Assessing the Power of Introgression Detection Methods: A Comparative Guide for Genomic Researchers
Accurately detecting the direction of introgression—the transfer of genetic material between species or populations—is crucial for evolutionary biology, drug target discovery, and understanding disease genetics.
Assessing the Power of Introgression Detection Methods: A Comparative Guide for Genomic Researchers
Abstract
Accurately detecting the direction of introgression—the transfer of genetic material between species or populations—is crucial for evolutionary biology, drug target discovery, and understanding disease genetics. This article provides a comprehensive assessment of the power and limitations of modern introgression detection algorithms. We explore the foundational principles of 12 representative methods, including tree-based, statistical, and signal-processing approaches like S*, D-statistics, IBDmix, and IntroMap. For a research-focused audience, we detail methodological applications, troubleshoot common pitfalls like false positives from homoplasy, and present a rigorous validation framework. A key finding from recent research is that downstream analyses can yield different conclusions depending on the introgression map used, underscoring the need for a multi-method approach to ensure robust, reproducible results in biomedical research.
The Introgression Landscape: Core Concepts and Methodological Foundations
Introgression, the transfer of genetic material between species or distinct populations through hybridization and repeated backcrossing, represents a powerful evolutionary force with far-reaching implications across the tree of life. Once considered primarily a homogenizing process, research over the past decade has revealed that introgression serves as a significant mechanism for adaptation, enabling species to acquire beneficial alleles that facilitate rapid response to environmental challenges [1]. This process has been documented extensively in eukaryotes—most famously through Neanderthal introgression in modern humans—and increasingly in bacteria, where it challenges traditional concepts of species boundaries [2] [3].
The detection and analysis of introgressed genomic regions have become sophisticated endeavors, employing diverse methodological approaches including summary statistics, probabilistic modeling, and supervised learning [4]. Each method offers distinct advantages and limitations, with performance varying across evolutionary scenarios, taxonomic groups, and genomic contexts. This guide provides a systematic comparison of introgression detection methods, their experimental protocols, and their applications across biological systems—from archaic hominin DNA to bacterial core genomes—enabling researchers to select optimal approaches for their specific study systems.
Methodological Approaches for Detecting Introgression
Categories of Detection Methods
Current methods for identifying introgressed sequences fall into three primary categories, each with distinct theoretical foundations and implementation requirements. Summary statistics-based methods utilize population genetic metrics such as D-statistics and fd statistics to detect signatures of introgression from patterns of allele sharing [4]. These approaches benefit from computational efficiency and minimal demographic assumptions but offer limited power for pinpointing exact introgressed tracts. Probabilistic modeling methods employ hidden Markov models (HMMs) and related frameworks to infer introgression based on explicit demographic models. diCal-admix exemplifies this category, modeling the genealogical process along genomes to detect introgressed tracts while accounting for population history [5]. Supervised machine learning approaches such as VolcanoFinder, Genomatnn, and MaLAdapt leverage training datasets to classify genomic regions as introgressed or non-introgressed based on multiple features [6]. These methods can capture complex patterns but require extensive training data and may be sensitive to model misspecification.
Performance Comparison Across Methods
Comprehensive evaluations reveal that method performance varies significantly across evolutionary scenarios. A recent benchmark study tested VolcanoFinder, Genomatnn, and MaLAdapt on simulated datasets reflecting diverse divergence and migration times inspired by human, wall lizard (Podarcis), and bear (Ursus) lineages [6]. The results, summarized in Table 1, indicate that methods based on the Q95 summary statistic generally offer the best balance of power and precision for exploratory studies, particularly when accounting for the hitchhiking effects of adaptively introgressed mutations on flanking regions [6].
Table 1: Performance Comparison of Introgression Detection Methods
| Method | Category | Optimal Scenario | Strengths | Limitations |
|---|---|---|---|---|
| diCal-admix | Probabilistic modeling | Model-based detection in known demographic histories | Explicit demographic modeling; accurate tract length estimation | Performance depends on correct demographic model [5] |
| VolcanoFinder | Supervised learning | Adaptive introgression detection | Effectiveness in detecting selective sweeps from introgression | Variable performance across divergence times [6] |
| Genomatnn | Supervised learning | Complex introgression scenarios | Handles various introgression scenarios | Performance varies across evolutionary scenarios [6] |
| MaLAdapt | Supervised learning | Limited training data | Efficient with limited data | Lower power in some scenarios [6] |
| Q95-based methods | Summary statistics | Exploratory studies | Balanced performance; minimal assumptions | Less precise for tract boundary identification [6] |
Performance depends critically on evolutionary parameters including divergence time, migration rate, population size, selection strength, and recombination landscape [6]. Methods generally perform better with recent introgression events and stronger selection coefficients, while performance declines with increasing divergence between source and recipient populations. The genomic context of introgressed regions also significantly impacts detection power, with methods struggling more in low-recombination regions and near selective sweeps [6].
Introgression Across Biological Systems
Hominin Introgression
The study of Neanderthal introgression in modern humans represents a paradigm for understanding archaic introgression patterns and functional consequences. Genomic analyses reveal that 1-4% of genomes of present-day people outside Africa derive from Neanderthal ancestors, with these introgressed regions exhibiting distinct evolutionary fates [7]. Some Neanderthal alleles facilitated human adaptation to novel environments, including climate conditions, UV exposure levels, and pathogens, while others had deleterious consequences and were selectively removed [7].
Application of diCal-admix to 1000 Genomes Project data has revealed long regions depleted of Neanderthal ancestry that are enriched for genes, consistent with weak selection against Neanderthal variants [5]. This pattern appears driven primarily by higher genetic load in Neanderthals resulting from small effective population size rather than widespread Dobzhansky-Müller incompatibilities [5]. Notably, the X-chromosome shows particularly low levels of introgression, though the mechanistic basis for this pattern remains debated [5] [7]. Conversely, Neanderthal ancestry shows significant enrichment in genes related to hair and skin traits (keratin pathways), suggesting adaptive introgression helped modern humans adapt to non-African environments [5] [7].
Bacterial Introgression
While bacteria reproduce asexually, homologous recombination facilitates pervasive gene flow that shapes their evolution. Quantitative analyses across 50 major bacterial lineages reveal that introgression—defined here as gene flow between core genomes of distinct species—averages 2% of core genes but reaches 14% in highly recombinogenic genera like Escherichia-Shigella [2]. This challenges operational species definitions based solely on sequence identity thresholds (e.g., 95% ANI), as interruption of gene flow occurs across a range of sequence identities (90-98%) depending on the lineage [3].
Table 2: Introgression Patterns Across Bacterial Lineages
| Bacterial Group | Average Introgression Level | Notable Features | Implications for Species Definition |
|---|---|---|---|
| Escherichia–Shigella | Up to 14% of core genes | High recombination frequency | Porous species boundaries [2] |
| Campylobacter | ~20% of genome in some species | Gene flow between highly divergent species | Fuzzy species borders [2] |
| Neisseria | Variable | Recombinogenic nature | Historically noted "fuzzy" species [2] |
| Cronobacter | High levels | Extensive introgression | Challenges species delimitation [2] |
| Endosymbionts | Minimal | Clonal evolution | Clear species borders [3] |
Truly clonal bacterial species are remarkably rare, with only 2.6% of analyzed species showing no evidence of recombination [3]. These exceptional cases primarily include endosymbionts like Buchnera aphidicola with restricted access to exogenous DNA [3]. For most bacteria, homologous recombination maintains species cohesiveness while occasional introgression introduces adaptive variation across species boundaries, analogous to processes in sexual organisms [2] [3].
Experimental evolution studies with Escherichia coli demonstrate that high rates of conjugation-mediated recombination can sometimes overwhelm selection, with donor DNA segments fixation due to physical linkage to transfer origins rather than selective advantage [8]. This highlights how the mechanistic features of bacterial gene transfer can produce evolutionary outcomes distinct from eukaryotic introgression.
Experimental Protocols for Introgression Analysis
Probabilistic Modeling with diCal-admix
The diCal-admix method employs a hidden Markov model framework to detect introgressed tracts while explicitly incorporating demographic history [5]. The protocol begins with data preparation, requiring genomic sequences from the target population, putative source population, and outgroup. For Neanderthal introgression studies, this typically includes modern non-African individuals, Neanderthal reference genomes, and African individuals as an outgroup [5].
