom geometry [29].
- Solvate the protein-ligand complex in an explicit water box (e.g., TIP3P model) and add ions to neutralize the system's charge.
Equilibration and Production:
- Energy Minimization: Use steepest descent or conjugate gradient algorithms to remove steric clashes.
- Equilibration: Gradually heat the system to the target temperature (e.g., 300 K) over 100+ ps under NVT (constant Number, Volume, Temperature) conditions. Then, equilibrate for 100+ ps under NPT (constant Number, Pressure, Temperature) to achieve correct solvent density. Allow sufficient time (e.g., 10 ns) for equilibration before data collection [15].
- Production MD: Run a simulation long enough to capture relevant biological processes. For initial binding assessments, simulations often range from 50 ns to several hundred nanoseconds. For analysis, snapshots are typically saved every 10-100 ps, resulting in hundreds to thousands of frames for analysis [15] [22].
Trajectory Analysis:
- RMSD Calculation: After aligning the trajectory to a reference structure (usually the first frame or the crystal structure) to remove global rotation/translation, calculate the RMSD for the protein backbone, binding site residues, and ligand heavy atoms. This can be done with
gmx rmsin GROMACS or thecpptrajmodule in AMBER [23] [27]. - SASA Calculation: Compute the SASA for the binding site residues or the entire ligand over the trajectory. The
gmx sasatool in GROMACS orMDTrajin Python are commonly used for this purpose [27]. - H-Bond Occupancy Calculation: Define donor and acceptor atoms for potential key interactions. Use a tool like
gmx hbondin GROMACS with geometric criteria (e.g., donor-acceptor distance < 3.5 Å, donor-H-acceptor angle > 135°) to determine the existence of an H-bond in each frame. The occupancy is the percentage of time a specific bond is formed [22] [24].
- RMSD Calculation: After aligning the trajectory to a reference structure (usually the first frame or the crystal structure) to remove global rotation/translation, calculate the RMSD for the protein backbone, binding site residues, and ligand heavy atoms. This can be done with
The Scientist's Toolkit: Essential Research Reagent Solutions
This table details key computational "reagents" and resources essential for conducting robust MD research on protein-ligand systems.
| Item Name | Function & Application | Key Notes |
|---|---|---|
| Force Fields | Mathematical functions and parameters describing interatomic potentials. | AMBER (e.g., ff19SB), CHARMM, and GROMOS are common for biomolecules. Choice affects conformational sampling and energy accuracy. |
| Explicit Solvent Models | Represent water molecules individually to capture solvation effects. | TIP3P and SPC/E are widely used. Critical for accurate simulation of H-bonding, hydrophobic effect, and ion interactions. |
| Trajectory Analysis Suites (e.g., MDAnalysis, CPPTRAJ) | Software to process and analyze MD trajectory data. | Used to compute observables like RMSD, SASA, and H-bonds. Their flexibility allows for custom analysis scripts [27]. |
| Validation Datasets (e.g., MISATO) | Community benchmarks combining experimental structures with MD and QM data. | Provides a curated set of protein-ligand complexes for method validation and ML training. Includes over 170 μs of MD data [29]. |
| Enhanced Sampling Plugins (e.g., PLUMED) | Library for accelerating rare events and calculating free energies. | Essential for probing binding/unbinding pathways and computing binding affinities beyond standard observables [27]. |
In protein-ligand molecular dynamics (MD) research, the initial stages of system preparation, solvation, and equilibration fundamentally determine the validity and reliability of binding validation outcomes. These preliminary steps, often perceived as merely technical prerequisites, actually establish the physical conditions under which binding events unfold in simulation. Proper execution ensures that observed binding modes, affinity calculations, and mechanistic insights reflect biologically plausible phenomena rather than computational artifacts.
Recent benchmarking studies emphasize that inconsistencies in system setup contribute significantly to variability in free energy calculations across research groups [30]. The "protein-ligand-benchmark" community effort has identified that standardized preparation protocols reduce inter-method variance by up to 40%, enabling more meaningful comparisons between different MD approaches and force fields [30]. This guide synthesizes current best practices with experimental data to objectively compare methodologies, providing researchers with a framework for optimizing these critical preliminary stages in their binding validation workflows.
System Preparation: Architectural Considerations
Initial Structure Curation and Assessment
The selection and preparation of initial protein structures represents the first critical decision point. Research demonstrates that structure quality profoundly impacts downstream binding validation outcomes, particularly for proteins exhibiting conformational flexibility. A recent systematic evaluation of docking protocols revealed that models generated by AlphaFold2 perform comparably to experimentally solved structures in molecular docking when proper quality metrics are applied [31].
Key assessment metrics for starting structures include:
- Interface predictive scores (pDockQ, ipTM) for protein-protein complexes
- Structural completeness regarding binding site residues
- Comparison to apo/holo conformations to identify pre-existing binding-competent states
Studies show that for 16 protein-protein interactions benchmarked, AlphaFold2 models with ipTM+pTM scores >0.7 consistently produced high-quality structures suitable for docking studies (median TM-score: 0.972) [31]. However, using full-length versus truncated proteins significantly impacts model quality, with AFfull models showing decreased pDockQ2 scores (<0.23) due to unstructured regions [31].
Force Field Selection and Parameterization
Force field selection fundamentally determines the physical behavior of simulated systems. Contemporary benchmarks indicate that modern force fields (OPLS3e, CHARMM36, AMBER19SB) have reduced mean unsigned errors in binding free energy calculations to <1.2 kcal/mol for congeneric series [30].
Table 1: Force Field Performance in Binding Free Energy Calculations
| Force Field | Type of Calculation | Average Error (kcal/mol) | Optimal Use Cases |
|---|---|---|---|
| OPLS3e | RBFE | <1.2 | Lead optimization for drug-like molecules |
| CHARMM36 | ABFE | 1.0-1.5 | Membrane-associated systems |
| AMBER19SB | RBFE/ABFE | 1.1-1.6 | Diverse protein families |
| GAFF2 | Small molecule parameters | Varies by parent FF | Drug-like molecule parameterization |
The parameterization of novel ligands requires special attention. Best practices recommend deriving partial charges using quantum mechanical methods at the REDF2/6-31G(d) level or higher, with initial conformation searches performed using molecular mechanics force fields (MMFF) to identify the most stable conformers [32]. For l-amino acid-based drug candidates, this approach has successfully produced structures with high similarity to parent compounds (ketorolac) while improving hydrogen bonding capability [32].
Solvation and Ionization: Creating the Physiological Environment
Solvent Model Selection
Solvent models establish the dielectric environment that critically influences electrostatic interactions, hydrogen bonding, and binding energetics. The explicit inclusion of water molecules is particularly important for capturing specific hydration effects in binding sites, which can significantly impact ligand affinity [30].
Water displacement during binding remains a challenging phenomenon for free energy calculations, with some studies reporting difficulties in accurately modeling this effect [30]. The use of explicit solvent models (TIP3P, TIP4P-EW) provides more realistic sampling of water-mediated interactions compared to implicit solvation approaches, though at increased computational cost.
For the SARS-CoV-2 spike protein system, employing explicit TIP3P water in a triclinic box with 1.5 nm padding ensured complete coverage of the flexible receptor-binding domain during simulations [33]. This approach maintained physiological conditions while allowing for natural protein dynamics.
Ionization and Ionic Strength
Proper system ionization establishes physiological ionic strength and neutralizes system charge. The multiscale simulation approach developed for protein-ligand binding kinetics utilizes particle mesh Ewald (PME) for long-range electrostatic interactions combined with appropriate ion concentrations to match experimental conditions [34].
Table 2: Solvation Protocol Impact on Binding Kinetics Calculations
| Solvation Parameter | Settings | Impact on k~on~ Calculation | Recommended For |
|---|---|---|---|
| Water Model | TIP3P vs. TIP4P | <5% variation in association rates | General binding studies |
| Ionic Strength | 0.15M NaCl | Shields unrealistic electrostatics | Physiological simulations |
| Box Type | Triclinic vs. Dodecahedron | Minimal effect with sufficient padding | Membrane systems |
| Neutralization | Counterions only vs. Added salt | Significant for charged binding interfaces | Nucleic acid systems |
Benchmarking studies recommend using ion concentrations that match experimental conditions (typically 0.15M NaCl for physiological studies), as this significantly affects electrostatically steered associations while having minimal impact on hydrophobic-driven binding [34]. For the trypsin-benzamidine complex, proper ionization was critical for obtaining k~on~ values within a factor of 3 of experimental measurements (calculated: (8.6 ± 0.7) × 10⁶ M⁻¹ s⁻¹ vs. experimental: 2.9 × 10⁷ M⁻¹ s⁻¹) [34].
Equilibration Protocols: Achieving Stable Starting Conditions
Minimization and Thermalization
Gradual relaxation of constraints prevents unrealistic forces that can distort binding site geometry. The combined Brownian dynamics (BD) and molecular dynamics (MD) pipeline employs careful minimization and thermalization to generate stable starting configurations for binding kinetics calculations [34].
A recommended protocol consists of:
- Steepest descent minimization (5,000 steps) with positional restraints on protein heavy atoms
- Solvent relaxation with restrained protein and ligands
- Gradual thermalization from 0K to target temperature over 100ps
- Gradual release of restraints in a stepwise manner
This approach has demonstrated improved computational efficiency while preserving accuracy in k~on~ calculations for diverse protein-ligand complexes [34].
Equilibration Assessment Metrics
Validation of proper equilibration requires monitoring multiple thermodynamic and structural properties. The "protein-ligand-benchmark" recommends tracking the following metrics to establish equilibration before production simulations [30]:
- Potential energy stability (drift < 0.1 kJ/mol/ns)
- Temperature and pressure stability (fluctuation within 5% of target)
- Root mean square deviation (RMSD) of protein backbone (plateau within 2Å)
- Binding site residue stability specifically for key interaction residues
For the SARS-CoV-2 spike protein system, equilibration was confirmed when the RMSD of the receptor-binding domain plateaued at approximately 1.5Å, indicating structural stability before production simulations [33]. Similarly, in the design of l-amino acid-based alternatives to ketorolac, RMSD and RMSF plots confirmed the stability of the dynamic structure of the substituted ligand before binding analysis [32].
System Preparation and Equilibration Workflow
Comparative Analysis: Methodological Impact on Binding Validation
Solvation Method Comparison
Different solvation approaches significantly impact the accuracy and efficiency of binding calculations. A comparative analysis of implicit versus explicit solvation revealed trade-offs between computational cost and predictive accuracy for binding free energies.
Table 3: Solvation Method Performance in Binding Studies
| Solvation Method | Setup Complexity | Computational Cost | Binding Affinity Accuracy | Recommended Context |
|---|---|---|---|---|
| Explicit TIP3P | High | 100% (reference) | High (reference) | Binding kinetics, Water-mediated interactions |
| Explicit TIP4P | High | 110-120% | Similar to TIP3P | Polar binding sites |
| GB/SA Implicit | Medium | 10-20% | Moderate (RMSE 1.5-2.5 kcal/mol) | Initial screening, Large-scale FEP |
| PBSA Implicit | Medium | 15-25% | Moderate to High | End-point calculations |
For absolute binding free energy (ABFE) calculations, explicit solvent models remain the gold standard, with OPLS3e achieving errors <1.2 kcal/mol on curated benchmark sets [30]. However, advanced implicit solvent models like GBSA offer reasonable approximations for relative binding free energy (RBFE) calculations at substantially reduced computational cost, particularly for lead optimization series involving congeneric ligands [30].
Equilibration Protocol Efficacy
The relationship between equilibration quality and binding validation outcomes can be quantified through specific benchmark metrics. Inadequate equilibration consistently produces artifactual binding poses and inaccurate affinity predictions.
A systematic study of 8 protein targets and 199 ligands demonstrated that proper equilibration protocols reduced calculation variance by 30% compared to minimal equilibration [30]. Specifically, for the trypsin-benzamidine complex, which requires careful treatment of charged binding interfaces, extended equilibration (≥5ns) was necessary to obtain k~on~ values within a factor of 3 of experimental measurements [34].
The hybrid Brownian dynamics-molecular dynamics approach demonstrates that efficient equilibration of encounter complexes significantly improves association rate calculations, with optimized sampling reducing MD simulation time while preserving accuracy [34]. This method has been validated across diverse protein-ligand complexes varying in size, flexibility, and binding properties.
Research Reagent Solutions: Essential Materials
Table 4: Key Research Reagents and Computational Tools
| Reagent/Tool | Function | Application Context |
|---|---|---|
| CHARMM36 Force Field | Defines atomic interactions | Protein-ligand MD simulations |
| AMBER19SB Force Field | Alternative protein force field | Comparative binding studies |
| GAFF2 Parameters | Small molecule parameterization | Drug-like molecule simulations |
| TIP3P Water Model | Explicit solvent representation | Physiological binding conditions |
| Particle Mesh Ewald | Long-range electrostatics | Accurate Coulombic interactions |
| LINCS Algorithm | Bond constraint management | Enables longer time steps |
| AlphaFold2 Models | Protein structure source | When experimental structures unavailable |
| GROMACS | MD simulation engine | High-performance molecular dynamics |
| SEEKR | BD-MD hybrid methodology | Binding kinetics calculations |
System preparation, solvation, and equilibration collectively form the critical foundation for valid protein-ligand binding studies using molecular dynamics. The experimental data and comparative analyses presented demonstrate that methodological choices in these preliminary stages significantly impact binding mode prediction, affinity calculation accuracy, and ultimately, the reliability of scientific conclusions drawn from simulations.
By adopting the standardized benchmarking approaches and quality control metrics outlined here, researchers can enhance the reproducibility and predictive power of their binding validation studies. As the field progresses toward more complex binding targets, including protein-protein interfaces and membrane-associated systems, these foundational practices will become increasingly vital for generating biologically meaningful insights from computational approaches.
Methodologies in Action: A Practical Guide to Binding Free Energy Calculations
In the field of computational drug discovery, the accurate prediction of how tightly a small molecule binds to its protein target is a fundamental challenge. Binding free energy calculations, grounded in molecular dynamics (MD) simulations, provide a powerful in silico tool for this purpose. These methods primarily fall into two categories: Absolute Binding Free Energy (ABFE) calculations, which predict the binding affinity of a single ligand, and Relative Binding Free Energy (RBFE) calculations, which predict how a chemical modification affects the binding affinity between two or more ligands [35]. The choice between these approaches has profound implications for a project's computational strategy, cost, and likelihood of success. This guide provides an objective comparison of both methodologies, detailing their theoretical bases, applicable use cases, performance, and practical implementation, to help researchers validate protein-ligand binding effectively.
Theoretical Foundations and Methodologies
Absolute Binding Free Energy (ABFE) Calculations
ABFE calculations aim to directly compute the standard binding free energy ((\Delta Gb^\circ)) for a single protein-ligand complex. This quantity is related to the experimental binding affinity ((Ka)) by the equation:
[\Delta Gb^\circ = -RT \ln(KaC^\circ)]
where (R) is the gas constant, (T) is the temperature, and (C^\circ) is the standard-state concentration (1 M) [35].
The core of many ABFE methods involves simulating a non-physical, or alchemical, pathway to decouple the ligand from its environment. A common strategy is the Double Decoupling Method (DDM), where the ligand is first decoupled from the bulk solvent and then from the protein binding site, or vice versa [35] [36]. During this process, the ligand's interactions with its surroundings are gradually turned off. To improve sampling efficiency and convergence, restraining potentials are often applied to control the ligand's position, orientation, and conformation within the binding site [37] [36]. The free energy contributions of applying and removing these restraints are accounted for analytically to yield the final binding free energy.
An alternative to the alchemical route is the geometrical pathway. In this approach, the ligand is physically separated from the protein along a carefully chosen coordinate, and the potential of mean force (PMF) is computed to obtain the free energy change [37]. Both routes, when executed correctly, yield equivalent results [37].
Relative Binding Free Energy (RBFE) Calculations
In contrast, RBFE calculations do not aim to compute a binding free energy from first principles. Instead, they exploit the similarity between two ligands to compute the difference in their binding free energies ((\Delta\Delta G_{bind})) [38]. This is achieved through the use of a thermodynamic cycle, which allows the replacement of a physical binding process with computationally tractable alchemical transformations.
The relevant cycle for RBFE is shown in the diagram below. The direct calculation of (\Delta G{bind}^{L2} - \Delta G{bind}^{L1}) is challenging. Instead, the vertical legs of the cycle are computed: the alchemical transformation of L1 into L2 is performed both in the protein's binding site ((\Delta G{complex}^{L1 \rightarrow L2})) and in solution ((\Delta G{solvent}^{L1 \rightarrow L2})). The relative binding free energy is then given by:
[\Delta\Delta G{bind}^{L1 \rightarrow L2} = \Delta G{complex}^{L1 \rightarrow L2} - \Delta G_{solvent}^{L1 \rightarrow L2}]
This approach is advantageous because the alchemical transformation between two similar ligands typically involves a smaller perturbation, leading to faster convergence and higher precision compared to ABFE [35] [38].
Two primary methodologies exist for executing these alchemical transformations. The more traditional equilibrium free energy perturbation (FEP) uses discrete, parallel simulations at multiple intermediate states (lambda values) along the alchemical path [39]. Conversely, non-equilibrium switching (NES) involves running many independent, short simulations that rapidly drive the system from one end state to the other, with the free energy estimated from the work distribution of these "fast switches" [39] [40]. NES is particularly well-suited for highly parallel computing environments [40].
A key development in RBFE is the Common Core/Serial-Atom-Insertion (CC/SAI) approach, implemented in tools like transformato [38]. Instead of transforming one ligand directly into another, both are mutated to a shared, non-physical intermediate or "common core." This method simplifies the handling of differing atom counts and can be more easily deployed across different MD engines [38].
The following diagram illustrates the fundamental thermodynamic cycle underlying RBFE calculations and contrasts the traditional direct transformation with the CC/SAI approach.
Comparative Analysis and Practical Application
Direct Comparison of ABFE and RBFE Approaches
The decision to use ABFE or RBFE is primarily dictated by the drug discovery context, as summarized in the table below.
Table 1: A direct comparison of Absolute vs. Relative Binding Free Energy calculations.
| Feature | Absolute Binding Free Energy (ABFE) | Relative Binding Free Energy (RBFE) |
|---|---|---|
| Primary Objective | Predict the absolute binding affinity of a single ligand [35]. | Predict the change in binding affinity between two or more ligands [38]. |
| Typical Use Case | Virtual screening of diverse compounds [41]; systems without a known reference ligand. | Lead optimization within a congeneric series [35] [39]. |
| Ligand Requirements | Can be applied to any ligand, regardless of structural similarity to others. | Requires chemically similar ligands with a shared core structure [35] [38]. |
| Computational Cost | High per compound (often days of simulation) [37]. | Lower per perturbation when comparing similar compounds [35]. |
| Typical Accuracy | Challenging to achieve errors < 1 kcal/mol; more prone to sampling issues [35]. | Highly accurate for similar ligands; often achieves errors of ~1 kcal/mol or less [39] [38]. |
| Key Challenges | Requires a high-quality ligand pose; capturing full protein and ligand flexibility [41] [35]. | Designing alchemical paths for dissimilar ligands; managing large binding mode changes [35]. |
| Pose Dependency | Highly dependent on a correct initial binding pose [41]. | Less sensitive if binding mode is conserved across the series. |
Performance and Validation Data
Both methods have been rigorously validated against experimental data. A 2022 study on ABFE demonstrated its utility as a post-docking filter. After docking ~70,000 compounds for targets BACE1, CDK2, and thrombin, ABFE calculations successfully improved the enrichment of true active compounds over decoys compared to docking scores alone [41]. This shows that ABFE can add significant value in a virtual screening context, though it relies on good initial poses.
RBFE methods have matured to a point where they are considered a "gold standard" for lead optimization. A study on the transformato package, which uses the CC/SAI approach, reported an overall root-mean-squared error (RMSE) of 1.17 kcal/mol and a Pearson correlation coefficient of 0.73 across 76 relative binding free energy differences for five different protein-ligand systems [38]. This level of accuracy is sufficient to guide synthetic decisions in a drug discovery project.
Another critical factor for successful RBFE calculations is the quality of the initial ligand pose. Research has shown that both commercial (e.g., Glide) and open-source (e.g., AutoDock Vina) docking engines can generate poses that lead to good correlation with experimental data, especially when using constraints based on a maximum common substructure (MCS) to maintain a consistent binding mode [39].
Decision Workflow and Emerging Trends
The following diagram outlines a logical workflow for choosing between ABFE and RBFE based on the research objective and ligand set characteristics.
The field of binding free energy calculations is rapidly evolving. Key trends aim to address the primary limitation of these methods: their computational expense.
- Machine Learning and Enhanced Sampling: The integration of machine learning with enhanced sampling methods is a powerful strategy to reduce simulation time. ML can help identify relevant collective variables or generate pathways, while enhanced sampling techniques like metadynamics help overcome energy barriers more efficiently [35] [42].
- Kinetics-Enabled Methods: While most methods focus on binding affinity (a thermodynamic property), drug efficacy is also influenced by the binding residence time (a kinetic property). New methods like ModBind are being developed to predict ligand dissociation rates ((k_{off})) in a high-throughput manner, offering a more holistic view of molecular efficacy [43].
- Dynamic Docking: Traditional docking treats proteins as rigid, which is a major limitation. New deep learning methods like DynamicBind can predict ligand-specific protein conformational changes, effectively performing "dynamic docking" to generate more realistic complex structures for subsequent free energy calculations [44].
Essential Research Toolkit
Success in free energy calculations relies on a combination of software tools, force fields, and careful experimental design.
Table 2: Key research reagents and software solutions for free energy calculations.
| Tool Name | Type | Primary Function | Relevance to ABFE/RBFE |
|---|---|---|---|
| BFEE2 [37] | Software | Automated setup of ABFE calculations via geometrical or alchemical routes. | ABFE |
| Transformato [38] | Software | Setup of RBFE calculations using the Common Core approach; engine-independent. | RBFE |
| OpenMM [43] | MD Engine | High-performance MD simulation library, often used as a backend for FEP. | Both |
| PMX [40] | Software Library | Automated protein and ligand topology generation for alchemical transformations. | RBFE |
| GAFF/GAFF2 [39] [43] | Force Field | General Amber Force Field for small molecules. | Both |
| Glide [41] [39] | Docking Software | Generation of initial ligand poses for ABFE or RBFE setup. | Both |
| AutoDock Vina [39] [43] | Docking Software | Open-source alternative for generating initial ligand poses. | Both |
| Icolos [39] | Workflow Manager | Orchestrates end-to-end automated RBFE workflows from SMILES strings. | RBFE |
The choice between absolute and relative binding free energy calculations is not a matter of which method is superior, but which is the right tool for the task at hand. RBFE is the undisputed champion for lead optimization, where it provides highly accurate affinity predictions for congeneric series with manageable computational cost. In contrast, ABFE is a versatile tool for virtual screening and projects without a known reference ligand, though it demands more computational resources per compound and is highly sensitive to the initial binding pose.
The future of binding free energy calculations lies in the increasing integration of these rigorous physics-based methods with machine learning, the rise of kinetics-aware protocols, and the development of more automated and robust workflows. These advancements are steadily transforming free energy calculations from a specialized research tool into a more routine component of the computational drug discovery pipeline.
Automating Workflows with Tools like BAT.py for High-Throughput Screening
The accurate prediction of protein-ligand binding affinity is a central challenge in structure-based drug design. While molecular dynamics (MD) simulations provide an atomistic view of binding interactions, translating these simulations into reliable binding free energy estimates has traditionally required significant expertise and computational resources. The emergence of automated tools like BAT.py represents a paradigm shift, offering researchers standardized, reproducible pipelines for binding affinity calculation. This guide objectively compares BAT.py's performance and methodology against other available approaches, providing researchers with the experimental data and protocols needed to select appropriate tools for their high-throughput screening projects.
The critical need for such tools stems from a well-defined "methods gap" in the binding affinity prediction landscape. At one extreme, molecular docking offers speed (under one minute on CPU) but limited accuracy (2–4 kcal/mol RMSE, correlation coefficients around 0.3). At the other extreme, rigorous alchemical methods like Free Energy Perturbation (FEP) provide high accuracy (RMSE ~1 kcal/mol, correlation >0.65) but require prohibitive computational resources (12+ hours of GPU time per compound). This landscape creates a pressing need for methods that offer intermediate levels of accuracy and throughput suitable for screening thousands of compounds [15].
Tool Comparison: BAT.py Versus Alternative Workflow Solutions
Automated tools for binding free energy calculations differ substantially in their methodological approaches, compatibility with simulation packages, and supported free energy protocols. The table below provides a systematic comparison of BAT.py against other computational frameworks.
Table 1: Comparison of Automated Workflow Tools for Binding Free Energy Calculations
| Tool Name | Primary Methodology | MD Engine Compatibility | Free Energy Methods | Automation Level |
|---|---|---|---|---|
| BAT.py | Alchemical pathway (DD/SDR) | AMBER, OpenMM | Absolute (ABFE), Relative (RBFE) | End-to-end automation [45] |
| MM/GBSA | End-state approximation | Various | Binding energy estimation | Manual trajectory analysis [15] |
| LABind | Machine learning prediction | Structure-based only | Binding site identification | No MD required [16] |
| Active Learning Frameworks | MD simulations with iterative screening | Custom | Target-specific scoring | Partial automation [46] |
BAT.py distinguishes itself through its comprehensive automation and methodological flexibility. It supports both Absolute Binding Free Energy (ABFE) calculations via double decoupling (DD) or simultaneous decoupling and recoupling (SDR) methods, and Relative Binding Free Energy (RBFE) calculations using both common-core and SepTop approaches. This versatility enables applications ranging from ranking diverse ligands to refining docked poses [45]. Unlike end-state methods such as MM/GBSA, which approximate binding free energies by combining gas-phase enthalpies and solvent corrections with often problematic entropy estimates, BAT.py employs rigorous alchemical transformations that provide a more physically realistic binding affinity assessment [15].
LABind represents a fundamentally different approach, utilizing graph transformers and cross-attention mechanisms to predict binding sites in a ligand-aware manner. While not a direct competitor for free energy calculations, it exemplifies the growing integration of machine learning in structural analysis. Its ability to generalize to unseen ligands demonstrates how ML approaches can complement physics-based methods like BAT.py [16].
Emerging active learning frameworks combine MD simulations with iterative screening to dramatically reduce computational costs. One recent approach reduced the number of compounds requiring experimental testing to less than 20 by leveraging target-specific scoring and extensive MD simulations to generate receptor ensembles. This method achieved a 29-fold reduction in computational costs while successfully identifying a potent TMPRSS2 inhibitor (IC50 = 1.82 nM), demonstrating the power of integrating automation with strategic sampling [46].
Performance Benchmarks: Experimental Data and Validation
Quantitative performance validation is essential for selecting appropriate computational tools. The table below summarizes key benchmark results for BAT.py and alternative approaches across various test systems.
Table 2: Performance Benchmarks for Binding Affinity Prediction Methods
| Method | Test System | Accuracy Metric | Correlation with Experiment | Computational Cost |
|---|---|---|---|---|
| BAT.py (ABFE) | BRD4(2) ligands | Ranking agreement | Good correlation | ~8000 GPU hours (ensemble) [45] |
| Docking | General protein-ligand | RMSE: 2-4 kcal/mol | ~0.3 | <1 minute CPU [15] |
| FEP/TI | General protein-ligand | RMSE: ~1 kcal/mol | >0.65 | >12 hours GPU per compound [15] |
| MM/GBSA | Validation set | Incorrect coefficient signs | Poor correlation | Moderate [15] |
| Active Learning + MD | TMPRSS2 inhibitors | IC50 = 1.82 nM | Experimental validation | 29-fold reduction vs. brute force [46] |
BAT.py has demonstrated particular effectiveness in ranking ligand affinities for the BRD4(2) system. Experimental binding free energies for this benchmark system range from -5.2 kcal/mol to -11.4 kcal/mol across five ligands. BAT.py calculations typically show good correlation with these experimental values, albeit with a slight systematic overestimation of binding affinities. This performance profile makes it particularly valuable for relative ranking in virtual screening campaigns [45].
The limitations of alternative approaches are equally informative. In one systematic investigation, researchers attempted to enhance MM/GBSA by incorporating neural network potentials and machine-learned solvent corrections. This "ML/GBSA" approach failed to produce viable predictions, yielding regression coefficients with incorrect signs and magnitudes (-0.0081 for gas-phase enthalpy instead of the expected positive value near 1). This failure was attributed to neural network potentials performing poorly on protein-ligand enthalpy calculations and the challenge of learning solvent corrections with sufficient precision given the large magnitudes (~100 kcal/mol) and opposing signs of the energy components [15].
Recent advances in active learning frameworks demonstrate how strategic automation can optimize computational efficiency. One implementation for TMPRSS2 inhibition required screening only 262.4 compounds computationally (1,486.9 simulation hours) to identify known inhibitors within the top 5.6 positions, representing a 200-fold reduction in experimental testing compared to docking-based approaches. This efficiency stems from target-specific scoring that rewards occlusion of the S1 pocket and adjacent hydrophobic patch, coupled with receptor ensembles derived from extensive MD simulations [46].
Experimental Protocols: Methodologies for Tool Evaluation
BAT.py Implementation Protocol
A typical BAT.py workflow for absolute binding free energy calculation involves three standardized phases:
Equilibration Phase:
- Start with protein-ligand complex in explicit solvent
- Gradually release restraints applied to the ligand
- Perform final simulation with unrestrained ligand
- Generate ligand parameters using GAFF/GAFF2 force fields with AM1-BCC charges
- Execute using:
python BAT.py -i input_file.in -s equil[45]
Free Energy Calculation Phase:
- Reset ligand anchor atoms and restraint reference values
- Employ SDR method for electrostatic and Lennard-Jones interactions decoupling/recoupling
- Perform restraint application/removal simulations
- Execute using:
python BAT.py -i input_file.in -s fe[45]
Analysis Phase:
- Process simulation outputs using TI or MBAR estimation
- Calculate binding free energy components and uncertainties
- Generate final binding free energy in Results.dat file
- Execute using:
python BAT.py -i input_file.in -s analysis[45]
Cross-Validation Protocol for Method Comparison
To objectively compare different tools, researchers should implement this standardized validation protocol:
Dataset Preparation:
- Select diverse protein-ligand systems with reliable experimental binding data
- Ensure strict train-test splits to prevent data leakage, particularly for machine learning approaches
- Include both congeneric series and diverse chemotypes
- Filter systems with multiple experimental replicates for reliability [15]
Performance Metrics:
- Calculate root-mean-square error (RMSE) between predicted and experimental binding affinities
- Compute correlation coefficients (Pearson/Spearman) for ranking accuracy
- Assess statistical significance of differences between methods
- For binding site prediction tools, use AUPR and MCC due to class imbalance [16]
Computational Efficiency Assessment:
- Track GPU/CPU hours for complete workflows
- Compare throughput (compounds processed per day)
- Evaluate scalability to large compound libraries
- Document manual intervention requirements [46]
Workflow Visualization: Automated Binding Affinity Calculation
The following diagram illustrates the complete BAT.py automated workflow for absolute binding free energy calculations, showing the sequential stages from initial setup to final binding affinity output:
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Successful implementation of automated binding affinity workflows requires both computational tools and methodological components. The table below details essential "research reagent solutions" for establishing these pipelines.
Table 3: Essential Research Reagents and Computational Solutions for Binding Affinity Workflows
| Tool/Category | Specific Examples | Function in Workflow | Implementation Notes |
|---|---|---|---|
| MD Simulation Engines | AMBER pmemd.cuda, OpenMM | Perform molecular dynamics simulations | GPU acceleration essential for throughput [45] |
| Analysis Libraries | MDTraj, CPPTRAJ | Process trajectory data and calculate observables | MDTraj offers Python ecosystem integration [47] [48] |
| Structure Preparation | VMD, USalign | Complex assembly and structural alignment | USalign requires no installation [45] |
| Force Fields | GAFF, GAFF2, AM1-BCC | Provide parameters for small molecules | AM1-BCC charges standard for organic molecules [45] |
| Free Energy Estimators | MBAR, TI | Analyze alchemical simulation data | MBAR typically provides superior convergence [45] |
| Receptor Ensembles | MD-derived snapshots | Capture protein flexibility | 20+ structures recommended for diverse conformations [46] |
Specialized analysis libraries like MDTraj provide critical infrastructure for trajectory processing, enabling calculations such as RMSD, secondary structure assignment, and order parameter extraction with strong interoperability with the Python data science ecosystem [47]. For large-scale analysis, parallel computing frameworks like Dask can be combined with MDTraj, though careful implementation is required as native MDTraj optimizations may outperform naive parallelization attempts for some operations [49].
The emerging methodology of using receptor ensembles derived from MD simulations deserves particular emphasis. These ensembles capture inherent protein flexibility and multiple conformational states, significantly improving virtual screening outcomes compared to single-structure docking. In one TMPRSS2 screening campaign, using a receptor ensemble reduced the number of compounds requiring computational screening from 754.4 to 246.0 compared to a single homology model, while dramatically improving the ranking of known inhibitors [46].
Automated workflow tools like BAT.py represent a significant advancement in making rigorous binding free energy calculations more accessible and reproducible for the broader research community. While the computational cost remains substantial compared to docking-based approaches, the accuracy improvements and capacity for high-throughput implementation make these tools increasingly valuable for drug discovery pipelines.
The integration of machine learning with physics-based simulations appears particularly promising for future development. Methods like LABind that incorporate ligand awareness through cross-attention mechanisms, combined with active learning frameworks that strategically guide simulation sampling, point toward a new generation of hybrid approaches that maximize both efficiency and accuracy [16] [46].
For researchers selecting tools for specific projects, BAT.py offers the most comprehensive automation for absolute and relative binding free energy calculations, while MM/GBSA remains a faster but less accurate option for preliminary screening. Machine learning tools like LABind provide complementary capabilities for binding site identification, particularly for novel targets or orphan ligands. As these tools continue to mature, their systematic validation against experimental data will remain essential for establishing appropriate applications and limitations in the context of high-throughput drug discovery.
Within the framework of validating protein-ligand binding using molecular dynamics (MD), absolute binding free energy calculations are crucial for estimating binding affinities and accelerating drug discovery. These methods are broadly categorized into alchemical pathways, which use non-physical transformations, and physical pathways, which model the actual unbinding process. The Attach-Pull-Release (APR) method is a prominent physical pathway approach that computes the work of mechanically pulling a ligand out of the binding site along a defined coordinate. This guide provides an objective comparison of APR against leading alchemical alternatives, detailing their performance, optimal use cases, and implementation protocols to inform researchers' selection of appropriate binding validation tools. [50]
Method Comparison: APR vs. Alchemical Alternatives
The core distinction between APR and methods like Double Decoupling (DD) lies in their treatment of the ligand. APR uses a physical pathway, while DD and its variants use an alchemical decoupling process. The Simultaneous Decoupling-Recoupling (SDR) method was developed to combine advantages of both approaches. [50]
The table below summarizes the fundamental characteristics of these three primary methods.
| Feature | APR (Attach-Pull-Release) | DD (Double Decoupling) | SDR (Simultaneous Decoupling-Recoupling) |
|---|---|---|---|
| Pathway Type | Physical Pulling [50] | Alchemical Decoupling [50] | Alchemical Transfer [50] |
| Core Mechanism | Ligand is physically pulled from binding site to bulk solvent along a reaction coordinate [50] | Ligand is alchemically decoupled from both binding site and pure solvent in separate, non-physical steps [50] | Ligand is decoupled from binding site while simultaneously recoupled to bulk solvent in a single simulation box [50] [51] |
| Handling of Charged Ligands | Avoids net charge change in system; no numerical artifacts [50] | Changes net charge of system; requires corrections for numerical artifacts [50] | Net charge of system remains constant; avoids numerical artifacts [50] |
| Best For | Solvent-exposed binding sites, charged ligands [50] | Buried binding sites, neutral ligands [50] | Charged or neutral ligands, buried sites without a clear physical pathway [50] |
| Key Limitation | Can disrupt protein structure for buried binding sites [52] [50] | Artifacts from system charge changes for charged ligands [50] | newer method; less extensively validated [50] |
Performance and Experimental Data
Validation studies have tested these methods across various protein-ligand systems. A key benchmark for relative binding free energy (RBFE) methods, like the Alchemical Transfer Method (ATM), involves comparing their predictions to experimental data across diverse protein targets. [51]
Performance is typically measured by the Pearson correlation coefficient (R) between calculated and experimental binding free energies, and the Mean Absolute Error (MAE) which indicates average prediction accuracy.
The following table summarizes published performance data for several methods on a benchmark set of 330 ligand pairs across eight protein targets (including MCL-1, TYK2, and BACE). [51]
| Calculation Method | Pearson Correlation (R) | Mean Absolute Error (MAE, kcal/mol) | Key Characteristics |
|---|---|---|---|
| FEP+ [51] | ~0.80 | ~0.90 | Established commercial protocol; requires significant expertise [51] |
| ATM (RBFE) [51] | ~0.80 | ~1.00 - 1.20 | Open-source; avoids leg splitting and softcore potentials; simpler setup [51] |
| Amber (TI) [51] | ~0.70 | ~1.10 | Traditional thermodynamic integration [51] |
| pmx [51] | ~0.60 | ~1.30 | Based on non-equilibrium trajectories [51] |
For absolute binding free energy calculations, APR has been extensively validated on host-guest systems, showing "moderate to strong correlations" between calculated and experimental binding thermodynamics for complexes with cucurbit[7]uril (CB7), octa acid (OA), and cyclodextrins. [52] However, its application to proteins with deeply buried binding sites is cautioned, as convergence issues may arise from substantial protein conformational changes during pulling. [52]
Detailed Experimental Protocols
APR Workflow and Setup
The APR method calculates the work of unbinding a ligand along a physical pathway. The implementation in software such as BAT.py automates this process using the AMBER simulation package. [50]
Core Workflow:
- System Preparation: The input protein-ligand structure is protonated, force field parameters are assigned, and the system is solvated in a water box with ions for neutrality. [50]
- Restraint Setup: A set of restraints is applied to maintain the ligand's orientation and the protein's binding site geometry during the pulling process. This involves defining anchor atoms on the protein and ligand, as well as dummy atoms to guide the pulling direction. [50]
- Pulling Simulation: The ligand is mechanically pulled from its bound position to a point in the bulk solvent through a series of simulation windows. In each window, the ligand is harmonically restrained at a different point along the pull coordinate, and the restraint's force constant is also varied. [50]
- Free Energy Calculation: The potential of mean force (PMF) along the pull coordinate is constructed by integrating the average forces on the ligand across all windows. The absolute binding free energy is derived from this PMF. [50]
Protocol for Alchemical Methods (DD/SDR)
Alchemical methods like DD and SDR estimate the binding free energy through non-physical pathways where the ligand's interactions are turned on or off.
Core Workflow for DD and SDR:
- System Preparation: Similar to APR, the complex is solvated and parameterized. For SDR, two copies of the ligand are often used—one in the binding site and one in the solvent. [51]
- Alchemical Transformation: The calculation proceeds along an alchemical coordinate (λ). In DD, this involves two separate legs: decoupling the ligand from the binding site and decoupling it from solvent. [50] In SDR, the ligand is simultaneously decoupled from the binding site and recoupled to the solvent in a single process, keeping the system charge constant. [50] [51]
- Free Energy Estimation: The free energy change for the alchemical transformation is computed using methods like Free Energy Perturbation (FEP) or Thermodynamic Integration (TI). Hamiltonian Replica Exchange MD (HREMD) is often used to improve sampling between λ-states. [51]
- Analysis: The binding free energy for DD is the difference between the two legs. For SDR, it is obtained directly from the transfer process. [50]
The Scientist's Toolkit: Essential Research Reagents and Software
Successful implementation of these advanced simulation methods relies on a suite of software tools and computational resources.
| Tool Name | Type/Function | Key Utility in Workflow |
|---|---|---|
| BAT.py [50] | Python Software Package | Automates absolute binding free energy calculations for a series of ligands using APR, DD, or SDR methods; interfaces with AMBER. |
| AMBER [50] | Molecular Dynamics Suite | Provides the pmemd.cuda engine for running high-performance GPU-accelerated MD simulations and AmberTools for system preparation. |
| AToM-OpenMM [51] | OpenMM Plugin | Implements the Alchemical Transfer Method (ATM) for relative binding free energy calculations within the OpenMM ecosystem. |
| OpenBabel [50] | Chemical Toolbox | Used for ligand file format conversion and protonation state assignment. |
| VMD [50] | Visualization & Analysis | Used for visualizing molecular structures, trajectories, and conducting various analyses. |
| GAFF2/AM1-BCC [51] | Force Field & Charges | A common combination for generating force field parameters and partial atomic charges for small molecule ligands. |
| GPU Cluster [50] | Computing Hardware | Essential for achieving practical computation times (e.g., on NVIDIA RTX cards). [51] |
For researchers validating protein-ligand binding, the choice between path-based and alchemical methods is not one of superiority but of context. APR offers a more intuitive, physical pathway that is robust for charged ligands in solvent-accessible sites. In contrast, alchemical methods like DD and SDR are often more efficient for buried binding pockets, with SDR specifically overcoming DD's limitations with charged ligands.
The field is moving toward increased automation, as seen in tools like BAT.py and AToM-OpenMM, lower costs via GPU computing, and robust validation against expanding benchmark sets. For practical applications, researchers should select a method based on their specific system's characteristics: binding site accessibility, ligand charge, and the desired balance between physical intuitiveness and computational efficiency.
In the field of computational drug discovery, the accurate prediction of protein-ligand binding affinities is crucial for reducing the time and costs associated with experimental trial-and-error [53]. Alchemical free energy calculations have emerged as powerful molecular dynamics (MD) based approaches that enable researchers to estimate binding strengths from first principles [51]. These methods employ non-physical (alchemical) pathways to compute free energy differences, avoiding the need to simulate the actual binding process, which often occurs on timescales too long for conventional MD simulations [54]. Among these techniques, the Double Decoupling Method (DD) represents a well-established standard, while the Simultaneous Decoupling-Recoupling Method (SDR) has more recently been developed to address some of DD's limitations [53].
Both methods aim to compute the absolute binding free energy (ABFE) of a ligand to a protein target, a critical metric in early-stage drug discovery when screening diverse compounds [53]. The accuracy and reliability of these calculations depend significantly on the choice of methodology, force field parameters, and sufficient sampling of the molecular configurations [51]. This guide provides an objective comparison of DD and SDR methodologies, supported by experimental data and implementation protocols, to assist researchers in selecting appropriate approaches for validating protein-ligand interactions.
Theoretical Foundations and Methodologies
Double Decoupling Method (DD)
The Double Decoupling Method (DD), also known as the "standard double-decoupling" approach, computes the absolute binding free energy through a thermodynamic cycle that decouples the ligand from its environments [53] [55]. The method calculates the work of decoupling the ligand from the binding site and the work of decoupling the ligand from pure solvent [53]. This process effectively transfers the ligand from the bound state to the unbound state through an alchemical pathway.
The DD method is particularly well-suited for charge-neutral ligands in both surface and deeper binding sites [53]. However, a significant limitation arises when handling ligands with nonzero net charge, as the decoupling processes cause changes in the net charge of the entire simulation system [53]. These changes can lead to undesirable numerical artifacts whose correction requires additional, time-consuming calculations [53]. The binding free energy in DD is obtained as the difference between two large numbers (the decoupling free energies in solution and in the receptor environment), and the statistical errors of these two terms combine additively, making convergence challenging, especially for larger ligands like peptides [55].
Simultaneous Decoupling-Recoupling Method (SDR)
The Simultaneous Decoupling-Recoupling (SDR) method was developed as a variant of double decoupling that avoids numerical artifacts associated with charged ligands [53]. Unlike DD, SDR uses nonphysical, alchemical pathways to extract the ligand from the binding site while simultaneously recoupling the ligand with bulk solvent at a distance from the protein [53].
This approach maintains a constant net charge of the system throughout the transfer process by recoupling the ligand to solvent as it is decoupled from the binding site [53]. The SDR method combines the advantages of both DD and physical pathway methods, being suitable for both neutral and charged ligands, regardless of whether clear access to solvent exists [53]. By maintaining charge neutrality throughout the alchemical transformation, SDR eliminates the need for complex correction factors that DD requires for charged ligands [53] [51].
Single-Decoupling Method (SDM)
Although not the focus of this guide, the Single-Decoupling Method (SDM) represents an alternative alchemical approach that addresses some convergence difficulties of conventional double-decoupling protocols [55]. SDM employs a λ-dependent potential energy function that interpolates directly between the dissociated and associated states of the complex in solution, avoiding the intermediate gas-phase state required by DD [55]. This method computes the binding free energy directly rather than as a difference between two large values, potentially offering favorable convergence properties for challenging systems like protein-peptide complexes [55].
Table 1: Core Methodological Differences Between DD and SDR
| Feature | Double Decoupling (DD) | Simultaneous Decoupling-Recoupling (SDR) |
|---|---|---|
| Thermodynamic Pathway | Two separate decoupling steps | Simultaneous decoupling and recoupling |
| Charge Handling | Changes system net charge for charged ligands | Maintains constant system net charge |
| Implementation Complexity | Moderate | Moderate to high |
| Ligand Suitability | Ideal for neutral ligands | Suitable for both neutral and charged ligands |
| Numerical Artifacts | Possible with charged ligands, requires corrections | Minimized through charge conservation |
| Computational Cost | Two separate free energy calculations | Combined transformation |
Comparative Performance Analysis
Accuracy and Applicability
The performance of alchemical binding free energy methods is typically evaluated through metrics such as Pearson correlation coefficient (R), mean absolute error (MAE), and root mean square error (RMSE) between computational predictions and experimental measurements [51]. While comprehensive head-to-head comparisons between DD and SDR are limited in the literature, available data provides insights into their relative performance.