Next, model parameterization establishes key demographic parameters including divergence times, population sizes, migration rates, and introgression timing. For human-Neanderthal analyses, standard parameters include: divergence time of 26,000 generations (650 kya), Neanderthal-modern human split at 4,000 generations (100 kya), introgression event at 2,000 generations (50 kya), and introgression coefficient of 3% [5]. The HMM implementation then computes the probability of introgression along genomic windows based on patterns of haplotype sharing and differentiation, generating posterior probabilities for Neanderthal ancestry across the genome [5].
Validation through extensive simulations confirms method robustness to parameter misspecification, though accurate demographic modeling significantly enhances performance [5]. The output consists of genomic tracts with high posterior probability of introgression, which can be further analyzed for functional enrichment and selective signatures.
Figure 1: Workflow for model-based introgression detection using diCal-admix and related probabilistic approaches
Bacterial Introgression Detection
Protocols for detecting introgression in bacteria differ significantly from eukaryotic approaches due to distinct genetic system properties. The standard workflow begins with genome collection and core gene identification, assembling a comprehensive dataset of bacterial genomes within a target lineage and identifying orthologous core genes present in most strains [2].
Next, species delineation employs Average Nucleotide Identity (ANI) thresholds (typically 94-96%) to classify genomes into operational species units, followed by phylogenomic reconstruction using maximum likelihood methods on concatenated core genome alignments [2]. The core analytical step involves phylogenetic incongruence analysis, where individual gene trees are compared against the species tree to identify potential introgression events [2].
A gene is considered introgressed when it satisfies two criteria: (1) it forms a monophyletic clade with sequences from a different species that is inconsistent with the core genome phylogeny, and (2) it is statistically more similar to sequences from a different species than to sequences from its own species [2]. Finally, biological species concept refinement adjusts initial ANI-based species boundaries based on patterns of gene flow, reducing inflated introgression estimates between recently diverged populations [2].
Research Reagent Solutions
Successful introgression studies require specialized analytical tools and genomic resources. Key reagents and their applications across study systems include:
Table 3: Essential Research Reagents for Introgression Studies
| Reagent/Tool | Category | Function | Application Examples |
|---|---|---|---|
| Reference Genomes | Genomic data | Provides basis for sequence alignment and variant calling | Neanderthal genome (Altai); Bacterial reference strains [5] [2] |
| Outgroup Sequences | Genomic data | Enables polarization of ancestral/derived alleles | African genomes for Neanderthal introgression; Distantly related bacterial species [5] [3] |
| diCal-admix Software | Analytical tool | HMM-based introgression detection | Neanderthal tract identification in 1000 Genomes data [5] |
| VolcanoFinder | Analytical tool | Machine learning approach for adaptive introgression | Performance testing across multiple lineages [6] |
| ANI Calculator | Bioinformatics tool | Species delineation in bacteria | Defining bacterial species boundaries [2] [3] |
| Phylogenetic Software | Analytical tool | Species and gene tree reconstruction | Detecting phylogenetic incongruence in bacterial genes [2] |
Introgression represents a fundamental evolutionary process with comparable importance across biological domains, from Neanderthal DNA in modern humans to core genome exchanges in bacteria. Detection methods perform variably across evolutionary scenarios, with summary statistics (particularly Q95-based approaches) offering robust exploratory power, while model-based methods like diCal-admix provide finer-scale inference when demographic history is well-characterized [6] [5]. Bacterial systems present unique challenges and opportunities, with homologous recombination maintaining species cohesion while permitting adaptive introgression across porous species boundaries [2] [3].
Future methodological development should focus on improving detection power for ancient introgression events, distinguishing adaptive from neutral introgression, and integrating across taxonomic divides to develop unified theoretical frameworks. The continued expansion of genomic datasets across diverse taxa, coupled with benchmarking studies under realistic evolutionary scenarios, will further refine our ability to decode genomic landscapes of introgression and understand its creative role in evolution [4].
Statistical power, defined as the probability that a test will correctly reject a false null hypothesis, is a foundational concept that critically influences the reliability of research in both evolutionary genetics and biomedical science. Low statistical power significantly increases the likelihood that statistically significant findings represent false positive results and inflates the estimated magnitude of true effects when they are discovered [9]. In evolutionary biology, this translates to uncertainty in detecting introgression and inferring evolutionary history, while in biomedical research, it undermines the validity of associations between biological parameters and disease. Evidence suggests that underpowered studies are widespread, with one analysis of biomedical literature revealing that approximately 50% of studies have statistical power in the 0-20% range, far below the conventional 80% threshold considered adequate [9]. This review compares the performance of various methods for detecting introgression, with a specific focus on their statistical power and practical applications, providing researchers with a framework for selecting appropriate methodologies based on their specific investigative needs and constraints.
Comparative Performance of Introgression Detection Methods
The detection of introgression—the transfer of genetic material between species or populations through hybridization—relies on identifying genomic regions that show unexpected similarity between taxa. Different methods have been developed to detect these patterns, each with varying strengths, power, and susceptibility to confounding factors. The table below summarizes the key characteristics of several prominent methods.
Table 1: Comparison of Methods for Detecting Introgression
| Method | Underlying Principle | Data Requirements | Power & Strengths | Limitations & Vulnerabilities |
|---|---|---|---|---|
| D-statistic (ABBA-BABA) | Compares frequencies of discordant site patterns to detect gene tree heterogeneity [10]. | A minimum of four lineages (e.g., P1, P2, P3, Outgroup); works with a single sequence per species [10]. | High power to detect introgression between non-sister lineages; robust to selection [10]. | Requires an outgroup; cannot be used for sister species pairs [11]. |
| dXY | Measures the average pairwise sequence divergence between two populations [11]. | Can use single or multiple sequences per species; does not require phased data or an outgroup. | Robust to the effects of linked selection; provides an intuitive measure of divergence [11]. | Low sensitivity to recent or low-frequency introgression; confounded by variation in mutation rate [11]. |
| dmin | Identifies the minimum sequence distance between any pair of haplotypes from two taxa [11]. | Requires phased haplotypes from multiple individuals per species. | High power to detect rare introgressed lineages, as it focuses on the most similar haplotypes [11]. | Highly sensitive to variation in the neutral mutation rate; requires accurate phasing. |
| Gmin | The ratio of dmin to dXY, normalizing for background divergence [11]. | Requires phased haplotypes from multiple individuals per species. | More robust to mutation rate variation than dmin alone while retaining sensitivity to recent migration [11]. | Still requires phased data; power can be reduced by high background divergence. |
| RNDmin | A modified dmin statistic normalized by divergence to an outgroup [11]. | Requires an outgroup species; works with phased data from multiple individuals. | Robust to variation in mutation rate and inaccurate divergence time estimates [11]. | Modest power increase over related tests; requires an outgroup. |
| Convolutional Neural Networks (CNNs) | Deep learning models trained on genotype matrices to identify complex patterns of introgression and selection [12]. | Genomic windows with data from donor, recipient, and outgroup populations; can use unphased data. | Very high accuracy (~95%); can jointly model introgression and positive selection (adaptive introgression) [12]. | "Black box" nature makes it difficult to interpret which features drive the prediction; requires extensive training data. |
Experimental Protocols for Key Methods
Protocol for D-Statistic Analysis
The D-statistic is a powerful, widely-used method for detecting introgression that is based on counting discordant site patterns in an alignment. The following workflow outlines its key steps [10]:
- Sequence Alignment and Filtering: Generate a whole-genome alignment for the three ingroup populations (P1, P2, P3) and an outgroup (O). P1 and P2 are the tested sister species, and P3 is the potential introgressing lineage. Filter for neutral, biallelic sites to avoid confounding effects from selection.
- Site Pattern Counting: For each genomic window or site, categorize the alleles into four patterns based on the derived (non-outgroup) state:
- ABBA: P1 and O share the ancestral allele, while P2 and P3 share the derived allele.
- BABA: P1 and P3 share the derived allele, while P2 and O share the ancestral allele.
- Calculate the D-statistic: Compute the statistic across all considered sites using the formula:
- D = (∑ABBA - ∑BABA) / (∑ABBA + ∑BABA) A significant deviation from zero (often assessed via block jackknifing) indicates an excess of gene tree discordance, which is consistent with introgression between P2 and P3 (if D > 0) or P1 and P3 (if D < 0).
- Account for Incomplete Lineage Sorting (ILS): The null hypothesis of the D-statistic is that all gene tree discordance is due to ILS. The test is powerful because it expects the two discordant topologies (ABBA and BABA) to be equally frequent under ILS alone; introgression causes a systematic skew in their frequencies [10].