The DD method has demonstrated good agreement with experimental values in various test systems. For instance, in a study predicting the binding affinity of MDM2 and a ligand, the DD approach yielded values in good agreement with experiments [56]. Similarly, the SDR method has shown promising results in initial test applications for both re-ranking docked poses and estimating overall binding free energies [53].
Recent benchmarking studies of alchemical methods in general have shown that they can achieve Pearson correlation coefficients of 0.65 or higher with experimental binding affinities, with RMSE values typically hovering around just below 1 kcal/mol [15]. This represents significant improvement over docking methods, which generally deliver results with 2-4 kcal/mol RMSE and correlation coefficients around 0.3 [15].
Challenges with Charged Ligands
A critical differentiator between DD and SDR emerges when handling ligands with nonzero net charge. The standard DD method leads to changes in the net charge of the simulated system when the ligand has a nonzero net charge, resulting in numerical artifacts that may be difficult and time-consuming to correct [53]. These artifacts stem from the treatment of long-range electrostatic interactions and require additional correction factors [51].
In contrast, the SDR method maintains a constant net charge throughout the simulation by design, as it recouples the ligand with bulk solvent at the same time as it decouples the ligand from the binding site [53]. This approach effectively circumvents the charge-related artifacts that plague DD calculations with charged ligands, making SDR particularly valuable for pharmaceutical compounds that often exist in charged states at physiological pH.
Table 2: Performance Comparison Across Method Types
| Method | Typical RMSE (kcal/mol) | Typical Pearson R | Charged Ligand Handling | Computational Cost |
|---|---|---|---|---|
| Docking | 2-4 [15] | ~0.3 [15] | Varies by scoring function | Low (minutes on CPU) |
| DD | ~1 [15] | ≥0.65 [15] | Challenging [53] | High (12+ hours GPU) [15] |
| SDR | Similar to DD [53] | Similar to DD [53] | Excellent [53] | High (comparable to DD) [53] |
| ATM | Marginally higher than FEP [51] | Matches FEP [51] | Good [51] | High [51] |
Implementation Protocols
Workflow for DD and SDR Calculations
The implementation of both DD and SDR methods follows a structured workflow that can be automated using tools like BAT.py, a Python package that invokes the AMBER simulation package to calculate binding free energies [53]. The overall process involves system preparation, equilibration, production simulations, and free energy analysis.
System Preparation and Equilibration
For both DD and SDR calculations, the initial step involves preparing the protein-ligand complex structure, which may come from experimental cocrystal structures or docking poses [53]. The BAT.py software automates many preparation steps, including:
- Protonation state assignment using OpenBabel for ligands [53]
- Protein alignment with a reference structure using MUSTANG [53]
- Force field parameter assignment using AMBERTools (ff14SB for proteins, GAFF2 for ligands) [53] [57]
- System solvation in an orthorhombic TIP3P water box with 10 Å extension from the protein surface [57]
- Addition of ions for electroneutrality and desired ionic strength [53]
Following system preparation, equilibration proceeds through several stages to ensure proper system relaxation before production simulations [57]:
- Minimization: 1000-2000 steps with harmonic restraints on protein backbone atoms [57]
- Heating: Gradual heating from 50K to 300K with backbone restraints [57]
- NVT equilibration: 1-2 ns at constant volume and temperature [57]
- NPT equilibration: 1-2 ns at constant pressure and temperature [57]
Production Simulations and Free Energy Analysis
For DD calculations, production runs involve separate alchemical transformations for the ligand in solution and in the receptor environment [53] [55]. These transformations typically use multiple λ windows (often 12 or more) to gradually decouple the ligand from its environment [51]. The binding free energy is calculated as the difference between these two transformations [55].
SDR simulations employ a different alchemical pathway where the ligand is simultaneously decoupled from the binding site and recoupled to bulk solvent [53]. This approach maintains a consistent system charge throughout the process. Both methods typically require extensive sampling, with modern implementations using Hamiltonian replica exchange (HREM) to enhance conformational sampling [51].
Free energy analysis is performed using methods such as the Multistate Bennett Acceptance Ratio (MBAR) or the Unweighted Histogram Analysis Method (UWHAM) to compute the free energy differences from the simulation data [51].
Research Reagent Solutions
Table 3: Essential Computational Tools for Alchemical Calculations
| Tool/Resource | Function | Application in DD/SDR |
|---|---|---|
| BAT.py [53] | Python package for automating binding free energy calculations | Implements DD, SDR, and APR methods; interfaces with AMBER |
| AMBER [53] | Molecular dynamics simulation package | Force field assignment, system building, MD simulations |
| OpenMM [51] | High-performance MD simulation toolkit | Implements ATM and other alchemical methods |
| GAFF2/AM1-BCC [51] | Force field and partial charge method for small molecules | Ligand parameterization |
| ff14SB [51] | Protein force field | Protein parameterization |
| PLAS-20k Dataset [57] | MD-based dataset of protein-ligand affinities | Benchmarking and method validation |
| AToM-OpenMM [51] | OpenMM plugin for alchemical transfer method | Implements ATM-RBFE and ATM-ABFE protocols |
The Double Decoupling and Simultaneous Decoupling-Recoupling methods represent sophisticated computational approaches for predicting protein-ligand binding affinities with accuracy superior to docking methods. DD serves as a well-established benchmark for neutral ligands but presents challenges for charged compounds due to charge-related artifacts. SDR extends the applicability of alchemical methods to charged ligands by maintaining system charge neutrality throughout the transformation, addressing a key limitation of DD.
Both methods require careful system preparation, sufficient sampling, and appropriate force field selection to yield reliable results. The development of automated tools like BAT.py has made these advanced calculations more accessible to researchers, potentially enabling broader adoption in early-stage drug discovery. As molecular dynamics simulations continue to benefit from hardware advances like GPU computing, alchemical binding free energy calculations stand to play an increasingly important role in reducing the time and cost of drug discovery pipelines.
The validation of protein-ligand binding mechanisms is a cornerstone of modern drug discovery, yet the occurrence of rare events in molecular dynamics (MD)—such as ligand dissociation or large-scale conformational changes in proteins—poses a significant challenge. These events occur on timescales vastly longer than what conventional MD simulations can routinely access. Enhanced sampling techniques have been developed to overcome this limitation, enabling the efficient calculation of crucial thermodynamic and kinetic parameters like binding free energies and dissociation rates. Among these methods, Weighted Ensemble (WE) and dissociation Parallel Cascade Selection Molecular Dynamics (dPaCS-MD) represent two powerful yet philosophically distinct approaches. This guide provides an objective comparison of their performance, experimental protocols, and applications, offering researchers a framework for selecting the appropriate method for validating protein-ligand interactions within their molecular dynamics research.
At their core, enhanced sampling methods accelerate the observation of rare events without distorting the underlying physics of the system. WE and dPaCS-MD achieve this through different strategic paradigms.
Weighted Ensemble (WE) is a path-sampling approach that does not alter the underlying energy landscape. It runs multiple parallel simulations (or "walkers") and periodically "resamples" them based on progress along user-defined collective variables (CVs). This resampling strategically allocates computational resources by splitting trajectories that progress toward rare events and merging those that do not, ensuring continuous exploration of the path from the bound to unbound state [58] [59]. Recent advancements include "binless" WE algorithms like WeTICA, which use a low-dimensional CV space to drive the simulation toward a target state, bypassing the need for complex binning schemes and improving efficiency [60].
Dissociation Parallel Cascade Selection Molecular Dynamics (dPaCS-MD), conversely, is a goal-oriented, brute-force method. It initiates numerous short MD simulations from the bound state. The trajectories that show the most progress toward dissociation (monitored by a simple CV like ligand-protein distance) are selected as starting points for the next cycle of simulations. This iterative process, akin to a directed evolutionary algorithm for pathways, rapidly generates multiple dissociation pathways [61].
The table below summarizes the fundamental characteristics of these two methods.
Table 1: Fundamental Comparison of WE and dPaCS-MD
| Feature | Weighted Ensemble (WE) | dPaCS-MD |
|---|---|---|
| Core Principle | Statistical resampling of unbiased trajectories in parallel; a path-sampling method. | Iterative selection and restart of simulations based on progress toward a goal state. |
| Energy Landscape | Unmodified; unbiased dynamics are preserved. | Unmodified; uses multiple short, unbiased MD runs. |
| Typical Collective Variables (CVs) | Requires carefully chosen CVs (e.g., protein-ligand contacts, angles). Progress coordinates can be derived from TICA or other dimension-reduction methods [62] [60]. | Often uses a simpler, one-dimensional CV, such as the distance between the ligand and the protein's binding pocket [61]. |
| Key Strength | Directly provides kinetic rate constants (e.g., koff). Efficiently explores conformational space and is well-suited for complex, multi-pathway processes [58] [59]. | Computationally efficient at generating dissociation pathways without requiring complex CVs. Easier to implement and automate. |
| Common Analysis Tool | Often combined with Markov State Models (MSM) to elucidate kinetics and metastable states. | Routinely analyzed with MSM to calculate free energy profiles and binding free energies [61]. |
The following workflow diagram illustrates the fundamental procedural differences between the two methods.
Diagram 1: Comparative Workflows of WE and dPaCS-MD
Performance and Experimental Data
The ultimate test for any computational method is its performance against experimentally validated systems. Both WE and dPaCS-MD have been applied to study the unbinding of ligands from proteins, providing quantitative data on kinetics and thermodynamics.
A key application of WE is in exploring conformational landscapes for drug discovery. In a study of Hepatitis B virus (HBV) capsid assembly modulators, WE simulations were employed to characterize the free-energy landscape of the tetrameric capsid building blocks. The results demonstrated that WE could sample conformations outside the reach of standard MD, including structures with enlarged binding pockets conducive to ligand binding. This highlights WE's utility in providing a more complete conformational ensemble for computational drug development [58].
For dPaCS-MD, a robust validation comes from its application to multiple protein/ligand complexes with diverse characteristics. The method, coupled with Markov state model analysis (dPaCS-MD/MSM), has been shown to accurately reproduce experimental binding free energies.
Table 2: Experimental Validation of dPaCS-MD/MSM on Model Systems
| Protein/Ligand Complex | Experimental Binding Free Energy (kcal/mol) | dPaCS-MD/MSM Calculated Free Energy (kcal/mol) | Key Interactions/Notes |
|---|---|---|---|
| Trypsin/Benzamidine | -6.6 [61] | -6.5 [61] | Primarily electrostatic interactions; a standard test case for binding. |
| FKBP/FK506 | -10.9 [61] | -11.1 [61] | A larger, clinically relevant immunosuppressant complex. |
| Adenosine A2A Receptor/T4E | -11.7 [61] | -11.8 [61] | A membrane-bound G-protein coupled receptor (GPCR) complex. |
The data in Table 2 shows that dPaCS-MD/MSM achieves remarkable agreement with experiment across systems of varying size and complexity, from the relatively simple trypsin/benzamidine complex to the challenging GPCR system [61]. This demonstrates the method's generalizability and accuracy for calculating binding free energies.
Detailed Experimental Protocols
To ensure reproducibility and provide a clear technical roadmap, this section outlines the standard protocols for implementing WE and dPaCS-MD simulations.
Protocol for Weighted Ensemble Simulations
- System Preparation: Obtain the initial protein-ligand complex structure (e.g., from PDB). Parameterize the ligand using a tool like GAFF and prepare the protein with a force field like AMBER14SB. Solvate the system in a water box and add ions to neutralize charge [58] [62].
- Equilibration: Perform energy minimization to remove steric clashes. Gradually heat the system to the target temperature (e.g., 300 K) and equilibrate under NVT and NPT ensembles.
- Define Progress Coordinate: Identify one or more collective variables that distinguish the bound from the unbound state. For protein-ligand dissociation, this could be the distance between the ligand and the binding pocket, or more complex coordinates derived from time-lagged independent component analysis (TICA) [62] [60].
- WE Simulation Setup: Use a WE software package like WESTPA. Define the "bins" or state boundaries along the progress coordinate. Specify the number of walkers (typically scores to hundreds) and the resampling time interval.
- Production Run: Launch the WE simulation. The software will manage the running of parallel trajectories, periodically check their progress coordinates, and perform resampling (splitting and merging walkers to maintain uniform sampling).
- Data Analysis: Analyze the resulting ensemble of trajectories to compute the dissociation rate constant (koff) directly from the flux of trajectories reaching the unbound state. The trajectories can also be used to build a Markov State Model for a more detailed understanding of the kinetics and metastable states [59].
Protocol for dPaCS-MD/MSM Simulations
- Initial Setup: Prepare the protein-ligand complex as described in Step 1 of the WE protocol [61].
- Define the Reaction Coordinate: Typically, a simple distance (d) between the centers of mass of the ligand and the protein binding site is used as the primary CV to monitor dissociation progress.
- Initial Sampling (Cycle 0): From the fully bound structure, run a set of independent, short MD simulations (e.g., 10-100 ps each).
- Trajectory Selection and Restart: After each cycle, rank all generated trajectories based on the maximum value of the distance
dachieved. Select the topNtrajectories that have progressed the farthest toward dissociation. - Iterative dPaCS-MD Cycles: Use the final snapshots from the selected trajectories as the initial structures for the next cycle of short MD simulations. Repeat this selection-and-restart process for multiple cycles until the ligand is fully solvated.
- MSM Construction and Analysis: Pool all generated trajectories from every cycle. Discretize the conformational space into microstates, often based on the ligand-protein distance
dand other relevant descriptors. Build a Markov State Model (MSM) to extract the underlying kinetic information and calculate the free energy profile along the dissociation coordinate. The standard binding free energy (ΔG°) is then computed from this profile [61].
The Scientist's Toolkit
Successfully implementing these advanced simulation techniques relies on a suite of software tools and force fields. The table below lists essential "research reagents" for the field.
Table 3: Essential Software and Force Fields for Enhanced Sampling
| Tool Name | Type | Primary Function |
|---|---|---|
| WESTPA [62] [60] | Software Package | A widely used, open-source implementation of the Weighted Ensemble algorithm for orchestrating and running WE simulations. |
| OpenMM [62] | MD Engine | A high-performance toolkit for molecular simulation that provides the computational backend for running the dynamics, often integrated with WESTPA. |
| PyEMMA / MSMBuilder [63] | Analysis Toolkit | Software packages for constructing and validating Markov State Models from MD trajectory data. |
| AMBER | Force Field | A family of force fields (e.g., AMBER14SB) and associated tools for simulating biomolecules, commonly used for protein parameters [62]. |
| GAFF | Force Field | The "General AMBER Force Field," used for parameterizing small organic molecules and drug-like ligands [59]. |
| BioLiP / PDBbind [64] | Database | Curated databases of protein-ligand complexes with binding affinity data, used for system selection and validation. |
Both Weighted Ensemble and dPaCS-MD are powerful validated methods for tackling the rare event problem in protein-ligand binding. The choice between them depends on the specific research question and available resources. Weighted Ensemble is particularly strong when the goal is to directly obtain accurate kinetic rates and explore complex, multi-pathway conformational changes in a highly efficient manner, though it may require more expertise in setting up progress coordinates. dPaCS-MD, with its simpler iterative restart mechanism, offers a more straightforward route to generating dissociation pathways and obtaining highly accurate binding free energies, as validated across diverse protein systems. For researchers engaged in validating protein-ligand interactions, integrating these enhanced sampling techniques into their molecular dynamics workflow provides a rigorous path from static structural data to a dynamic, mechanistic, and quantitative understanding of binding.
Markov State Models (MSMs) have emerged as a powerful computational framework for analyzing molecular dynamics (MD) simulations to elucidate protein-ligand binding mechanisms. As MD simulations now routinely access the microsecond timescale, direct sampling of ligand association events has become feasible, creating a critical need for analytical methods that can extract meaningful kinetic and mechanistic information from these extensive trajectory datasets [65]. MSMs address this need by providing a quantitative approach to model the long-timescale dynamics of biomolecular systems, including the complex processes of ligand binding and unbinding, by combining numerous short MD simulations [66]. This approach has fundamentally advanced our understanding of molecular recognition processes that are central to drug discovery and biological function.
The power of MSMs lies in their ability to identify metastable conformational states and quantify transition probabilities between them, thereby creating a kinetic network model of the binding process [67]. Unlike methods that focus solely on binding affinities, MSMs provide crucial information about binding pathways, rates, and mechanisms—key determinants of drug efficacy [68]. This comprehensive characterization enables researchers to move beyond static structural snapshots to a dynamic understanding of how ligands and proteins interact over time, revealing transient intermediate states and alternative binding pathways that would be difficult to capture experimentally [65] [68].
Fundamental Principles of Markov State Modeling
Theoretical Foundation
Markov State Models are built on the fundamental concept that the complex dynamics of biomolecular systems can be described as a memoryless process transitioning between discrete conformational states. The mathematical foundation of MSMs relies on the Markov assumption, which states that the probability of transitioning from state i to state j depends only on the current state i and not on the history of previously visited states [66]. This assumption holds when the chosen lag time (Δt) is long enough to allow for internal relaxation within states, making the dynamics Markovian at this timescale.
The core of an MSM is the transition probability matrix T(Δt), whose elements T¯ij represent the probability of transitioning from state i to state j after time Δt. This matrix enables the prediction of long-time dynamics through simple matrix multiplication: P(nΔt) = T¯(Δt)¯P(0), where P(t) is a vector of state populations at time t [66]. The eigenvalues and eigenvectors of T(Δt) provide crucial information about the slow dynamical processes of the system, with each eigenvector corresponding to a collective motion and its eigenvalue related to the timescale of this motion.
Key Advantages for Binding Studies
MSMs offer several distinct advantages for studying protein-ligand binding mechanisms compared to alternative approaches. First, they efficiently combine many short, distributed MD simulations to model processes occurring on timescales much longer than any individual simulation [66]. This makes it feasible to study binding events that may occur on millisecond timescales or longer using microsecond-length simulations. Second, MSMs provide a comprehensive kinetic picture that includes not only the dominant binding pathways but also rare events and alternative mechanisms [65]. Third, they enable the identification and characterization of metastable intermediate states that may be difficult to capture experimentally but could be crucial for understanding binding mechanisms or designing drugs with specific kinetic properties [68].
Perhaps most importantly, MSMs facilitate the distinction between different binding mechanisms, particularly conformational selection (where ligands bind to pre-formed receptor conformations) and induced fit (where binding induces conformational changes) [65] [67]. In reality, most binding processes involve aspects of both mechanisms, and MSMs provide a framework to quantify their relative contributions by analyzing the flow of probability between different conformational states in the presence and absence of ligands [68].
MSM Construction Workflow and Methodologies
Recommended Protocol for MSM Construction
Building a validated MSM for analyzing protein-ligand binding follows a systematic workflow that transforms raw MD trajectories into a quantitative kinetic model. The recommended protocol encompasses several stages from initial path generation to model validation [66]:
Initial Path Generation: Connecting known states (from crystallography or cryo-EM) using methods like targeted MD, action-based conformational state annealing (Action-CSA), Climber, or coarse-grained MD simulations, followed by path optimization with the String method or traveling-salesman-based automatic path searching (TAPS) [66].
Ensemble Simulation: Running extensive MD simulations initiated from conformations along the optimized initial pathways to ensure adequate sampling of the relevant conformational space [66].
Feature Selection: Identifying structural features (distances, angles, etc.) that best describe the functional conformational changes. Automatic selection methods like Spectral-oASIS, feature importance selection, or automatic mutual information noise omission (AMINO) are recommended [66].
Dimensionality Reduction: Projecting the high-dimensional feature space onto a few collective variables using algorithms such as time-lagged independent component analysis (TICA), VAMPNets, or state-free reversible VAMPNets (SRVs) [66].
Clustering and Discretization: Grouping MD conformations into microstates using clustering algorithms, then building and validating the microstate-MSM using the Chapman-Kolmogorov test [66].
The following workflow diagram illustrates this comprehensive protocol for constructing MSMs to study functional conformational changes:
Feature Selection and Dimensionality Reduction
Proper feature selection is critical for building informative MSMs, especially for functional conformational changes that are often localized rather than global. While early MSM studies relied on manual feature selection based on physical intuition, recent machine learning approaches enable more automated and objective selection [66]. Spectral-oASIS is one recommended method that identifies features most relevant to the slowest dynamical processes [66]. Similarly, time-lagged independent component analysis (tICA) has become a widely used approach for building informative models by identifying collective variables that maximize autocorrelation times [65] [67].
For large biomolecular complexes with many degrees of freedom, dimensionality reduction is essential to avoid the "curse of dimensionality" and focus on the most relevant conformational changes. Deep learning approaches like VAMPNets and SRVs have shown promise in learning optimal collective variables directly from simulation data [66]. These methods can automatically discover nonlinear combinations of features that best describe the slow dynamics of the system, potentially capturing complex conformational changes that might be missed by linear methods like tICA.
State Discretization and Model Validation
The discretization of conformational space into states represents a crucial step in MSM construction. States should be defined to be metastable, meaning intrastate transitions are fast compared to interstate transitions [66]. Common clustering algorithms include k-means, k-medoids, and regular spatial clustering, which group conformations based on structural similarity in the reduced dimensional space [66].
Model validation is essential to ensure the reliability of MSM predictions. The Chapman-Kolmogorov test serves as a key validation method, comparing the model's predicted state populations with those directly observed in simulations [66]. Additional validation approaches include examining the implied timescales to ensure they become constant at longer lag times (indicating Markovian behavior) and using cross-validation techniques to avoid overfitting [66].
Comparative Performance Analysis
Case Study: Trypsin-Benzamidine Binding
A comprehensive study of Trypsin binding with its competitive inhibitor Benzamidine illustrates the power of MSMs to reveal complex binding mechanisms. This analysis of 149.1 μs of cumulative simulation time identified seven metastable conformations of Trypsin with different binding pocket structures that interconvert at timescales of tens of microseconds [68]. These conformations exhibited strikingly different binding affinities and binding/dissociation rates, demonstrating how protein dynamics significantly impact ligand binding [68].
The MSM analysis revealed three key conformational switches regulating Benzamidine binding:
- S1 pocket opening and closing: The primary Benzamidine-binding pocket found in crystal structures
- S1* pocket opening and closing: A secondary binding pocket allowing binding to Asp189 from a different angle
- Conformational switch in the S1* pocket: Critical modulation of binding free energy depending on Asp189 loop position [68]
Interestingly, the MSM showed that the X-ray-like apo structure was one of the less stable conformations in solution, while states with closed S1 pockets were more stable—findings that align with known stabilities of similar conformations in related serine proteases like Thrombin [68]. This case study exemplifies how MSMs can provide insights that extend beyond what is apparent from static crystal structures alone.
Table 1: Kinetic Properties of Metastable States in Trypsin-Benzamidine System
| Metastable State | S1 Pocket | S1* Pocket | Relative Stability | Binding Affinity |
|---|---|---|---|---|
| Magenta (X-ray-like) | Open | Closed | Low | Medium |
| Red | Closed | Open | High | High |
| Green | Open/Closed | Open/Closed | High | Variable |
Comparison with Alternative Approaches
MSMs occupy a unique position in the landscape of computational approaches for studying protein-ligand interactions. The table below compares MSMs with other common methods across key performance metrics:
Table 2: Performance Comparison of Protein-Ligand Binding Analysis Methods
| Method | Timescale Access | Mechanistic Insight | Kinetic Information | State Resolution | Computational Cost |
|---|---|---|---|---|---|
| Markov State Models | Microseconds to milliseconds | High (pathways, intermediates) | Yes (rates, fluxes) | High (multiple states) | Medium-High |
| Standard MD Analysis | Nanoseconds to microseconds | Medium (limited by sampling) | Limited | Low | Medium-High |
| Docking | N/A | Low (single pose) | No | Very Low | Low |
| MM/PBSA/GBSA | N/A | Low (ensemble average) | No | Low | Medium |
| Free Energy Perturbation | N/A | Low (end states only) | No | Low | High |
MSMs provide significant advantages in elucidating binding mechanisms compared to endpoint methods like docking or free energy calculations. While docking offers speed and MM/GBSA provides affinity estimates, neither captures the dynamic process of binding or identifies intermediate states [15]. Free energy methods like FEP can provide accurate binding affinities but require significant computational resources and still lack detailed mechanistic information [15]. MSMs uniquely combine the ability to predict kinetics with detailed structural mechanisms, making them particularly valuable for understanding complex binding processes with multiple pathways or intermediate states.
Research Toolkit and Implementation
Essential Software and Tools
Implementing MSM analysis requires specialized software tools for various stages of the workflow. The following table outlines key resources available to researchers:
Table 3: Essential Research Tools for MSM Construction and Analysis
| Tool/Resource | Function | Key Features | Availability |
|---|---|---|---|
| pyEMMA | MSM construction and validation | Comprehensive MSM analysis, tICA, clustering | http://pyemma.org [68] |
| MDTraj | Trajectory analysis | Distance calculations, SASA, feature extraction | Open source |
| VAMPNets | Deep learning for MSMs | Neural networks for dimensionality reduction | Research code [66] |
| Spectral-oASIS | Feature selection | Automatic identification of relevant features | Research code [66] |
| HiQBind-WF | Dataset curation | High-quality protein-ligand complex preparation | Open source [64] |
| LABind | Binding site prediction | Graph transformer for binding site identification | Available online [16] |
Data Requirements and Preparation
The quality of input data fundamentally determines the success of MSM analysis. Recent work has highlighted issues with widely used datasets like PDBbind, which can contain structural artifacts that compromise accuracy and generalizability [64]. Tools like HiQBind-WF provide a semi-automated workflow to curate non-covalent protein-ligand datasets, addressing common problems including covalent binders, ligands with rare elements, steric clashes, and incorrect bond orders or protonation states [64].
For binding site identification, methods like LABind utilize graph transformers to capture binding patterns in the local spatial context of proteins, incorporating cross-attention mechanisms to learn distinct binding characteristics between proteins and ligands [16]. Such tools can enhance MSM studies by helping researchers focus on relevant structural features and binding sites.
Adequate sampling remains crucial for building reliable MSMs. While early studies required massive computational resources (e.g., 150 μs for Trypsin-Benzamidine [68]), methodological advances have improved sampling efficiency. Still, researchers should ensure sufficient coverage of both bound and unbound states, as well as potential intermediate states, to build comprehensive models.
Future Perspectives and Challenges
Methodological Advancements
The MSM field continues to evolve rapidly, with several promising directions emerging. Quasi-MSMs (qMSMs) represent an important advancement that addresses the challenge of large state counts in conventional MSMs [66]. By encoding non-Markovian dynamics via the generalized master equation, qMSMs can significantly reduce the number of states needed for accurate modeling, facilitating the interpretation of functional conformational changes [66].
Integration of machine learning approaches continues to transform MSM methodology. Deep learning architectures like VAMPNets and SRVs show promise for automatically learning optimal collective variables and features, reducing the reliance on expert knowledge and potentially discovering previously unrecognized important degrees of freedom [66]. These approaches may be particularly valuable for studying large biomolecular complexes where manual feature selection becomes impractical.
Another emerging trend is the combination of MSMs with experimental data. MSMs can incorporate constraints from experimental measurements such as NMR relaxation, FRET, or single-molecule experiments to improve model accuracy and validate predictions. This integrative approach helps bridge the gap between computational models and experimental observables.
Current Challenges and Limitations
Despite significant progress, several challenges remain in applying MSMs to protein-ligand binding. The feature selection problem persists, particularly for large systems where relevant motions may be localized and non-obvious [66]. While automatic feature selection methods help, they may not always capture biologically relevant motions, especially for systems with limited prior knowledge.
Validation remains challenging, particularly for predicting absolute kinetics rates. While the Chapman-Kolmogorov test provides internal validation, comparison with experimental kinetics data (when available) offers important external validation [66]. However, such experimental data may be scarce or difficult to measure precisely.
The timescale gap between simulation and biological function, though partially bridged by MSMs, persists for very slow processes (seconds or longer). Enhanced sampling methods that combine MSMs with techniques like accelerated MD or metadynamics may help address this challenge.
Finally, accessibility and usability present barriers for broader adoption of MSM methods. While software tools like pyEMMA have made MSM construction more accessible, the process still requires significant expertise and computational resources. Continued development of automated workflows and user-friendly interfaces will be important for expanding the application of MSMs in drug discovery and structural biology.
In conclusion, Markov State Models provide a powerful framework for extracting kinetic and mechanistic information from molecular dynamics trajectories of protein-ligand binding. By transforming high-dimensional simulation data into interpretable kinetic networks, MSMs reveal the dynamic nature of molecular recognition, including multiple pathways, intermediate states, and the interplay between conformational selection and induced fit. As methodology continues to advance, particularly through integration with machine learning approaches, MSMs are poised to play an increasingly important role in computational drug discovery and fundamental studies of biomolecular function.
Troubleshooting and Optimization: Ensuring Robust and Efficient Simulations
Common Pitfalls in Simulation Setup and How to Avoid Them
Validating protein-ligand binding through molecular dynamics (MD) is a cornerstone of modern drug discovery. However, the accuracy and reliability of these simulations are heavily dependent on the initial setup. This guide examines common pitfalls encountered during simulation setup, provides objective comparisons of methodological choices, and outlines protocols to ensure robust and reproducible results for drug development professionals.
The foundation of a meaningful MD simulation is laid before the first nanosecond of production run. Errors introduced during the setup phase—ranging from inadequate structure preparation to poor sampling strategies—can propagate through the entire simulation, leading to misleading conclusions about protein-ligand binding. A flawed setup can compromise the validation process, rendering even long and computationally expensive simulations unreliable. Adopting rigorous setup protocols is therefore not merely a technical formality but a fundamental requirement for producing scientifically valid results that can guide drug development decisions [69] [70].
Major Pitfalls and Comparative Solutions
The following section details specific setup challenges, compares common approaches, and provides data-driven recommendations.
Pitfall 1: Inadequate Protein-Ligand Structure Preparation
Using raw structural data from protein data banks without rigorous curation is a primary source of error. These structures often contain unresolved atoms, incorrect protonation states, and improper bond orders.
Table 1: Comparison of Dataset Preparation and Curation Workflows
| Criterion | Raw PDB Entry | Standard Curation (e.g., PDBbind) | Advanced Workflow (e.g., HiQBind-WF) |
|---|---|---|---|
| Ligand Bond Order | As provided in file; often incomplete | Corrected using chemical dictionaries | Corrected and validated [64] |
| Protonation States | Often missing or incorrect | Added for protein and ligand independently | Added to protein and ligand in complex for realistic intermolecular interactions [64] |
| Steric Clashes | Common in crystal structures | May remain unresolved | Identified and removed via constrained energy minimization [64] |
| Handling of Additives | Inconsistent | Varies | Systematic identification of ions, solvents, and co-factors within 4 Å [64] |
| Reproducibility | Low | Moderate, can rely on manual steps | High, due to open-source and semi-automated workflow [64] |
Recommendation: Employ a systematic, reproducible workflow like HiQBind-WF for high-quality starting structures. This is particularly crucial for training machine-learning scoring functions or performing free energy calculations [64].
Pitfall 2: Poor Handling of System Protonation
Incorrectly assigning the protonation states of ionizable residues and ligands, especially for environment-dependent lipids in lipid nanoparticles (LNPs), is a subtle but critical error.
- Challenge: The protonation state of ionizable lipids in LNPs is highly environment-dependent, influenced by local pH and the lipid's pKa, which can shift based on manufacturing conditions and helper lipids [71].
- Incorrect Approach: Using fixed protonation states throughout the simulation.
- Solution: Utilize constant pH molecular dynamics (CpHMD) methods. Scalable CpHMD models perform at speeds comparable to standard MD and can accurately reproduce apparent pKa values (e.g., with a mean average error of 0.5 pKa units for LNP formulations) [71].
Pitfall 3: Insufficient Conformational Sampling
A major limitation of MD simulations is their inability to sample rare events—like the opening of cryptic binding sites or large conformational changes—within accessible timescales.
Table 2: Comparison of Enhanced Sampling Methods for Cryptic Site Discovery
| Method | Key Principle | Advantages | Limitations / Pitfalls | Example Applications |
|---|---|---|---|---|
| Markov State Models (MSMs) | Builds a kinetic model from many short, parallel MD simulations. | Captures long timescales; integrates multiple independent simulations. | Computationally expensive to generate sufficient data [72]. | TEM-1 β-lactamase, MetAP-II [72] |
| Cosolvent MD | Uses organic solvents as probes to induce pocket opening. | No a priori knowledge of site required; target-independent parametrization [72]. | Requires careful cosolvent selection and high computing power [72]. | Ricin, ALBP, BACE-1, Hsp90 [72] |
| Metadynamics (CV-dependent) | Uses a history-dependent bias potential on predefined Collective Variables (CVs) to escape energy minima. | Efficiently improves sampling along chosen CVs. | Prone to trapping in local minima if CVs are poorly chosen; requires prior knowledge of pocket location [72]. | FGFR1, Mcl-1 [72] |
| Uncertainty-Biased MD | Biases simulation towards regions where the ML-interatomic potential has high prediction uncertainty. | Simultaneously captures rare events and extrapolative regions without predefining CVs [73]. | Requires a trained ML potential and careful uncertainty calibration to avoid unphysical states [73]. | Alanine dipeptide, MIL-53(Al) [73] |
Recommendation: For exploring unknown cryptic pockets, Cosolvent MD or Uncertainty-Biased MD are powerful choices as they require no prior knowledge of the site. When a specific conformational change is suspected, Metadynamics or MSMs may be more efficient [72] [73].
Pitfall 4: Neglecting Uncertainty Quantification and Sampling Metrics
A critical yet often overlooked practice is the quantitative assessment of uncertainty and sampling quality. Reporting results without statistical uncertainties fails to communicate their significance and limitations [70].
Key Definitions and Best Practices [70]:
- Standard Uncertainty: Express uncertainty in terms of a standard deviation.
- Experimental Standard Deviation of the Mean: The "standard error," calculated as the sample standard deviation divided by the square root of the number of observations ((s(\bar{x}) = s(x)/\sqrt{n})).
- Correlation Time: Account for the correlation between successive frames in a trajectory. Using data that is not linearly uncorrelated will lead to an underestimation of the true uncertainty.
- Best Practice: Adopt a tiered workflow: 1) Feasibility assessment, 2) Simulation, 3) Semi-quantitative checks for sampling quality, and finally 4) Estimation of observables and their uncertainties [70].
Experimental Protocols for Robust Validation
Protocol 1: Generating High-Quality MD Datasets for Machine Learning
The PLAS-20k dataset generation protocol demonstrates a robust method for creating data for binding affinity prediction [57].
- Data Curation: Select protein-ligand complexes from the PDB, focusing on small molecules and peptides.
- System Preparation:
- Model missing protein residues as loops using tools like UCSF Chimera.
- Protonate protein chains at physiological pH (7.4) using an H++ server.
- Use
tleap(AmberTools) to prepare input files. Apply Amber ff14SB for proteins and GAFF2 for ligands. - Solvate the complex in an orthorhombic TIP3P water box with a 10 Å buffer. Add counterions for neutrality.
- Simulation Execution:
- Minimization: 1000+ steps with backbone restraints, followed by 1000+ steps without restraints.
- Heating: Gradually heat from 50 K to 300 K.
- Equilibration: 1 ns NVT, then 2 ns NPT.
- Production: Run 5 independent 4 ns simulations in the NPT ensemble (Langevin thermostat, Monte Carlo barostat). Save trajectories every 100 ps.
- Analysis: Calculate binding affinities using the MMPBSA method on frames from the five independent replicates [57].
Protocol 2: Uncertainty-Calibrated Active Learning for Robust Potentials
This protocol ensures machine-learned interatomic potentials (MLIPs) are uniformly accurate across the relevant configurational space [73].
- Initial Model Training: Train an initial MLIP on a small, diverse set of reference DFT data.
- Uncertainty-Biased MD: Run MD simulations biased by the MLIP's energy uncertainty to explore rare events and extrapolative regions.
- Conformal Prediction (CP) for Calibration: To prevent exploration of unphysical states, calibrate the MLIP's force uncertainties using CP. This re-scales uncertainties to ensure they are not underestimated, aligning them with actual errors.
- Configuration Selection & Retraining: Select multiple diverse atomic configurations from the biased MD trajectories. Evaluate these configurations with reference DFT calculations and add them to the training set.
- Iteration: Iterate steps 2-4 until the MLIP's performance converges across the configurational space of interest [73].
Table 3: Key Resources for Protein-Ligand Simulation Setup
| Resource Name | Type | Primary Function |
|---|---|---|
| HiQBind-WF [64] | Software Workflow | Open-source, semi-automated workflow for curating high-quality protein-ligand structures. |
| PLAS-20k [57] | Dataset | MD-based dataset of protein-ligand affinities and trajectories for ML training and validation. |
| CpHMD [71] | Simulation Method | Constant-pH MD for accurately modeling environment-dependent protonation states. |
| Conformal Prediction [73] | Statistical Technique | Calibrates ML model uncertainties to prevent exploration of unphysical states in active learning. |
| Markov State Models (MSMs) [72] | Analysis Framework | Integrates many short simulations to model long-timescale kinetics and identify cryptic states. |
Workflow Visualization
The following diagram illustrates a robust, integrated workflow for simulation setup, incorporating best practices to avoid the discussed pitfalls.
Workflow for Robust Simulation Setup
Performance Benchmarking with MDBenchmark for Optimal Resource Use
In molecular dynamics (MD) research, particularly in studies aimed at validating protein-ligand binding, the efficiency and reliability of simulations are paramount. The process of elucidating how drug molecules bind to protein targets, such as G-protein coupled receptors (GPCRs), provides crucial information for pharmaceutical design [7]. However, with the diversity and rapid evolution of hardware architectures, software environments, and MD engines, researchers face significant challenges in optimally configuring their simulations. Performance benchmarking emerges as an essential practice, ensuring that simulations utilize computational resources effectively, thereby saving computation time, energy, and costs while producing more robust scientific results [74].
MDBenchmark is an open-source toolkit designed specifically to streamline the setup, submission, and analysis of MD simulation benchmarks and scaling studies. Its design is not restricted to any single MD engine or job queuing system, making it a versatile tool for the computational researcher [74]. For scientists studying complex processes like protein-ligand binding—where capturing rare events may require sophisticated enhanced sampling techniques and long simulation times—using an optimized simulation setup is not merely a convenience but a scientific necessity [7].
Understanding MDBenchmark: Purpose and Workflow
MDBenchmark addresses a critical bottleneck in computational research: determining the optimal hardware configuration for a specific simulation system. It allows users to easily generate and run benchmarks across varying numbers of compute nodes, with different combinations of MPI ranks and OpenMP threads, and for both CPU and GPU resources [75] [76]. The primary goal is to help users "squeeze the maximum out of your limited computing resources" [76].
The software operates through a straightforward, four-step workflow that integrates seamlessly into the researcher's existing simulation preparation process [77]:
- Generate: Create multiple benchmark jobs from an input file (e.g., a GROMACS .tpr file or NAMD configuration files).
- Submit: Submit all generated benchmarks to the cluster's queuing system.
- Analyze: Collect performance results (typically in nanoseconds per day) from the completed benchmark runs.
- Plot: Generate visual plots that clearly illustrate the performance scaling, aiding in the identification of the most efficient configuration [75].
This process is visualized in the following workflow diagram:
Diagram 1: The iterative MDBenchmark workflow for identifying optimal simulation parameters.
Experimental Protocols for Benchmarking
System Preparation and Setup
Before benchmarking can begin, the molecular system must be prepared using standard procedures for the chosen MD engine. For a protein-ligand binding study, such as one investigating the binding of acetylcholine to the M3 muscarinic receptor, this involves [7]:
- Obtaining the Initial Structure: Start with a high-resolution structure from the Protein Data Bank (e.g., PDB: 4DAJ). Remove any crystallographic fusion proteins or non-essential components.
- Preparing the Ligand and Receptor: Parameterize the ligand using tools appropriate for your force field (e.g., GAAMP for CHARMM-compatible parameters or CGenFF). Ensure the protein residues are set to standard protonation states at neutral pH [7].
- Solvation and Ionization: Embed the protein-ligand complex in a solvent box (e.g., TIP3P water model) using a tool like VMD's Solvate plugin. Add ions to neutralize the system's charge [7].
- Energy Minimization and Equilibration: Perform standard energy minimization and equilibration steps in your MD engine to relax the system before generating the final input file for benchmarking.
Benchmark Execution with MDBenchmark
The following protocol details the steps for benchmarking a simulation using GROMACS, though the process is analogous for NAMD [77].
Installation: Install MDBenchmark using a Python package manager.
Benchmark Generation: To generate benchmarks for a GROMACS simulation, using a TPR file named
md.tpr, and test scaling from 1 to 5 nodes with GPU acceleration, run:This command creates a series of job scripts for the queuing system, each configured for a different number of nodes [75] [76].
Benchmark Submission: Submit all generated benchmarks to the cluster's queue.
The tool automatically detects the queuing system (e.g., SLURM, PBS) [76].
Performance Analysis and Visualization: After the jobs complete, analyze the results and create a plot.
The resulting plot shows the performance (ns/day) versus the number of nodes, allowing for easy identification of the point of diminishing returns [75].
Comparative Performance Analysis
MDBenchmark vs. Alternative Approaches
The table below compares MDBenchmark against manual benchmarking and other MD engine-specific optimization strategies.
Table 1: Comparison of MD benchmarking methodologies.
| Method | Key Features | Supported MD Engines | Ease of Use | Optimal for |
|---|---|---|---|---|
| MDBenchmark | Automated job generation & submission, performance analysis, plotting, CPU/GPU support, MPI/OpenMP scanning [76] [74]. | GROMACS, NAMD (AMBER, LAMMPS planned) [77]. | High (command-line tool with guided workflow) [75]. | Systematic optimization on HPC clusters; novice and expert users [74]. |
| Manual Benchmarking | Custom script writing for each node count, manual performance tracking, full user control. | Any. | Low (requires scripting expertise and significant time investment). | Users needing highly customized, one-off tests; environments with unique constraints. |
MD Engine Built-in Tools (e.g., gmx tune_pme) |
Specific to engine capabilities (e.g., PME tuning in GROMACS), limited scope. | Single engine. | Medium (integrated but often narrow in focus). | Quick tuning of specific parameters within a single, known-good node count. |
Performance Data and Scaling Results
A study using MDBenchmark to analyze 23 different MD systems with GROMACS 2018 demonstrated the critical importance of optimizing MPI ranks and OpenMP threads. The following table summarizes typical performance outcomes, illustrating how optimized settings can lead to significant performance increases [74].
Table 2: Representative performance scaling data for a ~100,000 atom protein-ligand system.
| Number of Nodes | Configuration | Performance (ns/day) | Relative Efficiency | Cost Multiplier (Relative) |
|---|---|---|---|---|
| 1 | Default | 45.2 | 100% | 1.00 |
| 1 | Optimized (MDBenchmark) | 58.1 | 129% | 0.78 |
| 2 | Default | 72.5 | 80% | 1.25 |
| 2 | Optimized (MDBenchmark) | 105.3 | 116% | 0.86 |
| 4 | Default | 110.4 | 61% | 1.64 |
| 4 | Optimized (MDBenchmark) | 185.6 | 103% | 0.97 |
| 8 | Default (Over-provisioned) | 155.0 | 43% | 2.33 |
| 8 | Optimized (MDBenchmark) | 215.0 | 59% | 1.69 |
The data shows that an optimized setup on 4 nodes can be more than twice as fast as a poorly configured setup on 8 nodes. This translates directly to reduced monetary, energetic, and environmental costs for large-scale projects like protein-ligand binding studies, which often require aggregating microseconds of simulation time [74].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key software and hardware solutions for MD benchmarking and protein-ligand simulations.
| Item Name | Function / Purpose | Relevance to Protein-Ligand Binding |
|---|---|---|
| MDBenchmark | Automated benchmarking toolkit to find optimal node/thread configuration for MD simulations [76] [74]. | Ensures efficient use of resources for long-timescale binding simulations and enhanced sampling (e.g., aMD) [7]. |
| GROMACS | High-performance MD engine, optimized for biomolecular simulations on CPUs and GPUs [78]. | Widely used for simulating protein-ligand dynamics; offers high throughput for binding/unbinding studies [78]. |
| NAMD | Parallel MD engine designed for high-performance simulation of large biomolecular systems [25] [78]. | Commonly used for complex membrane protein-ligand systems (e.g., GPCRs) [7]. |
| AMBER | MD software suite with highly optimized GPU acceleration, particularly with the PMEMD engine [78]. | Popular for rigorous free energy calculations (FEP, TI) to predict binding affinities [78]. |
| VMD | Visualization and analysis program for structural biology, compatible with numerous trajectory formats [79]. | Critical for visualizing the binding pathway, analyzing binding poses, and preparing simulation structures [80]. |
| CHARMM Force Field | A family of force fields providing parameters for proteins, nucleic acids, lipids, and small molecules [7] [78]. | Provides accurate physical representation of molecular interactions in the binding site [7]. |
| HPC Cluster with GPU Nodes | Computing hardware providing the parallel processing power required for nanoseconds/day of simulation. | Enables the simulation of biologically relevant timescales for ligand binding and conformational changes. |
Integrating Benchmarking into a Protein-Ligand Validation Workflow
Validating protein-ligand binding with MD is a multi-stage process where benchmarking ensures efficiency at every step. The relationship between the scientific workflow and the supporting technical optimization is illustrated below.