Protocol for RNDminAnalysis
The RNDmin method is particularly useful for detecting introgression between sister species. The protocol below details its implementation [11]:
- Data Preparation and Phasing: Obtain whole-genome sequence data from multiple individuals for the two sister species (X and Y) and an outgroup (O). Phase the haplotypes to resolve individual chromosomes.
- Calculate Minimum Divergence (dmin): For a given genomic window, compute the number of sequence differences (dx,y) for every possible pairing of haplotypes from species X and Y. Find dmin, which is the smallest of these pairwise distances.
- dmin = minx∈X,y∈Y{dx,y}
- Calculate Outgroup Divergence (dout): Compute the average sequence distance between species X and the outgroup O (dXO) and between species Y and the outgroup O (dYO). Then calculate dout.
- dout = (dXO + dYO) / 2
- Compute RNDmin:
- RNDmin = dmin / dout
- Identify Outliers: Calculate RNDmin for windows across the genome. Genomic regions with exceptionally low RNDmin values are candidate introgressed loci, as they indicate haplotypes in the sister species that are more similar to each other than expected given their collective divergence from the outgroup. The significance of candidates is typically assessed by comparing them to a null distribution generated through coalescent simulations.
Protocol for CNN-Based Detection of Adaptive Introgression
Convolutional Neural Networks (CNNs) represent a model-free approach that can detect complex patterns of adaptive introgression. The following methodology is based on the genomatnn framework [12]:
- Data Matrix Construction:
- Windowing: Partition the genome from the donor, recipient, and an unadmixed outgroup population into windows (e.g., 100 kbp).
- Binning and Encoding: Within each window, divide the sequence into equally sized bins. For each bin in each individual, encode the data as a count of minor alleles, creating a genotype matrix.
- Sorting and Concatenation: Sort the pseudo-haplotypes or genotypes within each population by their similarity to the donor population. Concatenate the matrices from the three populations into a single input matrix for the CNN.
- Model Training (Pre-training): Train the CNN using simulated data. Simulations must encompass a wide range of evolutionary scenarios, including:
- Neutral evolution with ILS.
- Selective sweeps without introgression.
- Adaptive introgression (AI) with varying selection coefficients and times of selection onset.
- Application and Inference: Apply the trained CNN to real genomic data. The network takes the genotype matrix for each window as input and outputs a probability score that the region underwent adaptive introgression. High-probability regions are considered strong AI candidates.
- Model Interpretation (Saliency Maps): To interpret the "black box" model, generate saliency maps that highlight which parts of the input genotype matrix most influenced the CNN's prediction, providing biological insight into the features of AI [12].
Visual Guide to Method Selection
The following diagram illustrates the logical decision process for selecting an appropriate introgression detection method based on the research question and data availability.
Diagram 1: A flow chart for selecting an introgression detection method.
The Scientist's Toolkit: Key Research Reagents and Solutions
Successful detection of introgression relies on a combination of bioinformatic tools, genomic resources, and analytical frameworks. The table below details essential components of the modern introgression research toolkit.
Table 2: Research Reagent Solutions for Introgression Studies
| Tool/Reagent | Type | Primary Function |
|---|---|---|
| Whole-Genome Sequencing Data | Genomic Data | Provides the raw nucleotide variation data required for all downstream analyses. Can be derived from a single individual or multiple individuals per species/population. |
| Phased Haplotypes | Processed Data | Resolved sequences of alleles on individual chromosomes, which are essential for methods like dmin, Gmin, and RNDmin that rely on pairwise haplotype comparisons [11]. |
| Reference Genome & Annotation | Genomic Resource | Serves as a coordinate system for alignment and allows for the functional interpretation of candidate introgressed regions (e.g., identifying genes). |
| stdpopsim & SLiM | Simulation Software | Provides a standardized framework for generating realistic genomic data under complex evolutionary models, which is critical for training CNNs and creating null distributions for summary statistics [12]. |
| genomatnn (CNN Framework) | Software/Method | A dedicated convolutional neural network pipeline for detecting adaptive introgression from genotype data, offering high accuracy even on unphased genomes [12]. |
| Outgroup Genome | Genomic Data | A genome from a lineage known to not have hybridized with the study species, which is required for polarizing alleles (D-statistic) and normalizing divergence (RNDmin) [10] [11]. |
The choice of method for detecting introgression has profound implications for the power, accuracy, and biological validity of evolutionary inferences. Summary statistics like the D-statistic and RNDmin offer powerful, intuitive, and computationally efficient approaches for specific phylogenetic contexts, while emerging deep learning techniques like CNNs provide unparalleled ability to detect complex patterns of adaptive introgression by leveraging the full information content of genomic data. The pervasive issue of low statistical power in biological research underscores the necessity of selecting methods with high discriminatory power and of designing studies with adequate sample sizes and sequencing depth. By carefully matching the methodological approach to the biological question and available data, researchers can more reliably uncover the historical and adaptive significance of introgression in shaping biodiversity.
The detection of introgressed genomic regions—where genetic material has been transferred between species or populations through hybridization and backcrossing—has become a fundamental analysis in evolutionary genetics. As genomic datasets expand across diverse taxa, the methodological landscape for identifying introgression has diversified into three major algorithmic families: reference-based, reference-free, and simulation-based methods [4]. Each approach offers distinct advantages and limitations, with performance varying significantly across different evolutionary scenarios.
Understanding the power of these methods to correctly identify the direction of introgression—which population donated genetic material and which received it—is particularly crucial for reconstructing accurate evolutionary histories [13]. This guide provides a systematic comparison of these methodological families, focusing on their underlying principles, experimental requirements, and empirical performance based on published benchmarking studies.
Methodological Families for Introgression Detection
Reference-Based Methods
Reference-based methods require genomic data from the putative introgressing (donor) population, which is used as a reference to identify foreign haplotypes in a target population.
IntroMap exemplifies this approach by employing signal processing techniques on next-generation sequencing data aligned to a reference genome. The pipeline identifies introgressed regions by detecting significant divergence in sequence homology without requiring variant calling or genome annotation. The method converts alignment information into a binary representation of matches/mismatches, applies signal averaging to reduce noise, and uses statistical thresholding to call introgressed regions [14]. This method is particularly valuable in plant breeding programs where one parental genome is available as a reference.
Key advantage: High accuracy when suitable reference genomes are available. Primary limitation: Limited applicability to scenarios involving "ghost" lineages or unsampled extinct populations.
Reference-Free Methods
Reference-free methods detect introgression without direct comparison to archaic reference genomes, instead leveraging population genetic patterns characteristic of admixed haplotypes.
ArchIE (ARCHaic Introgression Explorer) employs a logistic regression model trained on population genetic summary statistics to infer archaic local ancestry. The method combines multiple features including the individual frequency spectrum (IFS), pairwise haplotype distances, and their statistical moments to distinguish introgressed from non-introgressed regions [15]. This approach is particularly valuable for detecting introgression from unknown or unsampled archaic populations.
The S*-statistic is another reference-free method that identifies introgressed regions by detecting clusters of highly diverged single nucleotide polymorphisms (SNPs) in high linkage disequilibrium [15]. However, its power is generally lower than model-based approaches, especially for ancient introgression events [15].
Key advantage: Applicable to cases where reference genomes from donor populations are unavailable. Primary limitation: Generally lower power compared to reference-based approaches.
Simulation-Based Methods
Simulation-based approaches use training data generated under explicit evolutionary models to distinguish different introgression scenarios.
genomatnn implements a convolutional neural network (CNN) framework that takes genotype matrices as input to identify regions under adaptive introgression. The method uses a series of convolution layers to extract features informative of both introgression and selection, outputting the probability that a genomic region underwent adaptive introgression [16]. The CNN is trained on simulated data encompassing a wide range of selection coefficients and timing parameters, enabling detection of complete or incomplete sweeps at any time after gene flow.
MaLAdapt is another machine learning method that employs a random forest classifier trained on summary statistics to detect adaptive introgression. Its performance varies across different evolutionary scenarios but shows particular strength in cases of strong selection [6].
Key advantage: Can jointly model complex processes like introgression and selection. Primary limitation: Performance depends on the match between simulated training data and real evolutionary history.
Comparative Performance Analysis
Performance Across Evolutionary Scenarios
Recent benchmarking studies have evaluated these method families across diverse evolutionary scenarios, including those inspired by human, wall lizard (Podarcis), and bear (Ursus) lineages [6]. These lineages represent different combinations of divergence times and migration histories, providing a robust framework for comparing methodological performance.