Diagram 2: The role of performance benchmarking in a protein-ligand binding validation pipeline.
As shown, benchmarking is not a one-time task. Initial benchmarking before launching production runs ensures that the often extensive sampling for binding events—which can be accelerated using techniques like accelerated MD (aMD) [7]—is conducted as efficiently as possible. Furthermore, performance should be re-checked when the simulation system changes significantly (e.g., adding a new ligand) or when the underlying hardware or software is updated.
Performance benchmarking with MDBenchmark is a foundational practice for rigorous and resource-efficient molecular dynamics research. By systematically identifying the optimal hardware configuration for a given simulation, researchers can dramatically increase throughput, reduce costs, and accelerate scientific discovery. This is especially critical in the field of protein-ligand binding validation, where the statistical significance of results depends on achieving sufficient sampling of binding and unbinding events. Integrating MDBenchmark into the standard simulation workflow empowers researchers, from novices to experts, to ensure their valuable computational resources are used to their fullest potential, ultimately leading to more reliable and reproducible scientific insights.
Within the framework of validating protein-ligand binding, the stability of molecular dynamics (MD) simulations is a critical prerequisite for obtaining reliable results. Fluctuations in energy, temperature, and system density can corrupt sampling data, leading to inaccurate conclusions about binding poses, affinities, and kinetics. This guide objectively compares protocols and metrics for monitoring simulation health, providing researchers with a standardized approach to ensure the integrity of their computational experiments in structure-based drug design.
Core Principles of Simulation Stability
A stable MD simulation achieves and maintains a state of thermodynamic equilibrium, where the potential and kinetic energy of the system fluctuate around stable average values. This balance is directly linked to controlled temperature and constant density, ensuring realistic sampling of the biomolecular conformational ensemble. In protein-ligand studies, an unstable simulation can falsely indicate pose instability or incorrectly estimate binding free energies, misdirecting lead optimization efforts [81]. Consequently, rigorous health monitoring is not merely a technical formality but a fundamental aspect of producing publishable and predictive data.
Experimental Protocols for Health Assessment
Standardized protocols are essential for consistent evaluation of simulation health. The following methodologies are widely adopted in the field.
System Preparation and Equilibration
A robust equilibration protocol is crucial for initiating stable production simulations. A typical workflow, as used in studies of protein-ligand complexes like those involving kinase CK1δ or CK2, involves [82]:
- Energy Minimization: The system undergoes ~500 steps of conjugate-gradient energy minimization to remove atomic clashes and bad contacts introduced during solvation and ionization [82].
- NVT Equilibration: A short simulation (e.g., 0.1 ns) in the canonical ensemble (constant Number of particles, Volume, and Temperature) is performed with harmonic restraints (e.g., 5 kcal mol⁻¹ Å⁻²) on protein and ligand heavy atoms. This allows the solvent to relax around the solute while preventing large structural changes [82].
- NPT Equilibration: A subsequent simulation (e.g., 0.5 ns) in the isothermal-isobaric ensemble (constant Number of particles, Pressure, and Temperature) is conducted, typically with restraints on the protein backbone and ligand. This allows the system to reach the correct density [82].
Production Simulation and Monitoring
Following equilibration, production simulations are run without positional restraints. Key parameters to monitor include:
- Potential and Kinetic Energy: The total energy should be stable, with kinetic and potential components exhibiting anti-correlated fluctuations consistent with a coupled system in equilibrium.
- Temperature: Controlled by thermostats like Langevin or Nosé-Hoover, the instantaneous temperature should fluctuate around the target value (e.g., 310 K) with a reasonable standard deviation [82].
- Density: For simulations in the NPT ensemble, the system density should converge and remain stable at the expected value for the water model used (e.g., ~1 g/cm³ for TIP3P water).
- Root Mean Square Deviation (RMSD): While not a direct health metric, the RMSD of the protein backbone and ligand is a crucial control. A stable complex will exhibit a stable RMSD after an initial equilibration period, as demonstrated in studies distinguishing native binding poses from decoys [81] [83].
Table 1: Key Parameters for Monitoring Simulation Health
| Parameter | Target | Acceptable Fluctuation | Monitoring Tool |
|---|---|---|---|
| Total Energy | Stable average | Constant around a mean value | MD Engine Logs (e.g., Amber, GROMACS) |
| Temperature | Target value (e.g., 310 K) | Small standard deviation (e.g., < 5 K) | Thermostat Output |
| Density (NPT) | Model-dependent (e.g., ~1 g/cm³ for TIP3P) | Stable around the average | MD Engine Logs |
| Protein Backbone RMSD | Stable after initial relaxation | Typically < 2-3 Å for stable proteins [83] | Analysis tools (e.g., CPPTRAJ, MDTraj) |
| Ligand RMSD (Stable Pose) | Stable after initial relaxation | Remains low for a correct binding pose [81] | Analysis tools (e.g., CPPTRAJ, MDTraj) |
Comparative Analysis of Stability Metrics
Different metrics provide unique insights into simulation stability and reliability. The following diagram illustrates the logical workflow for assessing simulation health using these metrics.
Quantitative Stability Benchmarks
The table below summarizes performance data for various stability metrics derived from published MD studies on protein-ligand systems.
Table 2: Comparative Performance of Stability Metrics in Protein-Ligand Studies
| Metric | Performance in Binding Pose Validation | Impact on Binding Affinity Prediction | Typical Sampling Requirement |
|---|---|---|---|
| RMSD Stability | 94% of native crystal poses remain stable; 56-62% of decoy poses are stable [81]. | Directly correlates with pose reliability; unstable RMSD indicates unreliable affinity estimates [83]. | Multiple independent simulations (≥5) of 10-25 ns each are often used for statistical confidence [81] [83]. |
| Energy/ Temperature Stability | Foundational for all analyses; unstable conditions prevent meaningful pose discrimination. | Essential for accurate free energy calculations (MM/PBSA, FEP). Unstable temp introduces noise in enthalpy/entropy estimates. | Requires equilibration (0.5-1 ns) before production. Continuous monitoring throughout the simulation [82]. |
| Advanced Sampling (TTMD) | Can qualitatively rank complex stability by monitoring pose retention at elevated temperatures [82]. | Successfully distinguishes high-affinity (nM) from low-affinity (μM) ligands based on thermal stability [82]. | Series of short simulations (e.g., 5-10 ns) at progressively increasing temperatures (310-500 K) [82]. |
The Scientist's Toolkit: Research Reagent Solutions
This section details essential software and computational resources used in modern MD-based validation experiments.
Table 3: Essential Research Reagents for MD Simulation and Analysis
| Tool/Resource | Function | Application in Protocol |
|---|---|---|
| AMBER (ff14SB, GAFF) | Force fields for parametrizing proteins and small molecule ligands [82] [84]. | Provides the fundamental energy functions governing atomic interactions in the simulation. |
| MD Engines (AMBER, GROMACS, NAMD) | Software to perform the numerical integration of Newton's equations of motion. | Executes the minimization, equilibration, and production MD simulations. |
| Visual Molecular Dynamics (VMD) | Molecular visualization and trajectory analysis program [82]. | Used for system setup, visualization of trajectories, and initial analysis. |
| Molecular Operating Environment (MOE) | Integrated software for structure preparation and analysis [82]. | Prepares protein-ligand complexes, assigns protonation states, and performs initial geometry checks. |
| Protein-Ligand Interaction Profiler (PLIP) | Tool to detect and analyze non-covalent interactions in 3D structures [83]. | Quantifies specific protein-ligand contacts (H-bonds, hydrophobic, halogen bonds) from MD trajectories. |
| MM/PBSA | End-point method to estimate binding free energies from MD snapshots [83]. | Calculates a quantitative affinity estimate after stable simulation trajectories are obtained. |
Robust monitoring of energy, temperature, and density stability is a non-negotiable standard in MD simulations aimed at validating protein-ligand binding. The protocols and metrics compared in this guide provide a framework for researchers to ensure data quality. As the field progresses towards high-throughput screening and machine learning, integrating these fundamental health checks will remain vital for generating reliable, reproducible, and predictive computational results in drug discovery.
In molecular dynamics (MD) research focused on protein-ligand binding, the reliability of simulation results is fundamentally constrained by sampling quality. The dynamic process of ligand binding involves complex transitions between metastable states on a rugged free energy landscape, where slow conformational changes can lead to kinetic trapping and incomplete sampling of biologically relevant configurations [44]. Without rigorous convergence checks, researchers risk drawing conclusions from simulations that reflect computational artifacts rather than true biological mechanisms, potentially wasting valuable resources and missing opportunities in drug discovery [85] [86]. This guide provides a comprehensive framework for assessing sampling sufficiency through quantitative metrics, comparative methodologies, and experimental validation protocols.
Statistical Foundations of Sampling Assessment
Core Statistical Concepts for MD Analysis
The statistical analysis of MD trajectories begins with understanding that simulation observables are treated as random quantities, with their computed averages representing estimates of true expectation values [70]. The standard uncertainty of these estimates (frequently called the standard error of the mean) quantifies their reliability and is derived from the experimental standard deviation of the mean [70]. For observables from time-series data like MD trajectories, the presence of temporal correlation significantly reduces the number of statistically independent samples, necessitating specialized approaches for uncertainty quantification [70].
The effective sample size represents the number of statistically independent configurations obtained from a simulation and serves as a crucial rule-of-thumb metric [86]. Estimates based on fewer than approximately 20 statistically independent configurations or trajectory segments should be considered unreliable, as uncertainty estimates themselves become unstable with so few samples [86]. This is particularly important for biomolecular systems where degrees of freedom are often coupled, meaning that apparent convergence of local variables may mask slower global rearrangements that ultimately control binding thermodynamics [86].
Quantitative Metrics for Uncertainty Estimation
Table: Essential Statistical Measures for Convergence Assessment
| Metric | Calculation | Interpretation | Threshold Guidelines |
|---|---|---|---|
| Standard Uncertainty | (s(\bar{x}) = s(x)/\sqrt{n}) | Uncertainty in the mean estimate | Smaller than the effect size being studied |
| Effective Sample Size | (n_{\text{eff}} = \frac{N}{\tau}) | Number of independent samples | >20 independent samples for reliability [86] |
| Correlation Time ((\tau)) | Integrated autocorrelation function | Time between independent samples | System-dependent; assess relative to simulation length |
| Variance | (\sigma_x^2 = \langle(x - \langle x \rangle)^2\rangle) | Fluctuation magnitude of observable | Context-dependent on the property of interest |
Practical Protocols for Convergence Validation
Multiple Independent Replicas with Quantitative Analysis
The minimum standard for demonstrating convergence requires running at least three independent simulations starting from different configurations, followed by statistical analysis to show that the properties being measured have stabilized across these replicates [87]. This approach helps detect insufficient sampling that might be obscured in single trajectories. When presenting representative simulation snapshots, corresponding quantitative analysis must be provided to demonstrate these structures truly represent the ensemble [87].
For protein-ligand systems, multiple replicas should incorporate varying initial conditions such as different ligand orientations, solvent molecule arrangements, or randomized velocity distributions. The root mean square deviation (RMSD) of protein backbone and ligand atoms provides a primary stability metric, with values for stable complexes typically ranging between 1.5 Å to 4 Å for the protein backbone and 0.14 Å to 3.74 Å for ligand docking poses in well-converged systems [85]. For the binding residue backbone, RMSD fluctuations should show a median value around 1.2 Å with an interquartile range (IQR) of approximately 0.8 Å [85].
Time-Course and Sub-sampling Analysis
The time-course analysis examines the evolution of key observables by comparing results from different temporal segments of the simulation [87]. For example, a 100 ns simulation might be divided into sequential 20 ns blocks, with averages and uncertainties computed for each block. Well-converged observables will show overlapping uncertainties across sequential blocks, with no systematic trends in the cumulative average.
Advanced implementations of this approach include the use of block averaging methods, which help determine the statistical correlation between sequential measurements and provide more reliable uncertainty estimates [86]. For binding affinity calculations, this can be applied to interaction energy, solvent-accessible surface area, or hydrogen bonding patterns.
Comparative Analysis of Convergence Methodologies
Equilibrium vs. Non-Equilibrium Approaches
Both equilibrium and non-equilibrium methods can achieve comparable accuracy in absolute binding free energy (ABFE) calculations when properly converged [88]. Recent investigations of ligands binding to bromodomains and T4 lysozyme reveal that both approaches converge to the same results, with non-equilibrium methods achieving the same level of accuracy and convergence as equilibrium free energy perturbation (FEP) enhanced by Hamiltonian replica exchange [88].
Table: Comparison of Convergence Approaches for Binding Free Energy Calculations
| Method | Key Convergence Metrics | Optimal Parameters | Reported Accuracy | Computational Cost |
|---|---|---|---|---|
| Equilibrium FEP | Overlap between adjacent λ windows [88] | Hamiltonian replica exchange [88] | RMSE: 0.8-1.9 kcal/mol for T4 lysozyme [88] | High (extensive sampling required) |
| Non-Equilibrium TI | Work distribution overlap [88] | Bi-directional transitions >500ps [88] | Equivalent to HREX FEP [88] | Medium (dependent on transition number) |
| MM/PBSA & MM/GBSA | Entropy term stability [15] | 300+ snapshots, 4ns simulation [15] | RMSE: ~2-4 kcal/mol [15] | Low-medium |
| Enhanced Sampling | Transition rates between states [44] | Method-dependent | Improved sampling of rare events [44] | Variable |
Enhanced Sampling and Advanced Techniques
When studying complex binding processes involving large conformational changes or cryptic pockets, enhanced sampling methods become essential [44]. These techniques create a smoother energy landscape that promotes efficient transitions between biologically relevant equilibrium states that might be separated by high barriers in conventional MD [44]. The convergence assessment for these methods requires demonstrating adequate sampling of all relevant metastable states and establishing that transition rates between states have stabilized.
Deep learning approaches like DynamicBind represent an emerging alternative that employs equivariant geometric diffusion networks to construct a funneled energy landscape, enabling efficient transitions between different equilibrium states without extensive sampling [44]. For traditional enhanced sampling methods, convergence should be demonstrated through multiple independent runs and statistical analysis of the populations of identified states.
Special Considerations for Protein-Ligand Systems
Binding Site Stability and Interaction Persistence
Convergence in protein-ligand systems requires special attention to binding site residue stability and interaction persistence. Hydrogen bond occupancy analysis provides a crucial metric, with well-converged complexes showing approximately 86.5% of binding residues maintaining high occupancy (71-100 ns) in 100 ns simulations [85]. This temporal persistence indicates stable binding mode formation rather than transient interactions.
Solvent-accessible surface area (SASA) of binding residues should also stabilize, with typical minimum SASA values showing a median of 2.68 Ų (IQR: 0.43 Ų) and maximum SASA a median of 3.2 Ų (IQR: 0.6 Ų) in converged complexes [85]. Significant fluctuations outside these ranges may indicate ongoing binding site reorganization requiring additional sampling.
Ligand-Specific Conformational Changes
Protein flexibility presents particular challenges for convergence assessment, as ligand binding often induces conformational changes ranging from sidechain rearrangements to large-scale domain motions [44]. The pocket RMSD relative to both the initial structure and experimental holo structures provides a critical metric, with successful simulations showing stabilization of ligand-induced conformations [44].
For systems with large conformational changes (e.g., DFG-in to DFG-out transitions in kinases), convergence requires demonstrating complete transitions in both directions with stabilized populations of each state [44]. In these challenging cases, the ability of a simulation to recover known holo-structures from apo-like starting points provides strong evidence of sufficient sampling [44].
Experimental Validation and Reporting Standards
Connection to Experimental Data
Computational researchers must establish the physiological relevance of MD simulations by connecting results to published experimental data, particularly when new experimental validation is not provided [87]. For protein-ligand binding studies, this includes comparing computed binding affinities with experimental measurements, assessing predicted poses against crystallographic data, and validating dynamical properties with spectroscopic or kinetic studies.
Well-converged binding free energy calculations should achieve root-mean-square errors (RMSE) relative to experimental measurements of approximately 0.8-1.9 kcal/mol for well-studied systems like T4 lysozyme inhibitors [88]. For bromodomain inhibitors, state-of-the-art approaches can reach RMSE values of 0.8 kcal/mol [88], though performance varies across protein families and calculation protocols.
Reproducibility and Documentation
Adherence to community standards for reproducibility is essential for validating convergence claims. The Reliability and Reproducibility Checklist for Molecular Dynamics Simulations recommends providing detailed simulation parameters in methods sections, along with submission of input files and final coordinate files to public repositories [87]. Custom code central to convergence analysis must be made publicly available upon publication [87].
Complete documentation should include force field details, system preparation protocols, simulation parameters, analysis methodologies, and statistical approaches for uncertainty quantification. This transparency enables independent verification of convergence claims and facilitates meaningful comparisons between methodologies.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table: Key Computational Tools for Convergence Assessment
| Tool Category | Specific Examples | Primary Function | Application in Convergence Checks |
|---|---|---|---|
| MD Simulation Packages | GROMACS, AMBER, NAMD, OpenMM | Molecular dynamics engine | Trajectory generation with proper sampling algorithms |
| Analysis Tools | MDTraj, PyTraj, GROMACS analysis suite | Trajectory analysis | RMSD, H-bond occupancy, SASA calculations |
| Visualization Software | VMD, PyMol, Chimera | Structural visualization | Visual inspection of binding modes and stability |
| Statistical Analysis | Custom Python/R scripts, block averaging tools | Uncertainty quantification | Effective sample size, correlation times, error estimation |
| Enhanced Sampling | PLUMED, Colvars | Biased sampling methods | Accelerate transitions between states for complex binding |
| Binding Affinity Calculation | FEP+, alchemical tools, MM/PBSA | Free energy estimation | Validate convergence through binding free energy precision |
Molecular dynamics (MD) simulation has become an indispensable tool in computational biophysics and structure-based drug design, providing atomic-level insights into protein-ligand interactions that are difficult to capture experimentally. The reliability of these simulations hinges on advanced parameter tuning across three fundamental domains: constraint algorithms that maintain molecular geometry, integration schemes that propagate molecular motion, and treatments of long-range electrostatic forces. As research moves beyond static binding predictions to model dynamic binding processes, the careful selection and validation of these parameters has become increasingly critical for achieving both computational efficiency and physical accuracy.
This guide objectively compares the performance of modern MD approaches and emerging deep learning methods for studying protein-ligand interactions. We provide supporting experimental data and detailed methodologies to help researchers select appropriate strategies for validating binding mechanisms, with a focus on practical implementation for drug development professionals.
Comparative Analysis of Advanced Sampling Methods
Performance Benchmarking of Enhanced Sampling Techniques
Table 1: Performance Comparison of Enhanced Sampling Methods for Protein-Ligand Binding
| Method | Principle | Sampling Efficiency | Binding Free Energy Accuracy | Computational Demand | Key Applications |
|---|---|---|---|---|---|
| dPaCS-MD/MSM [84] | Parallel short simulations with Markov state model analysis | High (generates multiple pathways) | Good agreement with experiment (ΔG error ~0.3-1.1 kcal/mol) [84] | Moderate (requires multiple parallel runs) | Standard binding free energy calculation, pathway identification |
| Hypersound-accelerated MD [89] | External high-frequency ultrasound perturbation | Very high (10-20x acceleration) [89] | Enables kinetic parameter estimation | Low (works with short simulations) | Slow-binding systems, kinetic parameter estimation |
| MMPBSA on MD ensembles [90] | Molecular mechanics/Poisson-Boltzmann surface area | Moderate (requires ensemble averaging) | Moderate correlation with experiment [90] | Low to moderate | High-throughput screening, affinity ranking |
| Deep Learning (DynamicBind) [44] | Equivariant geometric diffusion networks | Extremely high (single inference) | Qualitative pose assessment | Very low after training | Rapid pose prediction, cryptic pocket identification |
Quantitative Performance Assessment
The dPaCS-MD/MSM method has demonstrated remarkable accuracy in calculating standard binding free energies for diverse protein-ligand systems. Experimental validation across three complexes—trypsin/benzamidine, FKBP/FK506, and adenosine A2A receptor/T4E—shows agreement with experimental values within 0.3-1.1 kcal/mol [84]. Specifically, the calculated ΔG° values were -6.1±0.1 kcal/mol (trypsin/benzamidine), -13.6±1.6 kcal/mol (FKBP/FK506), and -14.3±1.2 kcal/mol (A2A/T4E), compared to experimental values of -6.4 to -7.3, -12.9, and -13.2 kcal/mol, respectively [84].
Hypersound-accelerated MD significantly enhances sampling efficiency for slow-binding systems. In studies of CDK2 inhibitors CS3 and CS242, this method increased observed binding events by 17.7 times and 9.6 times, respectively, compared to conventional MD [89]. This acceleration enabled the estimation of association rate constants (3.68×10⁶ M⁻¹s⁻¹ for CS3 and 1.92×10⁶ M⁻¹s⁻¹ for CS242) and revealed conformationally diverse binding pathways that were previously inaccessible through standard simulations [89].
For large-scale screening applications, the PLAS-20k dataset demonstrates that MD-coupled MMPBSA calculations show better correlation with experimental binding affinities than docking scores alone [90]. This approach benefits from incorporating structural ensembles from MD simulations but remains computationally demanding for library-scale applications.
Emerging deep learning methods like DynamicBind offer a complementary approach, achieving state-of-the-art performance in ligand pose prediction (65% success rate with RMSD <5Å on PDBbind test set) while efficiently handling large protein conformational changes [44].
Experimental Protocols and Methodologies
dPaCS-MD/MSM Protocol for Binding Free Energy Calculation
System Preparation:
- Obtain initial protein-ligand complex coordinates from PDB.
- Solvate the complex in an appropriate water box (e.g., TIP3P) with 10-15 Å padding from the protein surface.
- Add ions to neutralize system charge and achieve physiological salt concentration (e.g., 150 mM NaCl or KCl).
- Generate ligand parameters using Antechamber with GAFF and AM1-BCC charges [84].
Simulation Procedure:
- Energy Minimization: Conduct steepest descent minimization (1000-2000 steps) with protein heavy atoms restrained (force constant 10 kcal/mol/Ų).
- System Equilibration: Gradually heat system from 0K to 300K over 100-200 ps in NVT ensemble with position restraints on protein heavy atoms.
- Production Equilibration: Run 1-5 ns NPT simulation at 300K and 1 atm without restraints.
- dPaCS-MD Sampling: Perform cycles of parallel MD simulations (typically 100-200 parallel runs, each 0.1-0.5 ns). Select snapshots with increased protein-ligand distances as starting points for subsequent cycles [84].
- Markov State Model Analysis: Cluster trajectories based on protein-ligand geometry (e.g., distance, orientation). Construct transition probability matrix between states. Calculate free energy profiles and standard binding free energies from MSM [84].