Table 1: Performance Metrics Across Method Families
| Method Family | Example Tools | Power | False Discovery Rate | Direction Detection | Optimal Scenario |
|---|---|---|---|---|---|
| Reference-based | IntroMap | High [14] | Low [14] | High [14] | Donor genome available |
| Reference-free | ArchIE, S* | Moderate [15] | Variable [15] [17] | Limited [15] | Ghost introgression |
| Simulation-based | Genomatnn, MaLAdapt, VolcanoFinder | High [16] [6] | Low [16] | Moderate [16] | Complex introgression |
Impact of Genomic Context on Performance
A critical finding from comparative studies is that the genomic context of introgressed regions significantly impacts detection accuracy across all methods. The "hitchhiking effect" of an adaptively introgressed mutation affects flanking regions, making it challenging to discriminate between truly adaptive windows and adjacent neutral regions [6]. Performance metrics improve substantially when methods are trained to account for this effect by including adjacent windows in training data [6].
Table 2: Power Analysis Under Different Selection Strengths (Q95 Statistic) [6]
| Selection Coefficient | Divergence Time (Generations) | Power (Strongly Asymmetric Migration) | Power (Symmetric Migration) |
|---|---|---|---|
| 0.01 | 60,000 | 0.92 | 0.85 |
| 0.01 | 120,000 | 0.89 | 0.81 |
| 0.001 | 60,000 | 0.87 | 0.79 |
| 0.001 | 120,000 | 0.83 | 0.75 |
| 0.0001 | 60,000 | 0.75 | 0.68 |
| 0.0001 | 120,000 | 0.71 | 0.64 |
Performance in Direction Detection
Accurately determining the direction of introgression remains challenging for many methods. Full-likelihood approaches under the multispecies coalescent (MSC) framework generally provide the most reliable inference of directionality [13]. However, even these methods can produce biased estimates when gene flow is incorrectly assigned to ancestral rather than daughter lineages [13].
Summary statistic methods like the D-statistic (ABBA-BABA test) often struggle with direction detection, particularly for gene flow between sister lineages [13]. In comparative studies, the D-statistic demonstrated high false discovery rates, especially under scenarios with high incomplete lineage sorting [17].
Experimental Protocols and Workflows
Reference-Based Detection Workflow (IntroMap)
The IntroMap pipeline employs the following methodology [14]:
Sequence Alignment: NGS reads are aligned to a reference genome using standard tools (e.g., bowtie2) to produce BAM format alignment files.
Binary Representation: The MD tags in BAM files are parsed to create binary vectors for each read position, where 1 represents a match and 0 represents a mismatch/deletion.
Matrix Construction: Binary vectors are assembled into a sparse matrix C[d,l] where d represents read depth and l represents nucleotide position.
Signal Processing: Per-base calling scores are computed and smoothed using a low-pass filter convolution with a window vector of length w.
Homology Estimation: A locally weighted linear regression fit generates a homology signal hc, with values [0,1] representing the degree of homology at each position.
Threshold Detection: A threshold function T(hc,t) identifies regions where homology scores drop significantly, indicating potential introgression.
Reference-Free Detection Workflow (ArchIE)
The ArchIE methodology employs the following steps [15]:
Training Data Simulation: Coalescent simulations (e.g., using ms) generate genomic data under specified demographic models with known introgression events.
Feature Calculation: For each genomic window, multiple summary statistics are computed:
- Individual Frequency Spectrum (IFS)
- Pairwise haplotype distances and their moments (mean, variance, skew, kurtosis)
- Population differentiation metrics
Model Training: A logistic regression classifier is trained on the simulated data to distinguish introgressed from non-introgressed windows.
Application to Empirical Data: The trained model is applied to empirical genomic data to infer posterior probabilities of introgression.
Simulation-Based CNN Workflow (genomatnn)
The genomatnn framework implements the following protocol [16]:
Data Preparation: Genotype matrices are constructed from donor, recipient, and unadmixed outgroup populations for each genomic window (typically 100 kbp).
Matrix Sorting: Haplotypes within each population are sorted by similarity to the donor population.
Input Construction: Sorted matrices are concatenated into a single input matrix for the CNN.
CNN Architecture: The network uses:
- Multiple convolutional layers with 2×2 step size (instead of pooling layers)
- Feature extraction through successive dimensionality reduction
- Final classification layer outputting AI probability
Model Interpretation: Saliency maps identify genomic regions most influential to predictions.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Tool Category | Specific Tools | Function | Application Context |
|---|---|---|---|
| Sequence Aligners | bowtie2 | Alignment of NGS reads to reference genomes | Reference-based methods [14] |
| Coalescent Simulators | ms, msprime, stdpopsim | Generate simulated genomic data under evolutionary models | Training data for reference-free and simulation methods [15] [16] [18] |
| Population Genetics Frameworks | Python, R, Scientific Python | Compute summary statistics and implement custom analyses | All methodological families [14] [15] |
| Machine Learning Libraries | TensorFlow, PyTorch | Implement neural networks for classification | Simulation-based methods [16] [18] |
| Visualization Tools | matplotlib, ggplot2 | Create publication-quality figures | Results presentation and quality control [14] |
The three major algorithmic families for introgression detection each offer complementary strengths for different research scenarios. Reference-based methods provide the highest accuracy when reference genomes from donor populations are available. Reference-free approaches enable detection of introgression from unknown or unsampled populations. Simulation-based methods offer powerful frameworks for detecting complex evolutionary scenarios like adaptive introgression.
For researchers specifically interested in determining the direction of introgression, full-likelihood methods under the multispecies coalescent framework currently provide the most reliable inference, despite computational intensivity [13]. The choice of method should be guided by data availability, evolutionary context, and specific research questions, with particular attention to recent benchmarking studies that validate performance across diverse scenarios.
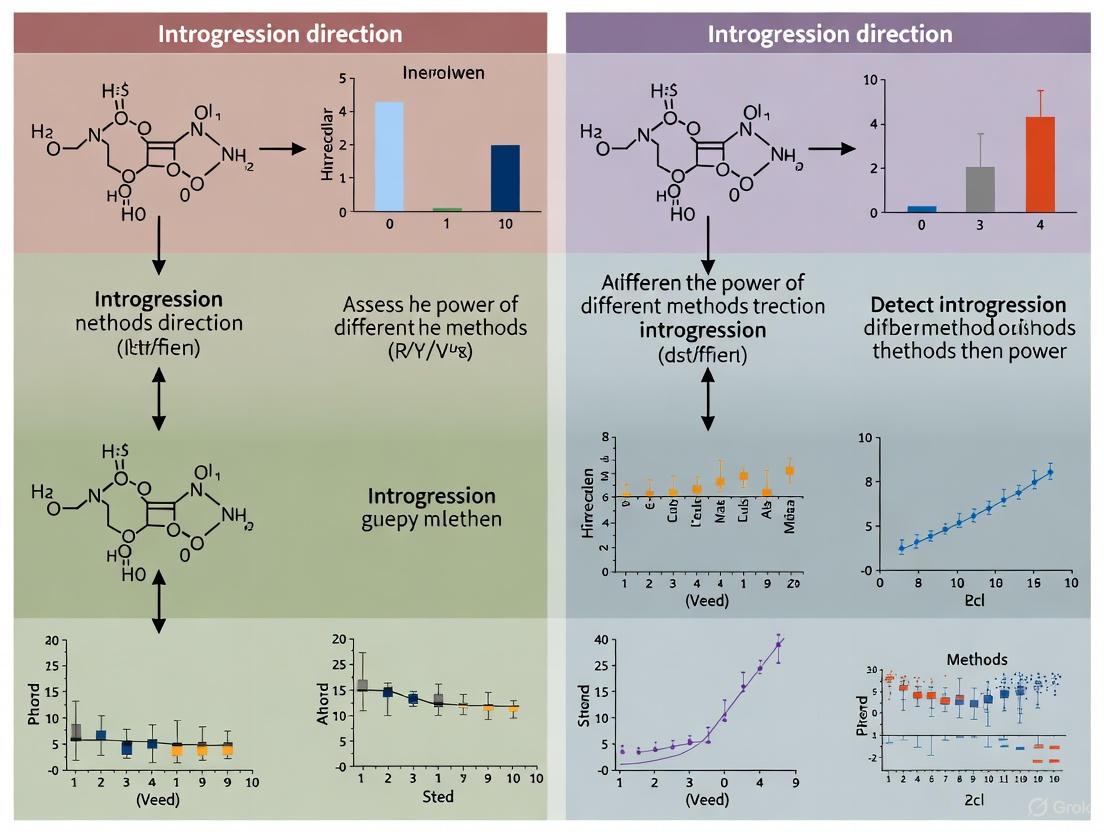
The accurate detection of introgression direction—the flow of genetic material between species—is fundamental to understanding evolutionary processes, adaptation, and speciation. However, validating methodological approaches in this field is critically hampered by the "ground truth problem": the fundamental lack of a perfectly known, real-world standard against which to benchmark performance. Without a biological gold standard, researchers must rely on comparative performance assessments using simulated datasets, well-established model systems, and internal consistency checks to evaluate the power and accuracy of different analytical techniques. This guide objectively compares the performance of leading methods for detecting introgression direction, providing researchers with a framework for selecting and applying these tools amidst the inherent uncertainties of evolutionary genomics.
Core Methodologies for Detecting Introgression Direction
Methods for detecting introgression direction can be broadly categorized into likelihood-based frameworks and summary-statistic approaches. The table below summarizes their fundamental characteristics and data requirements.
Table 1: Core Methodological Frameworks for Introgression Detection
| Method Category | Key Example(s) | Underlying Principle | Data Requirements | Primary Output |
|---|---|---|---|---|
| Likelihood / Bayesian | MSC-I (Multispecies Coalescent with Introgression) [19] | Computes the probability of the observed sequence data given a model of speciation and gene flow, including direction. | Multi-locus sequence alignments, a pre-specified species tree model. | Estimates of introgression probability (φ), its timing, and direction, with Bayesian posterior probabilities. |
| Summary Statistic | D-statistic (ABBA-BABA) [20] | Compares counts of discordant site patterns (ABBA vs. BABA) to detect gene flow, can be extended to infer direction. | Genotype or sequence data for a 4-taxon set (P1, P2, P3, Outgroup); can use allele frequencies. | A significant D-value indicates gene flow; direction is inferred from the specific taxon sharing derived alleles. |
| Summary Statistic | RNDmin, Gmin [11] | Uses minimum sequence divergence between populations (normalized by an outgroup) to identify recently introgressed haplotypes. | Phased haplotypes from two sister species and an outgroup. | A value significantly lower than the genomic background indicates introgression; direction can be inferred from haplotype pairing. |
The following diagram illustrates the logical workflow for applying and validating these methods in the absence of a perfect biological ground truth.
Quantitative Performance Comparison
The power and accuracy of these methods vary significantly based on evolutionary parameters such as population size, divergence time, and the strength and direction of gene flow. The following performance data is synthesized from simulation studies.
Table 2: Performance Comparison of Introgression Detection Methods Under Different Scenarios
| Method | Key Performance Metric | Scenario of High Power / Key Finding | Scenario of Reduced Power / Limitation |
|---|---|---|---|
| MSC-I (Bayesian) [19] | Accuracy in inferring direction (A→B vs. B→A). | Easier to infer gene flow from a small to a large population (power > 80% under simulated conditions). Easier with longer time between divergence and introgression. | Power is reduced when gene flow is from a large to a small population. Requires correct species tree and model specification. |
| D-Statistic [20] | Significance of D-value (Z-score > 3). | Robust across a wide range of divergence times. Effective at detecting recent and ancient gene flow. | Sensitivity is highly dependent on population size (scaled by generations). Power drops with smaller population sizes. Cannot detect gene flow between sister species without additional modification. |
| RNDmin / Gmin [11] | Proportion of true introgressed loci detected (True Positive Rate). | Offers a modest increase in power over related statistics (e.g., FST, dXY). Robust to variation in mutation rate. | Requires phased haplotypes. Power is contingent on the strength and recency of introgression; older, weaker events are harder to detect. |
Detailed Experimental Protocols
To ensure reproducibility and critical evaluation, this section outlines the core experimental and analytical protocols for the featured methods.
Protocol for MSC-I Analysis Using BPP
This protocol employs the Bayesian software BPP under the Multispecies Coalescent with Introgression model [19].
- Input Data Preparation: Compile a multi-locus sequence alignment file. Prepare a control file specifying the species tree topology with an introgression branch (e.g.,
A->B). Define prior distributions for parameters (e.g., divergence timestau, population sizestheta, introgression probabilityphi). - MCMC Analysis: Run a Markov Chain Monte Carlo (MCMC) analysis for a sufficient number of generations (e.g., 100,000-1,000,000), discarding the initial samples as burn-in. Monitor convergence using trace plots and effective sample size (ESS) diagnostics.
- Model Comparison: To test for introgression, compare the model with gene flow against a null model without gene flow using Bayes factors (calculated from marginal likelihoods, e.g., with stepping-stone sampling). A significant Bayes factor (e.g., > 10) supports the presence of gene flow.
- Inference of Direction: The direction of introgression is inherent in the specified model. The estimated introgression probability (
phi) and its Bayesian credibility interval are directly interpreted for the specified direction (e.g.,A->B). Analysis should be run with the alternative direction model (B->A) and the model with higher marginal likelihood is preferred.
Protocol for D-Statistic Analysis
This protocol details the steps for detecting and inferring the direction of gene flow using the D-statistic [20].
- Taxon Selection and Data Preparation: Select four taxa with a known phylogeny:
P1,P2(sister species),P3(the potential introgressing species), and anOutgroup. The phylogeny must be((P1,P2),P3),Outgroup). Generate a genome-wide SNP dataset or sequence alignment for these taxa. - Site Pattern Counting: For each informative site in the genome, count the occurrences of ancestral (
A) and derived (B) alleles. A site isABBAifP1andOutgrouphave the ancestral allele, whileP2andP3have the derived allele. A site isBABAifP1andP3have the derived allele, whileP2andOutgrouphave the ancestral allele. - Calculation and Testing: Calculate the D-statistic as
D = (Sum(ABBA) - Sum(BABA)) / (Sum(ABBA) + Sum(BABA)). Perform a statistical test (e.g., block jackknife) to determine ifDsignificantly deviates from zero. A significant positiveDsuggests gene flow betweenP3andP2; a significant negativeDsuggests gene flow betweenP3andP1.
Visualizing Method Workflows
The computational workflow for a comprehensive analysis, integrating multiple methods to overcome their individual limitations, is depicted below.
The Scientist's Toolkit: Essential Research Reagents & Materials
Successful research in this field relies on a combination of bioinformatic tools, genomic resources, and model systems.
Table 3: Key Research Reagent Solutions for Introgression Studies
| Tool / Resource | Type | Primary Function in Analysis | Relevance to Ground Truth |
|---|---|---|---|
| BPP Software Suite [19] | Bioinformatics Tool | Implements Bayesian MCMC analysis under the MSC and MSC-I models for estimating species trees, divergence times, and introgression parameters. | A primary method for likelihood-based inference of direction, performance of which is tested via simulation. |
| Phased Haplotype Data | Genomic Resource | High-quality reference genomes or population genomic data where the phase of alleles (which chromosome they reside on) is known. | Required for methods like RNDmin. The quality of phasing directly impacts the accuracy of the ground truth signal in empirical data. |
| Heliconius Butterfly Genomes [19] | Model System | A well-studied system with known and adaptive introgression, used as an empirical benchmark for method validation. | Serves as a "known-positive" empirical test case where methodological inferences can be compared to established biological knowledge. |
| Coalescent Simulators (e.g., ms, msprime) | Computational Tool | Generates synthetic genomic sequence data under user-specified evolutionary models (divergence times, population sizes, migration). | Creates a controlled "synthetic ground truth" where the history of gene flow is known exactly, enabling rigorous power assessments and false positive rate calculations. |
The precise identification of introgressed genomic regions is a fundamental challenge in evolutionary biology, with significant implications for understanding adaptation, speciation, and disease. As genomic datasets expand across diverse taxa, researchers are presented with an array of methodological approaches for detecting introgression, each with distinct strengths, limitations, and underlying assumptions [4]. This comparison guide provides an objective evaluation of current methods for identifying a core set of introgressed regions, focusing on areas of consensus across different analytical frameworks. The performance assessment is framed within the broader thesis of evaluating statistical power across different methodological approaches, providing researchers with evidence-based guidance for selecting appropriate tools based on their specific study systems and evolutionary questions. With the growing recognition that introgression serves as a crucial evolutionary force promoting adaptation across taxonomic groups [1], the need for robust and reliable detection methods has never been more pressing. This guide synthesizes recent benchmarking studies to illuminate the conditions under which different methods achieve consensus and where their interpretations diverge, thereby empowering researchers to make informed decisions in their introgression detection workflows.
Methodological Approaches to Introgression Detection
Current methods for detecting introgression can be broadly categorized into three major frameworks: summary statistics, probabilistic modeling, and supervised learning approaches [4]. Each category operates on different principles and makes different assumptions about the underlying evolutionary processes.