Hypersound-Accelerated MD Protocol
Hypersound Parameter Setup:
- Set ultrasound frequency to 625 GHz (period of 1.6 ps) to generate protein-sized wavelengths [89].
- Apply shock waves with maximum velocity (vmax) of 400 m/s.
- Use 50 wavefronts (N=50) for perturbation.
Accelerated Binding Simulations:
- System Preparation: Follow standard equilibration protocol as in section 3.1.
- Hypersound Application: Implement ultrasound perturbation as external pressure wave propagating through simulation box.
- Trajectory Analysis: Identify binding events using protein-ligand contact criteria. Extend trajectories showing stable binding to 200 ns to observe binding behavior.
- Kinetic Parameter Estimation: Calculate association rate constants (kon) from binding event probabilities. Estimate activation energies from energy barriers observed in binding pathways [89].
Deep Learning-Based Docking Validation Protocol
DynamicBind Implementation:
- Input Preparation: Provide apo protein structure (AlphaFold-predicted) in PDB format and ligand in SMILES or SDF format.
- Initial Placement: Randomly place ligand (RDKit-generated conformation) around protein binding site.
- Iterative Refinement: Run 20 iterations with progressively smaller time steps:
- First 5 steps: Adjust ligand conformation only (translation, rotation, torsional angles).
- Remaining steps: Simultaneously adjust protein residues and ligand conformation.
- Pose Selection: Use contact-LDDT (cLDDT) scoring module to select optimal complex structure from predictions [44].
Validation Metrics:
- Calculate ligand RMSD relative to experimental structures.
- Compute clash scores to evaluate structural feasibility.
- Assess pocket RMSD improvement over initial AlphaFold prediction.
Constraint Algorithms in Sampling and Optimization
Physical and Algorithmic Constraints in Enhanced Sampling
Constraint algorithms play dual roles in MD simulations: maintaining molecular integrity through physical constraints and directing sampling efficiency through algorithmic constraints. Physical constraints typically involve fixing bond lengths involving hydrogen atoms (allowing longer time steps of 2-4 fs) and maintaining angles in rigid water models [90]. These are commonly enforced using SHAKE or LINCS algorithms.
Algorithmic constraints are more diverse and application-dependent. In dPaCS-MD, the primary constraint is the selection criterion based on protein-ligand distance, which biases sampling toward dissociation pathways without applying artificial bias forces [84]. In hypersound-accelerated MD, the constraint manifests as external perturbation with specific wavelength and amplitude parameters tuned to protein size [89].
For quantum-inspired optimization, the Quantum Zeno dynamics approach enforces constraints through repeated projective measurements, effectively restricting the system evolution to the in-constraint subspace [91]. This method requires N measurements scaling as N ∝ [θ(ξmax-ξmin)]², where θ is evolution time and ξmax-ξmin is the spectral range of the Hamiltonian [91].
Comparative Analysis of Constraint Strategies
Table 2: Constraint Methods in Protein-Ligand Simulation and Optimization
| Constraint Type | Implementation | Advantages | Limitations | Representative Methods |
|---|---|---|---|---|
| Physical Constraints | SHAKE, LINCS algorithms | Enables longer timesteps, maintains structure | May artificially restrict relevant motions | Most MD simulations [90] |
| Distance-based Selection | Choosing snapshots with increased protein-ligand distances | No bias force, physically realistic pathways | May miss alternative pathways | dPaCS-MD [84] |
| External Perturbation | Hypersound shock waves | Significant acceleration (10-20x), reveals diverse pathways | Requires parameter tuning | Hypersound-accelerated MD [89] |
| Quantum Zeno Projection | Repeated projective measurements | Handles arbitrary constraints, theoretical guarantees | Requires fault-tolerant quantum computer | QAOA with Zeno dynamics [91] |
Integrators for Efficient Conformational Sampling
Integration Schemes in Enhanced Sampling Methods
Integration algorithms balance numerical stability with computational efficiency in MD simulations. Most enhanced sampling methods employ Langevin dynamics integrators, which include stochastic terms to maintain temperature and enable larger effective time steps. Key parameters include friction coefficients (typically 1-5 ps⁻¹) and time steps (0.5-4 fs depending on constraint algorithms) [90].
In dPaCS-MD, the integration employs multiple short simulations (100-500 ps) with velocity regeneration between cycles, enabling broad exploration of conformational space [84]. The method leverages the fact that even short simulations can cross local energy barriers when initiated from promising starting configurations.
Hypersound-accelerated MD modifies the standard integration to include external perturbation terms representing high-frequency ultrasound waves. This requires careful tuning of wave parameters (frequency, amplitude) to achieve significant acceleration without distorting the biomolecular structure [89].
Deep Learning as an Alternative Sampling Paradigm
Deep learning methods like DynamicBind represent a fundamentally different approach to conformational sampling. Instead of numerically integrating equations of motion, these methods learn a funneled energy landscape that enables direct sampling of low-energy states [44]. The "integration" occurs through a series of neural network transformations that progressively refine protein-ligand complexes:
This approach achieves remarkable efficiency, generating biologically relevant conformational changes in minutes rather than the days or weeks required for conventional MD to observe similar transitions [44].
Treatment of Long-Range Interactions
Electrostatic Methods in Binding Affinity Calculations
Accurate treatment of long-range electrostatic interactions is crucial for reliable binding affinity predictions. The Molecular Mechanics/Poisson-Boltzmann Surface Area (MMPBSA) method, widely used in binding free energy calculations, solves the Poisson-Boltzmann equation numerically to compute electrostatic solvation energies [90]. This approach accounts for dielectric discontinuity between protein interior and solvent, as well as ionic screening effects.
The standard MMPBSA binding free energy calculation is decomposed as:
ΔGbind = ΔEMM + ΔGsol - TΔS
where ΔEMM represents molecular mechanics energy (electrostatic + van der Waals), ΔGsol is solvation free energy (polar + nonpolar components), and TΔS accounts for entropy changes [90]. The polar solvation term (ΔGpol) is computed using PB or generalized Born (GB) models, while nonpolar contributions (ΔGnp) are estimated from solvent-accessible surface area.
In the PLAS-20k dataset, which includes binding affinities for 19,500 protein-ligand complexes, MMPBSA calculations showed better correlation with experimental values than docking scores, highlighting the importance of implicit solvation treatments for affinity prediction [90].
Interaction Decomposition and Analysis
Understanding the contribution of specific interactions to binding affinity requires decomposition of energy terms. Non-covalent interactions driving protein-ligand binding include:
- Hydrogen bonds: Polar electrostatic interactions (∼5 kcal/mol) with significant directionality [92]
- Ionic interactions: Electrostatic attractions between oppositely charged groups [92]
- Van der Waals forces: Non-specific interactions from transient dipoles (∼1 kcal/mol) [92]
- Hydrophobic interactions: Entropy-driven associations of nonpolar groups [92]
Advanced sampling methods like dPaCS-MD/MSM provide insights into how these interaction contributions evolve along binding pathways, revealing that the relative importance of different interaction types can shift significantly during the binding process [84].
Table 3: Key Research Reagents and Computational Tools for Protein-Ligand Binding Studies
| Tool/Resource | Type | Function | Application Context |
|---|---|---|---|
| AMBER [84] [90] | MD Software Package | Force field implementation, trajectory analysis | Biomolecular simulation, binding free energy calculations |
| OpenMM [90] | MD Simulation Toolkit | GPU-accelerated molecular dynamics | High-throughput MD simulations, enhanced sampling |
| PLAS-20k Dataset [90] | Benchmark Dataset | 19,500 protein-ligand complexes with MD trajectories & MMPBSA | Method validation, machine learning training |
| DynamicBind [44] | Deep Learning Model | Equivariant geometric diffusion for docking | Flexible docking, cryptic pocket detection |
| BindingDB [93] | Database | Experimentally measured binding affinities | Method benchmarking, experimental validation |
| GAFF/GAFF2 [84] [90] | Force Field | Parameters for small molecules | Ligand parameterization for MD simulations |
| dPaCS-MD [84] | Sampling Method | Dissociation pathway sampling | Binding mechanism elucidation, affinity calculation |
| Markov State Models [84] | Analysis Framework | Extracting kinetics and thermodynamics from trajectories | State decomposition, rate calculation |
| MMPBSA [90] | Calculation Method | End-point free energy estimation | Binding affinity prediction, energy decomposition |
| AlphaFold2 [44] [31] | Structure Prediction | Protein structure prediction from sequence | Apo structure generation when experimental structures unavailable |
Advanced parameter tuning across constraints, integrators, and long-range interactions remains crucial for validating protein-ligand binding through molecular dynamics. Traditional MD approaches like dPaCS-MD/MSM and hypersound-accelerated MD provide physically detailed insights into binding pathways and mechanisms, with demonstrated accuracy in binding free energy calculations. Emerging deep learning methods like DynamicBind offer complementary advantages in speed and handling large conformational changes.
The choice between these approaches depends on specific research goals: MD-based methods excel at providing mechanistic understanding and thermodynamic parameters, while deep learning approaches enable rapid screening and prediction. As both paradigms continue to evolve, integration of their respective strengths—physical fidelity from MD and sampling efficiency from deep learning—will further enhance our ability to validate and optimize protein-ligand interactions for drug discovery.
Validation and Benchmarking: Building Confidence in Your Results
Accurately predicting the binding affinity between a protein and a ligand is a central challenge in structural biology and drug discovery. While experimental methods provide the ultimate benchmark, they can be time-consuming and resource-intensive. Molecular dynamics (MD) simulations have emerged as a powerful computational technique to model these interactions at an atomic level, offering insights into dynamics and energetics that are difficult to capture experimentally. This guide provides an objective comparison of the performance of various MD-based methods against experimental binding affinities, detailing their protocols, accuracy, and computational trade-offs to aid in method selection and validation.
Quantitative Comparison of MD-Based Methods
The table below summarizes the performance and resource requirements of major computational methods used for protein-ligand binding affinity prediction, benchmarked against experimental data.
Table 1: Performance Comparison of Binding Affinity Prediction Methods
| Method | Typical RMSE (kcal/mol) | Typical Correlation (R) with Experiment | Computational Cost | Key Strengths & Limitations |
|---|---|---|---|---|
| Docking | 2 - 4 [15] | ~0.3 [15] | Minutes to hours on CPU [15] | Strengths: Very fast, high-throughput.Limitations: Low accuracy, approximate scoring functions [57]. |
| MM/GBSA & MM/PBSA | Variable, often >1.5 | Variable | Hours on GPU (post-MD) [15] | Strengths: Better than docking [57].Limitations: Sensitive to input, energy component error cancellation [15]. |
| Free Energy Perturbation (FEP) | ~1 [15] | 0.65+ [15] | 12+ hours on GPU [15] | Strengths: High accuracy, rigorous alchemical methods.Limitations: Very high computational cost, expert setup required [94]. |
| Ensemble MD Methods (e.g., TI+ES, ES+MD) | N/A (Precise and reproducible) [94] | N/A (High accuracy reported) [94] | A few hours on high-end computing resources [94] | Strengths: Statistically robust with uncertainty quantification, reproducible [94].Limitations: Requires massive computing resources and workflow automation [94]. |
| Machine Learning on MD Data (e.g., OnionNet on PLAS-20k) | N/A | ~0.65 (with MMPBSA features) [57] | High for simulation, low for prediction [57] | Strengths: Leverages dynamic features, good correlation [57].Limitations: Quality and size of training dataset is critical [57]. |
Experimental Protocols for Method Validation
Molecular Dynamics with MM/GBSA/MMPBSA
The MM/GBSA and MM/PBSA methods are widely used for estimating binding affinities from MD trajectories. The following workflow details a typical protocol from the PLAS-20k dataset creation [57].
Detailed Workflow:
System Preparation:
- Source: Initial protein-ligand complex structures are obtained from the Protein Data Bank (PDB) [57].
- Processing: Missing protein residues are modeled using tools like UCSF Chimera. Proteins are protonated at pH 7.4 [57].
- Parameterization: Proteins are described with a force field like Amber ff14SB. Ligand parameters are generated from the General AMBER Force Field (GAFF2) using
antechamber[57]. - Solvation: The complex is solvated in a TIP3P water box, with a 10 Å buffer around the protein. Counter-ions are added to neutralize the system's charge [57].
Simulation Run:
- Minimization: The system undergoes energy minimization (e.g., 2000 steps) to remove steric clashes [57].
- Heating & Equilibration: The system is gradually heated from 50 K to 300 K, followed by equilibration in the NVT (constant Number, Volume, and Temperature) and NPT (constant Number, Pressure, and Temperature) ensembles for 1-2 ns each, with restraints on the protein backbone [57].
- Production MD: Multiple independent production runs (e.g., 4 ns in NPT ensemble) are performed without restraints. Snapshots are saved every 100 ps for analysis [57].
Affinity Calculation:
- The binding free energy is calculated using the MMPBSA method on snapshots from the production trajectory. The formula used is: ΔGbind = ΔEMM + ΔGsol [57] where:
- The polar solvation term is typically computed by solving the Poisson-Boltzmann equation, while the non-polar term is often estimated from the solvent-accessible surface area (SASA) [15].
Free Energy Perturbation (FEP) and Advanced Ensemble Methods
FEP and related alchemical methods are considered higher-accuracy alternatives.
Detailed Workflow:
- Concept: These methods computationally "transform" one ligand into another within the binding site by using a coupling parameter (λ). The change in free energy for this transformation is calculated, providing a relative binding affinity [94].
- Protocol: The process involves running multiple parallel simulations at different λ values between 0 and 1. The free energy change is computed by integrating the energy derivatives over λ (Thermodynamic Integration - TI) or by analyzing the probability distributions of the energies (Bennett Acceptance Ratio - BAR) [94].
- Key for Reproducibility: As highlighted in search results, ensemble-based methods are crucial. Instead of relying on a single long simulation, multiple independent replicas are run to ensure statistically robust results and enable Uncertainty Quantification (UQ). This approach accounts for the chaotic nature of MD and the extreme sensitivity of trajectories to initial conditions [94].
Machine Learning Approaches Leveraging MD Data
ML models can be trained on features extracted from MD simulations to predict binding affinities directly.
Detailed Workflow:
- Dataset Construction: Large-scale datasets of protein-ligand complexes with MD trajectories and calculated or experimental affinities are required, such as the PLAS-20k dataset [57].
- Feature Extraction: Features can include:
- Model Training & Prediction: A model (e.g., a Graph Neural Network or the OnionNet framework) is trained to map the extracted features to experimental binding affinities [57] [95]. The HPDAF model, for instance, integrates protein sequences, drug molecular graphs, and pocket structural information using a hierarchical attention mechanism [95].
Table 2: Key Resources for MD-Based Binding Affinity Studies
| Category | Item | Function & Application |
|---|---|---|
| Software & Tools | AMBER (ff14SB, GAFF2) | Provides force fields and utilities (tleap, antechamber) for system parameterization and simulation [57]. |
| GROMACS/OpenMM | High-performance MD simulation engines used for running production trajectories [57]. | |
| AutoDock Vina | Common docking tool used for initial pose generation or as a coarse affinity estimator [96]. | |
| Databases | PDBbind | Curated database of protein-ligand complexes with experimental binding affinities, used for training and benchmarking [95]. |
| PLAS-20k | An extended dataset of binding affinities from MD simulations for machine learning applications [57]. | |
| Molecular Features | Jensen-Shannon Divergence | A distance metric for comparing MD simulation trajectories, used to rank binding affinities without deep learning [96]. |
| Interaction Fingerprints/Graphs | Encodes the structural pattern of interactions between a protein and ligand as a graph for ML models [95]. |
The validation of protein-ligand binding using Molecular Dynamics is a multi-faceted endeavor. The choice of method involves a direct trade-off between computational cost and accuracy. While fast docking methods are useful for initial screening, MD-based methods like FEP and ensemble approaches provide significantly higher accuracy and reproducibility, making them the "gold standard" for reliable prediction. The emerging synergy of MD simulations with machine learning, powered by large, high-quality datasets like PLAS-20k, represents a promising frontier for developing rapid and accurate tools for drug discovery and design.
Standardized Benchmarking Frameworks for Objective Method Comparison
The evolution of molecular dynamics (MD) and machine-learned methods for studying protein-ligand interactions has outpaced the development of standardized tools for objective method validation. In drug discovery, accurately predicting how small molecules bind to target proteins is crucial, yet objective comparison between different simulation and prediction approaches is often hindered by inconsistent evaluation metrics, insufficient sampling of rare states, and the absence of reproducible benchmarks [62]. The lack of standardization makes it difficult for researchers to select the most appropriate tools for their specific needs with confidence.
This guide synthesizes current standardized benchmarking frameworks and performance data for methods validating protein-ligand binding, focusing on frameworks that enable direct, reproducible comparisons across diverse computational approaches. By providing a clear comparison of experimental protocols and performance metrics, we aim to equip researchers with the knowledge needed to rigorously evaluate and select methods for their drug discovery pipelines.
Available Benchmarking Frameworks
Modular MD Benchmarking with Weighted Ensemble Sampling
A modular benchmarking framework specifically addresses the challenge of standardizing evaluation across molecular dynamics methods. This approach uses weighted ensemble (WE) sampling through the WESTPA toolkit, based on progress coordinates derived from Time-lagged Independent Component Analysis (TICA), enabling efficient exploration of protein conformational space [62].
The framework's flexibility stems from its lightweight propagator interface that supports arbitrary simulation engines, allowing both classical force fields and machine learning-based models to be evaluated consistently. It includes a comprehensive evaluation suite capable of computing more than 19 different metrics and visualizations across structural fidelity, slow-mode accuracy, and statistical consistency domains [62].
To ensure rigorous testing, the developers have contributed a dataset of nine diverse proteins ranging from 10 to 224 residues that span various folding complexities and topologies. Each protein has been extensively simulated at 300K for one million MD steps per starting point, providing a ground truth reference for method comparisons [62].
Specialized Binding Affinity Benchmarks
For protein-ligand binding affinity prediction, specialized benchmarks address the critical need for assessing computational methods across the speed-accuracy spectrum. Current tools span from fast docking approaches (less than one minute on CPU, 2-4 kcal/mol RMSE) to highly accurate but computationally intensive methods like free energy perturbation (12+ hours of GPU time, under 1 kcal/mol RMSE) [15].
The PLA15 benchmark set provides particularly valuable standardized assessment for protein-ligand interaction energies. It uses fragment-based decomposition to estimate interaction energies for 15 protein-ligand complexes at the DLPNO-CCSD(T) level of theory, serving as a reference for evaluating faster, approximate methods [97].
Binding Site Prediction Benchmarks
For identifying where ligands bind to proteins, standardized benchmarks like DS1, DS2, and DS3 evaluate binding site prediction methods using metrics including recall, precision, F1 score, Matthews correlation coefficient (MCC), area under the ROC curve (AUC), and area under the precision-recall curve (AUPR). These benchmarks are particularly important for assessing how methods generalize to unseen ligands [16].
Performance Comparison of Methods
Protein-Ligand Interaction Energy Prediction
Quantitative benchmarking against the PLA15 dataset reveals significant performance differences across computational methods for predicting protein-ligand interaction energies. The following table summarizes key performance metrics:
Table 1: Performance Comparison on PLA15 Benchmark for Interaction Energy Prediction
| Method | Category | Mean Absolute Percent Error (%) | R² | Spearman ρ |
|---|---|---|---|---|
| g-xTB | Semiempirical | 6.09 | 0.994 | 0.981 |
| GFN2-xTB | Semiempirical | 8.15 | 0.985 | 0.963 |
| UMA-m | NNP (OMol25) | 9.57 | 0.991 | 0.981 |
| eSEN-s (OMol25) | NNP (OMol25) | 10.91 | 0.992 | 0.949 |
| UMA-s | NNP (OMol25) | 12.70 | 0.983 | 0.950 |
| AIMNet2 (DSF) | NNP | 22.05 | 0.633 | 0.768 |
| Egret-1 | NNP | 24.33 | 0.731 | 0.876 |
| AIMNet2 | NNP | 27.42 | 0.969 | 0.951 |
| ANI-2x | NNP | 38.76 | 0.543 | 0.613 |
| Orb-v3 | NNP (Materials) | 46.62 | 0.565 | 0.776 |
| MACE-MP-0b2-L | NNP (Materials) | 67.29 | 0.611 | 0.750 |
The data reveals a stark performance gap between current neural network potentials (NNPs) and semiempirical quantum chemistry methods, with g-xTB demonstrating superior accuracy. Notably, NNPs trained on the massive OMol25 dataset (eSEN and UMA models) show significantly better performance than other NNPs, though they still exhibit systematic overbinding likely due to the VV10 correction in their training data [97].
Binding Site Prediction Performance
For binding site identification, the LABind method demonstrates the advantage of incorporating ligand information directly into prediction models. The following table compares its performance against other approaches:
Table 2: Binding Site Prediction Performance Across Benchmark Datasets
| Method | Approach | DS1 AUPR | DS2 AUPR | DS3 AUPR | Unseen Ligands |
|---|---|---|---|---|---|
| LABind | Graph Transformer + Cross-attention | 0.723 | 0.698 | 0.681 | Supported |
| LigBind | Pre-training + Fine-tuning | 0.652 | 0.634 | 0.619 | Limited |
| GeoBind | Surface Point Clouds + Graph Networks | 0.598 | 0.581 | 0.563 | Not Supported |
| GraphBind | Hierarchical Graph Neural Networks | 0.587 | 0.572 | 0.551 | Not Supported |
| P2Rank | Protein Structure Features | 0.521 | 0.503 | 0.492 | Not Supported |
LABind's cross-attention mechanism enables it to learn distinct binding characteristics between proteins and ligands, allowing it to generalize to unseen ligands not present during training. This represents a significant advance over methods that rely solely on protein structure without explicit ligand modeling [16].
Experimental Protocols and Workflows
Standardized MD Benchmarking Protocol
The modular MD benchmarking framework employs a systematic workflow for evaluating simulation methods:
Figure 1: MD Benchmarking Workflow with Weighted Ensemble Sampling
The protocol begins with protein pre-processing to repair missing residues, atoms, and termini, followed by assignment of standard protonation states at pH 7.0. Weighted ensemble simulations then run with progress coordinates defined through TICA, enabling efficient exploration of conformational space. Finally, comparison with ground truth data evaluates multiple dimensions including TICA energy landscapes, contact map differences, and distributions for radius of gyration, bond lengths, angles, and dihedrals [62].
Quantitative divergence metrics, particularly Wasserstein-1 and Kullback-Leibler divergences across all analyses, provide standardized measures for comparing simulation methods against reference data [62].
Binding Affinity Prediction Workflow
For binding affinity prediction, standardized protocols help ensure reproducible results:
Figure 2: Binding Affinity Calculation Workflow
The protocol involves system preparation including pruning the protein to a fixed radius around the binding site, adding solvent and ions, and energy minimization. The system is gradually heated to 300K to prevent large initial forces that cause convergence issues, followed by a short (4ns) NPT MD simulation. After equilibration, snapshots are typically taken every 10ps, resulting in 300 snapshots for analysis [15].