Summary statistics represent some of the earliest approaches for detecting introgression and continue to evolve with new implementations that broaden their applicability across taxa [4]. These methods typically compute measures of genetic divergence, similarity, or allele frequency differences that are expected to deviate from neutral expectations in introgressed regions. Their relative simplicity and computational efficiency make them particularly valuable for initial exploratory analyses and for studying non-model organisms with less well-characterized demographic histories.
Probabilistic modeling approaches provide a powerful framework that explicitly incorporates evolutionary processes and has yielded fine-scale insights across diverse species [4]. These methods typically use coalescent-based or hidden Markov model frameworks to infer the probability of introgression given the observed genetic data and a specified demographic model. While often computationally intensive, they can provide more detailed insights into the timing, direction, and extent of introgression when appropriate demographic models are available.
Supervised learning represents an emerging approach with great potential, particularly when the detection of introgressed loci is framed as a semantic segmentation task [4]. These machine learning methods can capture complex, multi-dimensional patterns in genetic data that might be difficult to summarize with individual statistics. Their performance, however, is highly dependent on the quality and representativeness of training data, and they may struggle when applied to evolutionary scenarios different from those used in training [6] [21].
Table 1: Major Methodological Categories for Introgression Detection
| Category | Underlying Principle | Key Advantages | Common Tools |
|---|---|---|---|
| Summary Statistics | Measures deviation from expected patterns under neutrality | Fast computation; minimal assumptions; good for exploratory analysis | (f)-statistics; (D)-statistics; (Q_{95}) |
| Probabilistic Modeling | Explicit models of evolutionary processes incorporating gene flow | Provides detailed parameter estimates; model-based confidence intervals | VolcanoFinder; ∂a∂i |
| Supervised Learning | Pattern recognition trained on simulated or known introgressed regions | Can capture complex, multi-dimensional patterns; high accuracy in trained scenarios | MaLAdapt; Genomatnn |
Figure 1: Workflow for Identifying Consensus Introgressed Regions Across Multiple Methods
Performance Benchmarking Across Evolutionary Scenarios
Recent systematic benchmarking efforts have revealed that method performance varies significantly across different evolutionary scenarios, with no single approach universally outperforming others in all conditions. A comprehensive evaluation of adaptive introgression classification methods tested three prominent tools (VolcanoFinder, Genomatnn, and MaLAdapt) and a standalone summary statistic ((Q_{95})) across simulated datasets representing various evolutionary histories inspired by human, wall lizard (Podarcis), and bear (Ursus) lineages [6] [21]. These lineages were specifically chosen to represent different combinations of divergence and migration times, providing a robust test of method performance across diverse evolutionary contexts.
The benchmarking study examined the impact of multiple parameters on method performance, including divergence time, migration rate, population size, selection coefficient, and the presence of recombination hotspots [6]. Performance was evaluated based on both power (the ability to correctly identify truly introgressed regions) and false positive rates (the incorrect identification of non-introgressed regions as introgressed). Importantly, the study also investigated how different types of non-adaptive introgression windows affected performance, including independently simulated neutral introgression windows, windows adjacent to regions under selection, and windows from unlinked chromosomes [6].
Table 2: Performance Comparison of Introgression Detection Methods Across Scenarios
| Method | Approach Type | Human Model Performance | Non-Human Model Performance | Optimal Application Context |
|---|---|---|---|---|
| (Q_{95}) | Summary statistic | Moderate to high | High across scenarios | Exploratory studies; non-model organisms |
| VolcanoFinder | Probabilistic modeling | High | Variable depending on divergence | Well-characterized demographic histories |
| MaLAdapt | Supervised learning | High | Lower when training scenario mismatch | Scenarios similar to training data |
| Genomatnn | Supervised learning | High | Lower when training scenario mismatch | Human and primate studies |
One of the most notable findings from these benchmarking efforts was that (Q_{95}), a straightforward summary statistic, performed remarkably well across most scenarios and often outperformed more complex machine learning methods, particularly when applied to species or demographic histories different from those used in training data [21]. This surprising result suggests that simple summary statistics remain valuable tools, especially for initial exploratory analyses in non-model systems.
The performance of machine learning-based methods like MaLAdapt and Genomatnn was generally high when applied to evolutionary scenarios similar to their training data but decreased when applied to different demographic contexts [6] [21]. This highlights the importance of considering evolutionary context when selecting methods and suggests that retraining may be necessary when applying these tools to divergent study systems.
Experimental Protocols for Method Evaluation
Simulation Framework for Power Assessment
Benchmarking studies evaluating introgression detection methods typically employ sophisticated simulation frameworks that generate genomic data under known evolutionary scenarios with and without introgression. The protocol generally follows these key steps:
Scenario Definition: Researchers first define evolutionary parameters based on real biological systems, typically including divergence times, migration times, effective population sizes, selection coefficients, and recombination rates [6]. These parameters are often derived from well-studied systems such as humans, wall lizards (Podarcis), and bears (Ursus) to represent diverse evolutionary histories.
Data Simulation: Genomic data is simulated using coalescent-based approaches that incorporate the defined parameters. Studies often utilize tools such as msprime [6] to generate sequence data under realistic demographic models with specified gene flow events.
Method Application: Each detection method is applied to the simulated datasets using standardized parameters and thresholds. This includes both complex machine learning approaches (MaLAdapt, Genomatnn) and simpler summary statistics ((Q_{95})).
Performance Calculation: Power and false positive rates are calculated by comparing method outputs to the known simulated truth. Performance metrics typically include area under the curve (AUC) of receiver operating characteristic (ROC) curves, precision-recall curves, and true/false positive rates at specific thresholds [6] [21].
Evaluation of Different Neutral Backgrounds
A critical aspect of method evaluation involves testing performance against different types of neutral genomic regions, as the hitchhiking effect of an adaptively introgressed mutation can strongly impact flanking regions and complicate discrimination between adaptive and neutral introgression [6]. The experimental protocol typically includes:
- Independent neutral simulations: Genomic windows simulated without any selection or introgression.
- Adjacent windows: Regions flanking simulated adaptive introgression sites, which may be affected by linked selection.
- Unlinked chromosomal regions: Windows from chromosomes completely unlinked to those under selection.
This comprehensive approach helps researchers understand how different methods perform in distinguishing true adaptive introgression from neutral patterns and linked selection effects [6].
Successful detection and validation of introgressed regions requires careful selection of computational tools, statistical frameworks, and data resources. The following table summarizes key solutions available to researchers in this field.
Table 3: Research Reagent Solutions for Introgression Detection Studies
| Resource Type | Specific Tools/Resources | Function and Application |
|---|---|---|
| Simulation Tools | msprime [6]; SLiM | Generate synthetic genomic data under specified evolutionary scenarios for method testing and validation |
| Summary Statistics | (Q_{95}) [6] [21]; (f)-statistics | Calculate measures of genetic divergence and similarity to detect deviations from neutral expectations |
| Probabilistic Models | VolcanoFinder [6] [21]; ∂a∂i | Implement model-based approaches that explicitly incorporate demographic history and selection |
| Machine Learning Tools | MaLAdapt [6] [21]; Genomatnn [6] [21] | Apply trained classifiers to identify introgressed regions based on multi-dimensional patterns |
| Visualization & Analysis | R; Python; GENESPACE [22] | Visualize and interpret introgression results; analyze synteny and structural variation |
Consensus and Discordance in Detected Regions
The identification of a core set of introgressed regions requires careful consideration of consensus across methods, as different approaches may highlight distinct genomic intervals. Studies indicate that while different methods often show substantial overlap in regions with strong signals of introgression, the agreement decreases for weaker signals or in more complex evolutionary scenarios [6].
Areas of strongest consensus typically include regions with recent, strong selective sweeps and high-frequency introgressed haplotypes [6] [21]. These regions are more readily detected by multiple methodological approaches, providing greater confidence in their identification. Conversely, regions with older introgression events, weaker selection, or complex demographic histories often show greater discordance across methods, reflecting differences in statistical power and underlying assumptions.
The hitchhiking effect presents a particular challenge for establishing consensus, as methods vary in their ability to distinguish the core introgressed site from flanking regions [6]. This has practical implications for determining the precise boundaries of introgressed segments and for identifying the specific adaptive variants responsible for selection.
Guidelines for Method Selection and Application
Based on recent benchmarking studies, researchers can follow these evidence-based guidelines for selecting and applying introgression detection methods:
For exploratory studies in non-model organisms, begin with summary statistics like (Q_{95}), which show robust performance across diverse evolutionary scenarios without requiring extensive training data [21].