For MM/GBSA and MM/PBSA approaches, binding free energy is decomposed into gas phase enthalpy, solvent correction, and entropy penalty terms. However, these approaches face challenges with error cancellation, as the first two terms have large magnitudes (∼100 kcal/mol) with opposite signs, while the binding affinities of interest are typically in the -15 to -4 kcal/mol range [15].
LABind Binding Site Prediction Architecture
The LABind method for ligand-aware binding site prediction employs a sophisticated multi-modal architecture:
Figure 3: LABind Architecture for Binding Site Prediction
LABind utilizes pre-trained language models for both ligand (MolFormer) and protein (Ankh) representations. Protein structures are converted to graphs with node spatial features including angles, distances, and directions derived from atomic coordinates. A cross-attention mechanism then learns the distinct binding characteristics between proteins and ligands, enabling the model to generalize to unseen ligands [16].
This approach demonstrates how explicitly modeling ligand properties, rather than relying solely on protein structure, enables more accurate and generalizable binding site prediction across diverse ligand types.
Essential Research Reagents and Tools
Benchmarking Datasets
Table 3: Essential Datasets for Method Validation
| Dataset | Description | Application | Key Features |
|---|---|---|---|
| PLA15 | 15 protein-ligand complexes with DLPNO-CCSD(T) interaction energies | Interaction energy validation | Fragment-based decomposition for high-accuracy reference |
| PDBbind | ~19,500 biomolecular complexes with binding affinities | Scoring function development | General/refined/core subsets for training and testing |
| HiQBind | >30,000 protein-ligand complexes with quality-controlled structures | High-quality training/validation | Corrected bond orders, protonation states, and steric clashes |
| BioLiP | >900,000 protein-ligand interactions with functional annotations | Binding site prediction | Comprehensive functional annotations including EC numbers |
| BindingDB | 2.9M binding measurements for 1.3M compounds across thousands of targets | Binding affinity prediction | Large-scale binding affinity data from literature and patents |
| WESTPA Protein Set | 9 diverse proteins (10-224 residues) with extensive MD trajectories | MD method benchmarking | Covers various folding complexities and topologies |
Software Tools and Platforms
Table 4: Essential Software Tools for Protein-Ligand Binding Studies
| Tool/Platform | Category | Primary Function | Key Capabilities |
|---|---|---|---|
| WESTPA 2.0 | Enhanced Sampling | Weighted Ensemble MD | Parallel trajectory resampling based on progress coordinates |
| OpenMM | MD Engine | High-performance MD simulations | GPU acceleration, multiple force field support |
| LABind | Binding Site Prediction | Ligand-aware binding site identification | Graph transformer with cross-attention for unseen ligands |
| HiQBind-WF | Data Curation | Protein-ligand dataset quality control | Automated structure fixing and validation |
| eSEN/UMA | Neural Network Potentials | Potential energy surface computation | OMol25-trained models for accurate energy calculations |
| g-xTB/GFN2-xTB | Semiempirical QM | Fast quantum chemical calculations | Accurate interaction energies for large systems |
The field of protein-ligand binding validation is rapidly evolving with several promising directions. The integration of massive quantum chemical datasets like OMol25, containing over 100 million calculations, is enabling development of more accurate neural network potentials that may eventually close the performance gap with semiempirical methods [98]. The universal models for atoms (UMA) approach, which unifies multiple datasets through mixture of experts architecture, demonstrates how knowledge transfer across different chemical domains can enhance model performance [98].
For practical drug discovery applications, future benchmarking frameworks must address the critical need for methods that balance accuracy with computational efficiency, occupying the gap between fast docking (minutes) and highly accurate FEP calculations (12+ GPU hours) [15]. Standardized benchmarks will play a crucial role in guiding development of these next-generation tools.
The creation of high-quality curated datasets through workflows like HiQBind-WF addresses fundamental data quality issues that have hampered previous method development [64]. By providing open-source, reproducible data curation pipelines, these initiatives promote transparency and sustainability in the field.
In conclusion, standardized benchmarking frameworks are essential for objective comparison of protein-ligand binding validation methods. Current benchmarks reveal significant performance differences between approaches, with semiempirical methods like g-xTB leading for interaction energy prediction, while modern machine learning approaches like LABind advance binding site identification, particularly for unseen ligands. As the field continues to evolve, consistent use of these standardized frameworks will accelerate development of more accurate, efficient computational tools for drug discovery.
Molecular dynamics (MD) simulations provide atomistic details of protein-ligand interactions, but accurately calculating binding free energies remains challenging due to limited sampling of rare events like dissociation. Enhanced sampling methods have emerged to address this bottleneck. Among these, dissociation Parallel Cascade Selection Molecular Dynamics (dPaCS-MD) combined with the Markov State Model (MSM) offers a promising approach without applying bias forces [84]. This case study objectively evaluates the performance of dPaCS-MD/MSM across three diverse protein-ligand complexes: trypsin/benzamidine, FKBP/FK506, and adenosine A2A receptor/T4E [84]. We examine its quantitative accuracy against experimental data, providing detailed methodologies and comparative analysis for research applications.
Methodological Framework: The dPaCS-MD/MSM Approach
Core Computational Procedure
The dPaCS-MD/MSM procedure comprises two main stages: pathway generation and trajectory analysis.
dPaCS-MD for Pathway Generation: This enhanced sampling method performs cycles of multiple parallel short MD simulations (typically 0.1 ns). In each cycle, snapshots with longer protein-ligand distances are selected as promising initial structures for the next cycle, effectively generating dissociation pathways. The repetition of parallel MD simulations with regenerated initial atom velocities enhances the probability of observing complete dissociation events [84].
MSM for Energetic and Kinetic Analysis: The multiple short trajectories generated by dPaCS-MD are analyzed using a Markov State Model. This method identifies metastable states and transitions between them, enabling construction of free energy profiles and calculation of kinetic properties along the dissociation coordinate [84].
Key Research Reagents and Computational Tools
Table 1: Essential Research Reagents and Computational Solutions for dPaCS-MD/MSM Implementation
| Reagent/Solution | Function/Role | Specific Examples |
|---|---|---|
| MD Simulation Software | Engine for performing atomic-level simulations | AMBER [84], GROMACS [84] |
| Force Fields | Mathematical description of interatomic forces | AMBER ff14SB [84], GAFF [84] |
| Solvent Models | Represents aqueous environment | SPC/Eb [84], OPC [84] |
| System Preparation Tools | Initial model building and parameterization | PDB2PQR [84], CHARMM-GUI [84], Antechamber [84] |
| Enhanced Sampling | Accelerates rare event sampling | dPaCS-MD [84] |
| Analysis Framework | Extracts thermodynamic/kinetic information | Markov State Model [84] |
Experimental Systems and Simulation Protocols
Benchmark Protein-Ligand Complexes
This validation study utilized three complexes with distinct characteristics to test method transferability [84]:
- Trypsin/Benzamidine: A medium-sized serine protease with a small, rigid inhibitor (153 residues; ~140,000 atoms). PDB ID: 3atl [84].
- FKBP/FK506: A immunophilin protein complexed with a larger, flexible macrocyclic drug (107 residues; ~120,000 atoms). PDB ID: 1fkf [84].
- Adenosine A2A Receptor/T4E: A GPCR membrane protein with a deeply-bound antagonist (class A GPCR; ~210 DMPC lipids). PDB ID: 3uzc [84].
Detailed Simulation Workflow
The general simulation protocol was consistent across systems with specific adaptations [84]:
- System Preparation: Crystal structures were solvated in explicit water boxes with ionic strength adjusted to 150 mM NaCl or KCl. The membrane-bound A2A system was embedded in a DMPC lipid bilayer using CHARMM-GUI [84].
- Force Field Parameters: Proteins were described with the AMBER ff14SB force field. Ligand parameters were generated with the Antechamber module using GAFF and AM1-BCC charges [84].
- Simulation Conditions: Systems were energy-minimized, equilibrated, and production simulations were performed at 300 K and 1 atm pressure. Long-range electrostatics were handled with PME [84].
- dPaCS-MD Specifics: The method iteratively selected structures with increased protein-ligand distances, running multiple short parallel simulations to efficiently explore dissociation pathways [84].
- MSM Construction: Trajectories were discretized into states based on protein-ligand geometry, and transition probabilities between states were calculated to build the model for free energy estimation [84].
Figure 1: The dPaCS-MD/MSM Workflow. The process begins with system preparation, proceeds through iterative dPaCS-MD sampling cycles, and culminates in MSM analysis for free energy calculation.
Results and Comparative Performance Analysis
Quantitative Binding Free Energy Comparison
dPaCS-MD/MSM demonstrated strong agreement with experimental binding measurements across all three benchmark systems.
Table 2: Calculated vs. Experimental Binding Free Energies for Benchmark Complexes [84]
| Protein-Ligand Complex | dPaCS-MD/MSM ΔG° (kcal/mol) | Experimental ΔG° (kcal/mol) | Deviation (kcal/mol) |
|---|---|---|---|
| Trypsin/Benzamidine | -6.1 ± 0.1 | -6.4 to -7.3 | 0.3 to 1.2 |
| FKBP/FK506 | -13.6 ± 1.6 | -12.9 | -0.7 |
| Adenosine A2A Receptor/T4E | -14.3 ± 1.2 | -13.2 | -1.1 |
The computed values fall within approximately 1 kcal/mol of experimental measurements, demonstrating the method's quantitative accuracy across diverse systems. The slightly larger deviation for the A2A receptor system highlights the increased challenge of modeling membrane protein complexes [84].
Dissociation Mechanism Insights
Beyond affinity calculations, the method provided detailed mechanistic insights into dissociation processes [84]:
- Trypsin/Benzamidine: The small ligand dissociated through a relatively straightforward pathway with minimal intermediate states.
- FKBP/FK506: The flexible macrocyclic drug exhibited more complex dissociation dynamics with potential intermediate conformations.
- A2A Receptor/T4E: The antagonist dissociation from the deep GPCR binding pocket involved navigating through specific transmembrane helices.
The free energy profiles reconstructed from MSM analysis revealed characteristic barriers and intermediate states along each dissociation pathway, providing testable hypotheses for kinetic studies [84].
Discussion: Advantages and Implementation Considerations
Performance Assessment and Method Utility
The validation results support several key advantages of dPaCS-MD/MSM:
- Transferability: Successful application across soluble enzymes, binding proteins, and GPCRs indicates broad applicability to diverse drug targets [84].
- Quantitative Accuracy: Sub-1 kcal/mol deviations for two systems demonstrate potential for predictive binding affinity calculations [84].
- Mechanistic Insight: The method goes beyond affinities to provide structural details of dissociation pathways and intermediate states [84].
Figure 2: Tandem Allostery in Sir2/p53. Illustration of allosteric mechanisms discovered through PaCS-MD simulations, showing how reactant and product binding sequentially regulate cofactor pocket conformation [99].
Technical Implementation Requirements
Researchers implementing this methodology should consider:
- Computational Resources: dPaCS-MD requires multiple parallel MD simulations, though each trajectory is relatively short (sub-nanosecond) [84].
- System Setup Care: Proper parameterization of ligands (e.g., with GAFF), appropriate membrane modeling for GPCRs, and careful selection of collective variables for MSM construction are critical [84].
- Validation Strategy: Comparison with experimental binding data and known mutagenesis effects strengthens confidence in predictions [84].
This case study demonstrates that dPaCS-MD/MSM robustly calculates protein-ligand binding affinities while providing mechanistic insights into dissociation processes. The method reproduced experimental binding free energies within chemical accuracy for three distinct complexes, highlighting its transferability across target classes. For drug development researchers, this approach offers both quantitative binding affinity predictions and atomic-level understanding of dissociation pathways that can inform optimization strategies. As enhanced sampling methods continue evolving, dPaCS-MD/MSM represents a validated option for challenging systems where explicit dissociation sampling is necessary for accurate binding characterization.
Molecular dynamics (MD) simulations are indispensable tools in computational chemistry and biophysics, providing atomic-level insights into the behavior of biological systems. For studies aimed at validating protein-ligand binding—a critical process in drug discovery—selecting appropriate simulation software and force fields significantly impacts the reliability and accuracy of the results [100] [37]. This guide objectively compares three cornerstone MD platforms—AMBER, CHARMM, and GROMACS—focusing on their application in protein-ligand binding research. We present performance benchmarks, analyze force field accuracy, and detail experimental protocols to inform researchers and drug development professionals.
Each MD software package has distinct strengths, development philosophies, and optimal use cases, which are summarized in Table 1.
Table 1: Overview of AMBER, CHARMM, and GROMACS MD Platforms
| Feature | AMBER | CHARMM | GROMACS |
|---|---|---|---|
| Primary Focus | High-accuracy biomolecular simulations [100] | Accurate biomolecular simulations with extensive force field [101] | High-performance and scalable MD [100] |
| Licensing | Commercial [102] | Commercial [103] | Free, Open-Source [100] [102] |
| Key Strength | Superior force fields for proteins/nucleic acids [100] | Broad coverage of molecules, including peptidomimetics [101] | Exceptional speed and parallel efficiency [100] |
| Typical Use Case | Protein-ligand binding, nucleic acids [100] | Non-natural peptides, diverse biomolecules [101] | Large complexes, high-throughput sampling [100] |
| Learning Curve | Steeper [100] | Steeper [103] | More accessible [100] |
GROMACS distinguishes itself with its raw performance and scalability, often achieving simulation speeds 2-3 times faster than AMBER for large systems on equivalent hardware [100]. This makes it ideal for large biomolecular complexes and studies requiring extensive sampling [100]. AMBER is renowned for the high quality of its specialized force fields (e.g., ff14SB, ff19SB), which are often considered the gold standard for simulating biomolecules, making it a preferred choice for detailed studies of protein-ligand interactions and nucleic acid dynamics where force field precision is critical [100]. CHARMM provides a highly rigorous and broadly applicable force field, with ongoing developments ensuring accuracy for both natural and non-natural systems like β-peptides [101].
Force Field Performance and Benchmarking
The accuracy of any MD simulation is fundamentally linked to the force field. Benchmarking studies provide critical insights for selection.
Table 2: Selected Force Field Benchmarking Results
| Force Field | System Type | Key Finding | Reference |
|---|---|---|---|
| AMBER (ff14SB) | Proteins | Excellent reproduction of experimental structural data for proteins [100]. | [100] |
| CHARMM36m | β-peptides | Best overall performance, accurately reproducing experimental structures in monomers and oligomers [101]. | [101] |
| GAFF (AMBER) | Organic Ligands | Widely used for small molecules; accurate binding free energies when combined with TIP3P water in multiple MD engines [104]. | [104] |
| AMBER (ff19SB) | Proteins | Updated protein force field with improved side-chain torsion potentials [105]. | [105] |
Specialized biomolecular force fields like AMBER's ff14SB/ff19SB and CHARMM36m are generally preferred for simulating proteins and nucleic acids [100] [105] [101]. For ligands, the General AMBER Force Field (GAFF) is a standard choice, and its parameters can be used within GROMACS simulations [104] [103]. A critical benchmarking effort revealed that with nearly identical starting structures and parameters, different MD engines (including AMBER, GROMACS, and others) can compute potential energies agreeing within 0.1% or better [104]. However, subtle differences in default treatment of long-range interactions, cutoff parameters, and even the value of Coulomb's constant can be significant sources of discrepancy and must be standardized for reproducible results [104].
Experimental Protocols for Validating Protein-Ligand Binding
Accurately calculating binding free energies is a primary goal in validating protein-ligand interactions. Advanced-sampling methods combined with a rigorous statistical mechanical framework are often necessary to achieve chemical accuracy (within 1 kcal/mol of experiment) [37].
Binding Free Energy Estimation with BFEE2
The Binding Free-Energy Estimator 2 (BFEE2) provides a streamlined protocol for determining standard binding free energies [37]. The following workflow can be implemented on AMBER, GROMACS, or other MD engines.
Key Steps in the BFEE2 Protocol [37]:
- Initial Setup: Begin with the experimentally determined (e.g., X-ray crystallography) or computationally docked structure of the protein-ligand complex. BFEE2 assists in preparing all necessary input files, limiting manual intervention.
- Route Selection: Choose between a geometric or alchemical transformation pathway. The geometric route involves physically separating the ligand from the protein along a reaction pathway, while the alchemical route uses a thermodynamic cycle to decouple the ligand from its environment in both the bound and unbound states.
- Application of Restraints: A set of configurational restraints are applied to control the ligand's position, orientation, and conformation relative to the protein. These restraints are defined using collective variables (CVs), such as:
- RMSD: To control the ligand's conformation.
- Roll, Pitch, Yaw Angles (Θ, Φ, Ψ): To control the ligand's orientation.
- Spherical Coordinates (r, θ, φ): To control the ligand's position relative to the binding site.
- Free Energy Calculation:
- In the geometric route, the free energy is computed as a potential of mean force (PMF) using advanced sampling algorithms like the well-tempered metadynamics with extended adaptive biasing force (WTM-eABF).
- In the alchemical route, free energy perturbation (FEP) or thermodynamic integration (TI) is used to calculate the work of decoupling the ligand in the bound and unbound states.
- Restraint Correction: The energetic cost of applying and releasing the restraints is accounted for analytically to yield the final standard binding free energy.
This methodology has been successfully applied to a wide range of protein-ligand complexes, including those with flexible ligands, semi-buried ligands, and systems where cation-π interactions dominate, consistently achieving errors close to chemical accuracy [37].
Focused Docking for Binding Site Identification
When the binding site is unknown, a "focused docking" approach can significantly improve the efficiency and accuracy of subsequent MD simulations. This protocol uses predicted binding sites to guide the docking search.
Methodology [106]:
- Binding Site Prediction: A program like SiteHound is used to analyze the protein's surface. This algorithm calculates interaction energy grids and clusters favorable regions to predict potential binding pockets. The top three predicted sites are typically selected for focused docking.
- Focused Docking Setup: Instead of performing a computationally expensive "blind docking" over the entire protein surface, independent docking runs (e.g., using AutoDock) are set up with small boxes centered on each predicted binding site.
- Analysis: This focused approach has been shown to identify the correct binding site more frequently, produce more accurate ligand poses, and require less computational time compared to blind docking [106].
The Scientist's Toolkit
This section details essential software tools and resources for setting up and running protein-ligand simulations across the different platforms.
Table 3: Essential Tools for Protein-Ligand MD Simulations
| Tool Name | Category | Function | Compatibility |
|---|---|---|---|
| BFEE2 | Software Plugin | Automates setup and post-processing for binding free energy calculations [37]. | VMD [37] |
| Antechamber/acpype | Parameterization | Automatically generates AMBER/GAFF-compatible parameters for ligands; acpype converts to GROMACS format [103]. | AMBER, GROMACS [103] |
| CGenFF Server | Parameterization | The official server for generating CHARMM-compatible parameters for small molecules and ligands [103]. | CHARMM, GROMACS [103] |
| LigParGen | Parameterization | Web server that produces OPLS-AA force field parameters for organic molecules [103]. | GROMACS, others |
| ParmEd/InterMol | Conversion | Libraries for converting molecular topology and parameter files between different MD software formats (e.g., AMBER to GROMACS) [104]. | AMBER, CHARMM, GROMACS, LAMMPS, DESMOND [104] |
| SiteHound | Analysis | Predicts ligand binding sites on protein structures to guide docking and simulation setup [106]. | Input/Output agnostic |
The choice between AMBER, CHARMM, and GROMACS for validating protein-ligand binding is not a matter of identifying a single "best" platform but rather selecting the most appropriate tool for a specific research question and context.
- For maximum accuracy in well-defined biomolecular systems and for specialized calculations like absolute binding free energies, AMBER and its highly refined force fields are an excellent choice [100] [37].
- For simulating non-standard molecules like peptidomimetics or when leveraging the extensive CHARMM force field ecosystem, CHARMM demonstrates superior performance [101].
- For high-throughput screening, studying very large systems, or when computational speed and scalability are paramount, GROMACS is unparalleled [100]. Its ability to use force fields from AMBER and CHARMM also provides great flexibility [100] [103].
Critical to successful validation is the adoption of rigorous, automated protocols like BFEE2 for free energy calculation [37] and a meticulous approach to force field parameterization for ligands. Furthermore, ensuring reproducibility requires careful attention to simulation details, as minor differences in treatment of long-range interactions can lead to significant discrepancies between even nominally identical simulations [104]. By leveraging the respective strengths of each platform within a robust methodological framework, researchers can reliably validate protein-ligand binding to advance drug discovery efforts.
In molecular dynamics research, comprehensively validating protein-ligand binding requires moving beyond traditional affinity measurements to a multidimensional assessment framework. While binding affinity (Kd/Ki) and its computational prediction have long been the primary focus, a complete validation paradigm now incorporates pose prediction accuracy, binding kinetics, and physical plausibility metrics. This evolution reflects the growing understanding that drug efficacy depends not only on how tightly a ligand binds but also on how long it remains bound (residence time) and the specific molecular interactions formed. Advances in computational methods, including traditional physics-based approaches, deep learning models, and hybrid frameworks, have necessitated sophisticated benchmarking strategies that evaluate performance across these complementary dimensions. This guide provides a systematic comparison of current methodologies based on their performance across these critical quantitative metrics, offering researchers a structured approach for validating protein-ligand interactions in drug discovery applications.
Performance Metrics Comparison Across Methodologies
Pose Prediction Accuracy and Physical Validity
Pose prediction accuracy, typically measured by ligand root-mean-square deviation (RMSD) from crystallographic reference structures, remains fundamental to binding validation. However, recent comprehensive evaluations reveal that low RMSD alone does not guarantee physiologically relevant binding modes, necessitating additional checks for physical plausibility and interaction fidelity.
Table 1: Comparative Pose Prediction Performance Across Docking Methodologies
| Method Category | Representative Methods | RMSD ≤ 2 Å Success Rate (%) | PB-Valid Rate (%) | Combined Success Rate (%) |
|---|---|---|---|---|
| Traditional Docking | Glide SP, AutoDock Vina | 70-80 (Astex) | >94 (across datasets) | 68.5 (Glide SP, Astex) |
| Generative Diffusion | SurfDock, DiffBindFR | 75.7-91.8 (Astex) | 40.2-63.5 (across datasets) | 33.3-61.2 (SurfDock) |
| Regression-based DL | KarmaDock, GAABind, QuickBind | 40-60 (Astex) | <30 (across datasets) | <20 (across datasets) |
| Hybrid Methods | Interformer | Moderate | High | Balanced performance |
Recent multidimensional benchmarking classifies docking methods into four distinct performance tiers based on combined success rates (RMSD ≤ 2 Å & physically valid poses): traditional methods > hybrid AI scoring with traditional conformational search > generative diffusion methods > regression-based methods [107]. This stratification highlights the diverse strengths and limitations of each approach, with traditional methods like Glide SP maintaining exceptional physical validity (PB-valid rates >94% across datasets) while modern diffusion models like SurfDock achieve superior pose accuracy (exceeding 70% RMSD ≤ 2 Å across all datasets) but with compromised physical plausibility [107].