When working with well-characterized demographic histories, probabilistic approaches like VolcanoFinder can provide more detailed insights into the timing and strength of selection [6].
For systems similar to human evolutionary history, machine learning methods like Genomatnn and MaLAdapt show high performance but should be retrained or validated when applied to divergent taxa [6] [21].
To establish a high-confidence set of introgressed regions, prioritize regions identified by multiple methods with different underlying assumptions, as consensus across approaches provides stronger evidence [6].
Always consider adjacent genomic windows when interpreting results, as the hitchhiking effect can influence detection probabilities in flanking regions and lead to false positives if not properly accounted for [6].
These guidelines emphasize that method choice should be informed by biological context, and that a combination of approaches often yields the most reliable results [21]. As the field continues to evolve, systematic benchmarking across diverse evolutionary scenarios will remain essential for developing and validating new methods for detecting introgressed regions.
A Practical Guide to Prominent Introgression Detection Algorithms and Tools
The study of introgression, the transfer of genetic material between species or populations through hybridization and backcrossing, has been revolutionized by advances in genome sequencing and computational phylogenetics. The precise identification of introgressed loci is a rapidly evolving area of research, providing valuable insights into evolutionary history, adaptation, and the complex web of interactions between lineages [4]. For researchers and drug development professionals, understanding these genetic exchanges can illuminate pathways of disease resistance, environmental adaptation, and functional genetic diversity.
This guide focuses on three powerful tree-based methods for detecting introgression: the summary statistic-based approaches S* and Sprime, and the model-based method ARGweaver-D. Each offers distinct advantages for characterizing genomic landscapes of introgression across diverse evolutionary scenarios, including adaptive and "ghost" introgression from unsampled populations [4]. We objectively compare their performance, experimental requirements, and applicability to help researchers select the optimal tool for specific introgression detection challenges.
S* is a summary statistic designed to identify archaic introgression without reference panels from putative archaic populations. It leverages the principles of the D-statistic (ABBA-BABA test) but enhances sensitivity to older introgression events by incorporating information from a large number of individuals from the recipient population. The method scans the genome for regions with an excess of derived alleles and high divergence from an outgroup, which are characteristic signatures of archaic ancestry [23].
Sprime is an evolution of the S* method that uses a hidden Markov model (HMM) to better delineate the boundaries of introgressed segments. It improves upon S* by more accurately estimating the length of introgressed haplotypes, which is particularly valuable for studying older introgression events where recombination has broken down archaic segments into smaller pieces [23].
ARGweaver-D represents a fundamentally different approach, using a probabilistic framework to sample Ancestral Recombination Graphs (ARGs) conditional on a user-defined demographic model that includes population splits and migration events [23]. As a major extension of the ARGweaver algorithm, ARGweaver-D can infer local genetic relationships and identify migrant lineages along the genome, providing a powerful method for detecting even ancient introgression events [23].
Table: Comparison of S, Sprime, and ARGweaver-D Methodological Characteristics*
| Characteristic | S* | Sprime | ARGweaver-D |
|---|---|---|---|
| Methodological Category | Summary statistic | Summary statistic with HMM | Probabilistic modeling of ARGs |
| Underlying Principle | Excess of derived alleles and high divergence | HMM-refined haplotype identification | Bayesian sampling of genealogies with migration |
| Demographic Model Requirement | No | No | Yes (user-defined) |
| Key Advantage | No need for archaic reference panels | Better resolution of segment boundaries | Can detect older, more complex introgression |
| Computational Intensity | Moderate | Moderate | High |
Performance Comparison and Experimental Data
Each method demonstrates distinct strengths under different evolutionary scenarios. S* and Sprime excel at detecting relatively recent introgression into modern humans, having been optimized for this specific problem [23]. However, they face limitations for older proposed migration events, such as gene flow from ancient humans into Neanderthals (Hum→Nea) or from super-archaic hominins into Denisovans (Sup→Den) [23].
ARGweaver-D shows remarkable power for detecting both recent and ancient introgression. In simulation studies, it successfully identifies regions introgressed from Neanderthals and Denisovans into modern humans, even with limited genomic data [23]. More significantly, it maintains power for older gene-flow events, including Hum→Nea, Sup→Den, and introgression from unknown archaic hominins into Africans (Sup→Afr) [23].
Application of ARGweaver-D to real hominin genomes revealed that approximately 3% of the Neanderthal genome was putatively introgressed from ancient humans, with estimated gene flow occurring 200-300 thousand years ago [23]. The method also predicted that about 1% of the Denisovan genome was introgressed from an unsequenced, highly diverged archaic hominin ancestor, with roughly 15% of these "super-archaic" regions subsequently passing into modern humans [23].
Table: Empirical Performance on Hominin Introgression Detection
| Introgression Event | S*/Sprime Performance | ARGweaver-D Performance | Key Findings |
|---|---|---|---|
| Neanderthal→Modern Humans | Well-powered for detection | Successfully detects even with few samples | Identifies 1-3% of non-African genomes as Neanderthal-derived [23] |
| Denisovan→Modern Humans | Well-powered for detection | Successfully detects with high confidence | Identifies 2-4% of Oceanian genomes as Denisovan-derived [23] |
| Ancient Humans→Neanderthal | Limited power | Confidently detects | Predicts 3% of Neanderthal genome from ancient humans [23] |
| Super-Archaic→Denisovan | Limited power | Confidently detects | Predicts 1% of Denisovan genome from unsequenced archaic [23] |
Experimental Protocols and Workflows
S* and Sprime Implementation
The implementation of S* and Sprime typically follows a standardized workflow. For S, the analysis begins with genome-wide calculation of the S statistic, which identifies regions with an excess of derived alleles and high divergence. These candidate regions are then subjected to filtering based on predefined thresholds to eliminate false positives. Finally, the boundaries of putative introgressed segments are refined, and their lengths are estimated.
Sprime builds upon this foundation by incorporating a hidden Markov model to improve boundary detection. The workflow involves similar initial identification of candidate regions using the S* statistic, followed by application of an HMM to more precisely delineate segment boundaries. The HMM parameters are trained on the data, and the Viterbi algorithm is typically used to decode the most likely path of introgressed segments. Finally, posterior probabilities are calculated for each putative introgressed segment to assess confidence.
ARGweaver-D Implementation
The ARGweaver-D workflow is more complex due to its model-based nature. The initial critical step involves specifying a demographic model that includes population divergence times, effective population sizes, and potential migration events. The algorithm then employs a Markov Chain Monte Carlo (MCMC) approach to sample ancestral recombination graphs (ARGs) conditional on this demographic model. From these sampled ARGs, migrant lineages are identified, representing potential introgression events. Finally, probabilities of introgression are calculated along the genome, providing a fine-scale map of gene flow events.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of these introgression detection methods requires specific computational resources and data inputs. The following table outlines key components of the research toolkit for phylogenetic analysis of introgression.
Table: Essential Research Reagents and Materials for Introgression Analysis
| Tool/Resource | Function/Purpose | Implementation Notes |
|---|---|---|
| High-Coverage Genomes | Primary input data for analysis | Multiple individuals per population enhance power [23] |
| Demographic Model | Population history framework (for ARGweaver-D) | Required for ARGweaver-D; includes divergence times and migration events [23] |
| Outgroup Sequence | Rooting phylogenetic trees and polarizing alleles | Essential for D-statistics and S* calculation; e.g., chimpanzee for hominin studies [23] |
| Reference Panels | Context for allele frequency spectra | Useful for S* but not required; can leverage existing datasets like 1000 Genomes |
| Computational Cluster | High-performance computing resources | Essential for ARGweaver-D MCMC sampling; reduces runtime for all methods |
The comparison of S, Sprime, and ARGweaver-D reveals a fundamental trade-off between computational efficiency and analytical power in introgression detection. Summary statistic methods like S and Sprime offer accessible approaches for detecting recent introgression, while ARGweaver-D provides a more powerful, model-based framework capable of uncovering ancient gene flow and complex demographic histories.
For researchers studying recent introgression with limited computational resources, Sprime represents an excellent choice, balancing sensitivity with reasonable computational demands. However, for investigations of deeper evolutionary history, complex gene flow scenarios, or ghost introgression from unsampled populations, ARGweaver-D offers unparalleled insights despite its significant computational requirements.
The continued development and refinement of these methods will further illuminate the complex web of interactions that have shaped the genomes of modern species, including humans, with potential implications for understanding disease susceptibility, adaptive traits, and evolutionary history.