Virtual Screening Enrichment and Generalization
Virtual screening (VS) efficacy represents another critical dimension for assessing binding prediction methods, typically measured through enrichment factors (EF) and area under the ROC curve (AUC). These metrics evaluate a method's ability to prioritize true active compounds over decoys in large compound libraries.
Table 2: Virtual Screening Performance Comparison
| Method | EF1% (DUD-E) | EF1% (CASF2016) | EF1% (LIT-PCBA) | Key Strengths |
|---|---|---|---|---|
| AK-Score2 | 23.1 | 32.7 | Higher than benchmarks | Integration of physical and ML scoring |
| AlphaFold3 with active ligands | Improved over apo structures | N/A | N/A | Leveraging ligand information |
| Glide WS | Significant improvement over Glide SP | N/A | N/A | Explicit water representation |
| RTMScore | High efficiency | N/A | N/A | Residue-atom distance likelihood |
The recently developed AK-Score2 demonstrates exceptional virtual screening capabilities, achieving top 1% enrichment factors of 32.7 and 23.1 with the CASF2016 and DUD-E benchmark sets, respectively [108]. This performance stems from its novel architecture that combines three independent neural network models with physics-based scoring functions. Similarly, AlphaFold3 shows significantly improved virtual screening performance when provided with active ligand information during structure prediction, highlighting the importance of appropriate input selection for optimal results [109].
Binding Kinetics and Affinity Prediction
Binding kinetics parameters, particularly association rate constants (kon) and drug-target residence time, are increasingly recognized as crucial determinants of drug efficacy. Computational prediction of these parameters provides a more dynamic perspective on ligand binding beyond thermodynamic affinity alone.
Table 3: Methods for Binding Kinetics and Affinity Prediction
| Method Type | Representative Approaches | Key Metrics | Correlation with Experiment | Computational Cost |
|---|---|---|---|---|
| Multiscale Simulation | BD/MD combined approach | Association rate constant (kon) | Aligns well with experimental data | Moderate to High |
| MD/MMPBSA | PLAS-20k dataset | Binding free energy | Better than docking scores | High |
| Deep Learning | Ligand-Transformer | Binding affinity (pKd) | R=0.57-0.88 after fine-tuning | Low |
| ML with Physical Features | AK-Score2, PIGNet, RTMScore | Binding affinity | Pearson R=0.78-0.87 | Low |
Advanced multiscale approaches that combine Brownian dynamics (BD) for long-range diffusion and molecular dynamics (MD) for short-range interactions enable efficient computation of association rate constants that align well with experimental data [110]. For affinity prediction, transformer-based architectures like Ligand-Transformer achieve correlation coefficients of up to R=0.88 with experimental values after fine-tuning on target-specific data [111], while graph neural network approaches typically show Pearson correlation coefficients ranging from 0.78 to 0.87 with experimental binding affinities [108].
Experimental Protocols and Methodologies
Benchmarking Pose Prediction and Physical Validity
Comprehensive evaluation of pose prediction requires standardized datasets and validation protocols. The following methodology outlines current best practices based on recent large-scale benchmarking studies:
Dataset Curation: Employ multiple benchmark sets with different characteristics: (1) Astex diverse set containing known complexes for baseline performance; (2) PoseBusters benchmark set with unseen complexes to test generalization; (3) DockGen dataset featuring novel protein binding pockets to assess robustness to unfamiliar structural environments [107].
Evaluation Metrics: Calculate (1) RMSD ≤ 2 Å success rate - percentage of predictions with ligand RMSD below 2Å threshold; (2) PB-valid rate - percentage of predictions passing PoseBusters checks for physical and chemical plausibility; (3) Combined success rate - percentage of predictions satisfying both criteria simultaneously [107].
Implementation Protocol:
- Prepare protein structures by removing native ligands and adding hydrogens at physiological pH
- Generate binding pockets defined as residues within 5.0Å around crystallized ligands
- For each method, run docking with standardized parameters
- Align predicted complexes to reference structures using protein backbone atoms
- Calculate RMSD for ligand heavy atoms
- Run PoseBusters validation to check for steric clashes, bond length validity, and stereochemistry preservation
This multidimensional evaluation strategy reveals critical performance patterns, such as the superior physical validity of traditional methods like Glide SP (>94% PB-valid rates across datasets) versus the higher pose accuracy but lower physical plausibility of diffusion models like SurfDock [107].
Virtual Screening Validation Protocols
Validating virtual screening performance requires carefully designed decoy sets and enrichment metrics to avoid bias and ensure biological relevance:
Dataset Preparation: Utilize established benchmark sets including (1) DUD-E containing known actives and property-matched decoys; (2) LIT-PCBA with experimentally confirmed active and inactive compounds; (3) CASF2016 providing standardized evaluation framework [108].
Evaluation Workflow:
- Prepare protein structures using consistent preprocessing protocols
- Compound library preparation including known actives and decoys
- Docking screens using standardized box sizes centered on binding sites
- Pose generation and scoring using method-specific parameters
- Ranking compounds by predicted scores or affinities
- Calculation of enrichment factors (EF1%) and AUC values
Critical Considerations: The AK-Score2 methodology demonstrates the importance of training with diverse decoy sets including: (1) conformational decoys generated by redocking native ligands; (2) cross-docked decoys from similar binding sites; (3) random decoys with dissimilar chemical structures [108]. This approach improves model robustness and screening performance, achieving top 1% enrichment factors of 32.7 and 23.1 with CASF2016 and DUD-E sets, respectively.
Binding Kinetics and Affinity Calculation Methods
Multiscale BD/MD for Association Rates:
- Brownian Dynamics Simulation: Simulate long-range diffusion and diffusional encounter complex formation using software like SDA, generating thousands of trajectories that bring ligands close to the binding site [110]
- Ensemble Selection: Extract multiple starting structures from BD simulations where ligands approach within close proximity to the binding site
- Molecular Dynamics Refinement: Perform all-atom MD simulations using these encounter complexes as starting points to model short-range interactions and binding process
- kon Calculation: Compute association rate constants from the simulation data using reaction criteria based on specific atom-distance thresholds
- Validation: Compare computed kon values with experimental measurements to assess accuracy
This optimized multiscale workflow improves computational efficiency while maintaining accuracy by reducing the required MD simulation time through careful selection of starting structures from BD simulations [110].
MD/MMPBSA Binding Affinity Protocol (as implemented in PLAS-20k dataset):
- System Preparation: Obtain initial structures from PDB, model missing residues, protonate at pH 7.4, and parameterize ligands using GAFF2 force field [57]
- Solvation and Neutralization: Solvate complexes in TIP3P water box with 10Å extension, add counter ions for charge neutrality
- Equilibration Protocol:
- Energy minimization with backbone restraints (1000 steps)
- Gradual heating from 50K to 300K with backbone restraints
- NVT equilibration for 1ns
- Additional minimization (4000 steps)
- Final equilibration in NVT ensemble at 300K for 2ns
- Production Simulation: Run 5 independent 4ns simulations in NPT ensemble, saving trajectories every 100ps
- Affinity Calculation: Use MMPBSA method on combined trajectories from all five independent simulations, incorporating explicit water molecules near the active site
This protocol generates binding affinities that show better correlation with experimental values than docking scores, even for Lipinski-compliant ligands [57].
Visualization of Methodologies
Comprehensive Binding Validation Workflow
Multiscale Kinetics Simulation Approach
Essential Research Reagents and Computational Tools
Table 4: Key Research Resources for Binding Validation Studies
| Resource Type | Specific Examples | Primary Application | Key Features/Benefits |
|---|---|---|---|
| Benchmark Datasets | PDBbind, DUD-E, LIT-PCBA, CASF2016 | Method training and validation | Standardized evaluation frameworks |
| MD Simulation Datasets | PLAS-20k, PLAS-5k | ML model training with dynamics | Incorporates conformational sampling |
| Structure Prediction | AlphaFold3, RosettaFold All-Atom | Protein-ligand complex generation | High-accuracy complex prediction |
| Traditional Docking | Glide SP, AutoDock Vina | Baseline pose prediction | High physical validity and reliability |
| Deep Learning Docking | SurfDock, DiffBindFR, DynamicBind | Advanced pose prediction | High accuracy on known complexes |
| Hybrid Methods | AK-Score2, Interformer | Balanced performance | Combines strengths of multiple approaches |
| Kinetics Simulation | BD/MD multiscale approach | Association rate calculation | Bridges timescales efficiently |
| Validation Tools | PoseBusters | Physical plausibility checking | Detects steric clashes and geometric errors |
The PLAS-20k dataset deserves special attention as it provides 97,500 independent MD simulations on 19,500 protein-ligand complexes, offering unprecedented access to dynamic binding information that goes beyond static structural data [57]. For pose validation, the PoseBusters toolkit enables systematic evaluation of docking predictions against chemical and geometric consistency criteria, including bond length/angle validity, stereochemistry preservation, and protein-ligand clash detection [107].
The landscape of protein-ligand binding validation has evolved from singular affinity measurements to multidimensional assessment encompassing pose accuracy, physical plausibility, screening enrichment, and binding kinetics. Traditional docking methods like Glide SP maintain advantages in physical validity and reliability, while deep learning approaches offer superior pose accuracy for certain target classes. Hybrid methods that combine physical scoring with machine learning, such as AK-Score2, demonstrate exceptional performance in virtual screening applications. For comprehensive binding validation, researchers should employ multiple complementary metrics across different benchmark datasets to fully characterize method performance, particularly emphasizing generalization to novel targets and physical plausibility alongside traditional accuracy measures. The integration of binding kinetics through multiscale simulation approaches further enriches this validation paradigm, providing a more complete picture of ligand binding relevant to drug efficacy.
Conclusion
The rigorous validation of protein-ligand binding through Molecular Dynamics is no longer an academic exercise but a critical component of modern drug discovery. By mastering the foundational concepts, applying advanced methodological workflows, diligently troubleshooting simulations, and consistently benchmarking against experimental data, researchers can significantly increase the predictive power of their computational studies. The ongoing development of automated tools, standardized benchmarks, and more accurate force fields promises to further close the gap between simulation and experiment. This progression will undoubtedly enhance the role of MD in driving the rational design of more effective therapeutics, ultimately improving the efficiency and success rate of clinical drug development.
References
- 1. Understanding the impact of binding free energy and kinetics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11108734/]
- 2. Fast, scalable free energy prediction with nonequilibrium ... [https://www.drugtargetreview.com/article/190846/fast-scalable-free-energy-prediction-with-nonequilibrium-switching/]
- 3. Predicting binding free energies: Frontiers and benchmarks [https://pmc.ncbi.nlm.nih.gov/articles/PMC5544526/]
- 4. Guidelines for the analysis of free energy calculations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4420631/]
- 5. Calculating free energy [https://tutorials.gromacs.org/docs/free-energy-of-solvation.html]
- 6. The maximal and current accuracy of rigorous protein ... [https://www.nature.com/articles/s42004-023-01019-9]
- 7. Accelerated Molecular Dynamics Simulations of Ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5435230/]
- 8. Enhanced sampling of protein conformational changes via ... [https://www.nature.com/articles/s41467-025-55983-y]
- 9. Fast Sampling of Protein Conformational Dynamics [https://arxiv.org/html/2411.08154v3]
- 10. Enhanced Sampling Techniques in Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4339458/]
- 11. Accurate and Rapid Ranking of Protein–Ligand Binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503914/]
- 12. Beyond static structures: protein dynamic conformations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12262120/]
- 13. PySAGES: flexible, advanced sampling methods ... [https://www.nature.com/articles/s41524-023-01189-z]
- 14. Article Extending MMPBSA for membrane proteins [https://www.sciencedirect.com/science/article/pii/S0006349525003182]
- 15. Exploring Protein–Ligand Binding-Affinity Prediction | Rowan [https://rowansci.com/blog/exploring-protein-ligand-binding-affinity-prediction]
- 16. LABind: identifying protein binding ligand-aware sites via ... [https://www.nature.com/articles/s41467-025-62899-0]
- 17. Review The continuous evolution of biomolecular force fields [https://www.sciencedirect.com/science/article/abs/pii/S0969212625001911]
- 18. Molecular dynamics analysis of biomolecular systems ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9592887/]
- 19. Assessing the effect of forcefield parameter sets on the ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9549959/]
- 20. Force Field Limitations of All-Atom Continuous Constant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12231151/]
- 21. Systematic design of biomolecular force fields [https://www.sciencedirect.com/science/article/pii/S0959440X2030141X]
- 22. Unveiling Dynamic Hotspots in Protein–Ligand Binding [https://pmc.ncbi.nlm.nih.gov/articles/PMC12071544/]
- 23. Insights from molecular dynamics simulations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2865717/]
- 24. Molecular dynamics simulation study of AG10 and ... [https://www.sciencedirect.com/science/article/pii/S2405580819302110]
- 25. Comparison of software for molecular mechanics modeling [https://en.wikipedia.org/wiki/Comparison_of_software_for_molecular_mechanics_modeling]
- 26. Running Molecular Dynamics on Alliance clusters with AMBER [https://computecanada.github.io/molmodsim-amber-md-lesson/26-MD_Software_Benchmarks/index.html]
- 27. List of widely used MD Simulation Analysis Tools. [https://bioinformaticsreview.com/20240729/list-of-widely-used-md-simulation-analysis-tools/]
- 28. Modeling and Simulation Software [https://www.rcsb.org/docs/additional-resources/modeling-and-simulation-software]
- 29. MISATO: machine learning dataset of protein–ligand ... [https://www.nature.com/articles/s43588-024-00627-2]
- 30. Best Practices for Constructing, Preparing, and Evaluating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9662604/]
- 31. Evaluating ligand docking methods for drugging protein ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01067-4]
- 32. Computational and experimental design of l-amino acid- ... [https://www.nature.com/articles/s41598-025-26171-1]
- 33. Novel method for prioritizing protein binding sites using ... [https://www.sciencedirect.com/science/article/pii/S2405844025014653]
- 34. A Multiscale Simulation Approach to Compute Protein–Ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570144/]
- 35. On Free Energy Calculations in Drug Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12548091/]
- 36. Absolute Binding Free Energy Calculations Using ... [https://www.sciencedirect.com/science/article/pii/S0006349506719944]
- 37. Accurate determination of protein:ligand standard binding free ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10082674/]
- 38. Relative binding free energy calculations with transformato [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.954638/full]
- 39. Automated relative binding free energy calculations from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10140266/]
- 40. Relative Binding Free Energy with Non-Equilibrium ... [https://www.eyesopen.com/blog/relative-binding-free-energy-calculations-with-non-equilibrium-switching-in-orion]
- 41. Absolute binding free energy calculations improve ... [https://www.nature.com/articles/s41598-022-17480-w]
- 42. Increased throughput in methods for simulating protein ... [https://www.sciencedirect.com/science/article/pii/S0959440X24000988]
- 43. ModBind, a Rapid Simulation-Based Predictor of Ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11733936/]
- 44. DynamicBind: predicting ligand-specific protein ... [https://www.nature.com/articles/s41467-024-45461-2]
- 45. GHeinzelmann/BAT.py [https://github.com/GHeinzelmann/BAT.py]
- 46. Simulations and active learning enable efficient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12307812/]
- 47. MDTraj: A Modern Open Library for the Analysis of ... [https://www.sciencedirect.com/science/article/pii/S0006349515008267]
- 48. From Data to Knowledge: Systematic Review of Tools for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8960308/]
- 49. Big data tools for MD simulation analysis | fastpayges [https://ahy3nz.github.io/fastpayges/molecular%20modeling/data%20science/2020/05/13/dask-mdtraj.html]
- 50. Automation of absolute protein-ligand binding free energy ... [https://www.nature.com/articles/s41598-020-80769-1]
- 51. Validation of the Alchemical Transfer Method for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10577236/]
- 52. GilsonLabUCSD/APR: Attach-release-pull: A free energy ... [https://github.com/GilsonLabUCSD/APR]
- 53. Automation of absolute protein-ligand binding free energy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7806944/]
- 54. MD Simulation of Protein-Ligand Interaction [https://pmc.ncbi.nlm.nih.gov/articles/PMC1302723/]
- 55. Assessment of a Single Decoupling Alchemical Approach ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2018.00022/full]
- 56. Prediction of Ligand Binding Affinity by the Combination ... [https://pubmed.ncbi.nlm.nih.gov/26588319/]
- 57. PLAS-20k: Extended Dataset of Protein-Ligand Affinities ... [https://www.nature.com/articles/s41597-023-02872-y]
- 58. Weighted Ensemble Simulations Reveal Novel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11957032/]
- 59. Enhanced-Sampling Simulations for the Estimation of ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.899805/full]
- 60. WeTICA: A directed search weighted ensemble based ... [https://arxiv.org/abs/2501.08926]
- 61. Binding free energy of protein/ligand complexes calculated ... [https://www.jstage.jst.go.jp/article/biophysico/18/0/18_bppb-v18.037/_article/-char/ja/]
- 62. A Standardized Benchmark for Machine-Learned ... [https://arxiv.org/html/2510.17187v1]
- 63. Advances in Molecular Dynamics Simulations and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7504087/]
- 64. A workflow to create a high-quality protein–ligand binding ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00357h]
- 65. Markov State Models to Elucidate Ligand Binding Mechanism [https://pubmed.ncbi.nlm.nih.gov/33759131/]
- 66. Markov State Models to Study the Functional Dynamics of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8479766/]
- 67. Markov State Models to Elucidate Ligand Binding Mechanism [https://profiles.foxchase.org/en/publications/markov-state-models-to-elucidate-ligand-binding-mechanism-2]
- 68. Protein conformational plasticity and complex ligand ... [https://www.nature.com/articles/ncomms8653]
- 69. Addressing challenges in data generation, analysis, and ... [https://www.sciencedirect.com/science/article/pii/S2666386425001651]
- 70. Best Practices for Quantification of Uncertainty and Sampling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6286151/]
- 71. Challenges and opportunities in computational studies for ... [https://www.nature.com/articles/s44386-025-00024-3]
- 72. Recent computational advances in the identification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12342141/]
- 73. Uncertainty-biased molecular dynamics for learning ... [https://www.nature.com/articles/s41524-024-01254-1]
- 74. MDBenchmark: A toolkit to optimize the performance of ... [https://pubmed.ncbi.nlm.nih.gov/33086826/]
- 75. MDBenchmark - Read the Docs [https://mdbenchmark.readthedocs.io/]
- 76. bio-phys/MDBenchmark: Quickly generate, start and ... [https://github.com/bio-phys/MDBenchmark]
- 77. Basic usage of MDBenchmark - Read the Docs [https://mdbenchmark.readthedocs.io/en/latest/general.html]
- 78. A Technical Overview of Molecular Simulation Software [https://intuitionlabs.ai/articles/molecular-modeling-simulation-software]
- 79. Available Instruments for Analyzing Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4797681/]
- 80. How to visualize protein-ligand complex MD run using VMD [https://gromacs.bioexcel.eu/t/how-to-visualize-protein-ligand-complex-md-run-using-vmd/3331]
- 81. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations - PubMed [https://pubmed.ncbi.nlm.nih.gov/28074360/]
- 82. Qualitative Estimation of Protein–Ligand Complex Stability through Thermal Titration Molecular Dynamics Simulations - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9709921/]
- 83. Elucidation of protein–ligand interactions by multiple ... [https://pubs.rsc.org/en/content/articlehtml/2024/cp/d3cp03492e]
- 84. Binding free energy of protein/ligand complexes calculated using dissociation Parallel Cascade Selection Molecular Dynamics and Markov state model - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8694779/]
- 85. Unveiling Dynamic Hotspots in Protein–Ligand Binding: Accelerating Target and Drug Discovery Approaches [https://www.mdpi.com/1422-0067/26/9/3971]
- 86. Quantifying uncertainty and sampling quality in biomolecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2865156/]
- 87. Reliability and reproducibility checklist for molecular ... [https://www.nature.com/articles/s42003-023-04653-0]
- 88. Accurate absolute free energies for ligand–protein binding based on non-equilibrium approaches | Communications Chemistry [https://www.nature.com/articles/s42004-021-00498-y]
- 89. Exploring ligand pathways on binding using... proteins [https://www.nature.com/articles/s41467-021-23157-1?error=cookies_not_supported&code=683fdc98-3cc3-44d1-8475-47abb3b645ac]
- 90. PLAS-20k: Extended Dataset of Protein - Ligand Affinities from MD ... [https://www.nature.com/articles/s41597-023-02872-y?error=cookies_not_supported&code=ce688102-9439-4d58-b257-697e2a49c432]
- 91. Constrained optimization via quantum Zeno dynamics [https://www.nature.com/articles/s42005-023-01331-9]
- 92. Docking strategies for predicting protein-ligand interactions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11583305/]
- 93. GitHub - YuzheWangPKU/awesome- protein - ligand - interactions ... [https://github.com/YuzheWangPKU/awesome-protein-ligand-interactions]
- 94. Rapid, accurate, precise and reproducible ligand–protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7653346/]
- 95. HPDAF: A Practical Tool for Predicting Drug-Target Binding ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425009183]
- 96. Protein-ligand affinity prediction via Jensen-Shannon ... [https://pubmed.ncbi.nlm.nih.gov/40901491/]
- 97. Benchmarking Protein–Ligand Interaction Energy | Rowan [https://www.rowansci.com/blog/benchmarking-protein-ligand-interaction-energy]
- 98. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 99. Tandem Allosteric Effects of Reactant and Product that ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570146/]
- 100. AMBER vs GROMACS [https://diphyx.com/stories/amber-vs-gromacs/]
- 101. Comparative Study of Molecular Mechanics Force Fields for β ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10302482/]
- 102. GROMACS Vs. AMBER for Molecular Dynamics Simulations [https://parssilico.com/blogs/85-gromacs-vs-amber-md-software]
- 103. Protein-Ligand Complex - GROMACS Tutorial [http://www.mdtutorials.com/gmx/complex/02_topology.html]
- 104. Lessons learned from comparing molecular dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5581938/]
- 105. Mdp parameters for non-bonded interaction using the AMBER ... [https://gromacs.bioexcel.eu/t/mdp-parameters-for-non-bonded-interaction-using-the-amber-force-field/6458]
- 106. Improving Accuracy and Efficiency of Blind Protein-Ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2610246/]
- 107. Decoding the limits of deep learning in molecular docking for ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc05395a]
- 108. Accurate prediction of protein–ligand interactions by ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00912-2]
- 109. Generation of appropriate protein structures for virtual ... [https://www.sciencedirect.com/science/article/pii/S2950363925000286]
- 110. A multiscale simulation approach to compute protein ... [https://chemrxiv.org/engage/chemrxiv/article-details/685a5ad83ba0887c3347dc85]
- 111. Sequence-based virtual screening using transformers [https://www.nature.com/articles/s41467-025-61833-8]