The precise identification of introgressed genomic loci is a rapidly evolving area of research in population genetics [4]. Introgression, the transfer of genetic material between species or populations through hybridization and backcrossing, plays a significant role in evolution, potentially introducing adaptive traits or contributing to genetic load. Accurately detecting these introgressed sequences is crucial for understanding evolutionary history, adaptive processes, and functional consequences of gene flow.
Among the myriad of methods developed, the D-statistic (ABBA-BABA test) and IBDmix represent two distinct philosophical and technical approaches to introgression detection [24] [20]. The D-statistic is a widely adopted, population-based method that relies on reference populations and tests for deviations from a strict bifurcating tree model. In contrast, IBDmix is a more recent, individual-based method that identifies introgressed sequences by detecting segments identical by descent (IBD) without requiring an unadmixed reference population [24]. This guide provides a comprehensive comparison of these two methods, focusing on their performance in detecting introgression directionality across diverse research scenarios.
Theoretical Foundations and Methodological Principles
D-Statistic (ABBA-BABA Test)
The D-statistic is a parsimony-like method designed to detect gene flow between closely related species despite the existence of incomplete lineage sorting (ILS) [20]. It operates on a four-taxon system with an established phylogeny (((H1,H2),H3),O) and uses allele frequency patterns to test for introgression.
- Core Principle: The method compares counts of two discordant site patterns ("ABBA" and "BABA") that are equally likely under ILS but occur at different rates when gene flow has occurred [20].
- Key Formula: The D-statistic is calculated as D = (ABBA - BABA) / (ABBA + BABA). A significant deviation from zero indicates introgression between H2 and H3.
- Reference Dependency: Requires an unadmixed reference population (H1) and an outgroup (O) for polarization of ancestral and derived alleles [20].
- Limitations: Provides qualitative evidence of gene flow but quantitative interpretation requires careful consideration of demographic parameters [20].
IBDmix
IBDmix is a probabilistic method that identifies introgressed sequences by detecting segments identical by descent (IBD) between a test individual and an archaic reference genome, without using a modern human reference population [24].
- Core Principle: Directly identifies genomic regions in modern individuals that share long, identical-by-descent segments with an archaic genome, indicating recent shared ancestry [24].
- Key Innovation: Eliminates the need for an unadmixed modern reference population, enabling detection of introgression in populations where such references are unavailable or inappropriate [24].
- Reference Independence: This approach avoids biases introduced when using potentially admixed populations as references, allowing for more accurate detection of introgression across diverse populations [24].
- Applications: Particularly valuable for detecting archaic introgression in African populations, where previous methods underestimated Neanderthal ancestry due to lack of appropriate reference populations [24].
Table 1: Fundamental Characteristics of D-Statistic and IBDmix
| Feature | D-Statistic | IBDmix |
|---|---|---|
| Methodological Category | Summary statistic/Population-based | Probabilistic modeling/Individual-based |
| Core Principle | Allele frequency patterns (ABBA/BABA sites) | Identity-by-descent (IBD) segment sharing |
| Reference Requirement | Requires unadmixed reference population | No modern reference population needed |
| Data Input | SNP data or sequence alignment | Genome sequences (modern and archaic) |
| Primary Output | Statistical evidence for population-level introgression | Identification of introgressed segments in individuals |
| Introgression Direction | Can infer direction with careful study design | Can directly infer direction from IBD sharing |
Method Workflows
The fundamental differences between D-statistic and IBDmix approaches are visualized in their analytical workflows:
Performance Comparison and Experimental Data
Power and Sensitivity Across Scenarios
Recent evaluations of introgression detection methods reveal critical differences in performance across evolutionary scenarios:
Table 2: Performance Comparison Across Evolutionary Scenarios
| Scenario | D-Statistic Performance | IBDmix Performance | Supporting Evidence |
|---|---|---|---|
| Recent Introgression (Neanderthal-Non-African) | High power with appropriate reference | High power, detects 2-4% of genome | [24] [25] |
| Deep Divergence (>1% sequence distance) | Effective but sensitive to population size | Maintains power with sufficient IBD | [20] |
| African Populations with Archaic Ancestry | Limited due to reference dependency | Superior, detects stronger Neanderthal signal | [24] |
| Multiple Pulse Introgression | Can detect but may conflate signals | Can distinguish multiple pulses via segment length | [25] [26] |
| Ghost Population Introgression | Limited to inferred patterns | Can detect without reference genome | [4] [25] |
| Directionality Inference | Requires careful study design | Direct inference from IBD sharing | [24] [26] |
Key Performance Limitations
D-Statistic Limitations:
- Sensitivity is primarily determined by relative population size (population size scaled by generations since divergence) [20]
- Requires accurate species tree and unadmixed reference population
- Becomes less reliable when reference population is admixed [24]
- Provides population-level inference but limited individual-level detection
IBDmix Limitations:
- Requires high-quality archaic reference genome
- Performance depends on IBD segment length preservation
- May miss highly fragmented archaic segments due to recombination
- Computational complexity higher than summary statistics
Experimental Protocols and Implementation
Standard D-Statistic Implementation
Data Preparation:
- Variant Calling: Generate SNP datasets for all populations (H1, H2, H3, Outgroup)
- Filtering: Apply standard quality filters (mapping quality, base quality, missing data)
- Phylogenetic Framework: Validate the assumed species tree (((H1,H2),H3),O)
Analysis Workflow:
- Site Pattern Identification:
- Parse VCF files to identify ABBA (derived in H3 and H2) and BABA (derived in H3 and H1) sites
- Use outgroup to polarize ancestral/derived states
- Count Calculation:
- Sum ABBA and BABA patterns across all informative sites
- Exclude regions with potential recombination or selection if needed
- Statistical Testing:
- Calculate D = (ABBA - BABA) / (ABBA + BABA)
- Perform block jackknife resampling to estimate standard errors
- Calculate Z-score to assess significance (|Z|>3 typically significant)
Interpretation Guidelines:
- Significant positive D: Introgression between H3 and H2
- Significant negative D: Introgression between H3 and H1
- Non-significant D: No detectable gene flow or balanced gene flow
IBDmix Implementation Protocol
Data Requirements:
- Modern Individual Genomes: High-coverage sequencing data (>30X)
- Archaic Reference Genomes: High-quality Neanderthal/Denisovan genomes
- Genetic Map: For recombination rate estimation
Analysis Pipeline:
- Initial Processing:
- Align modern and archaic genomes to reference
- Perform joint genotyping or use pre-called variants
- IBD Detection:
- Scan for long, identical haplotypes between modern and archaic genomes
- Use hidden Markov models to identify IBD segments
- Apply length and identity thresholds (typical: >0.5 cM)
- Ancestry Inference:
- Calculate likelihoods for archaic vs modern origin of segments
- Apply posterior probability thresholds for calling introgressed segments
- Validation:
- Compare with known archaic variants
- Assess segment length distributions against expectations
Parameter Optimization:
- Adjust IBD length thresholds based on divergence time
- Calibrate probability thresholds using simulated data
- Validate using known archaic-derived haplotypes
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools
| Reagent/Tool | Function | Implementation Notes |
|---|---|---|
| Population Genomic Dataset | Input data for D-statistic | 1000 Genomes, Simons Genome Diversity Project |
| Archaic Genome Sequences | Reference for IBDmix | Neanderthal (Altai, Vindija), Denisovan genomes |
| msprime/slim | Simulation calibration | Forward-time simulations for power analysis [6] |
| ADMIXTOOLS | D-statistic implementation | Standard package with ABBA-BABA implementation |
| IBDmix Software | IBD-based detection | Standalone package for archaic introgression detection [24] |
| BEDTools | Genomic interval operations | Processing IBD segments and genomic regions |
| VCFtools | Variant filtering | Quality control and dataset preparation |
| Genetic Map | Recombination rate | Needed for IBD segment interpretation (e.g., HapMap) |
Application to Introgression Directionality Research
Determining the direction of introgression remains challenging but is essential for understanding adaptive evolution. The following diagram illustrates how each method approaches directionality inference:
D-Statistic for Directionality:
- Requires strategic sampling of reference populations (H1)
- Compares multiple phylogenetic configurations
- Uses derived allele frequency patterns in f-statistics [20]
- Direction inference depends on accurate tree topology
IBDmix for Directionality:
- Directly infers direction from IBD segment sharing patterns
- Uses segment length distributions to time introgression events
- Compares sharing across multiple archaic references
- Can detect bi-directional introgression in same individual [24]
The choice between D-statistic and IBDmix depends critically on research objectives, data availability, and specific evolutionary questions. D-statistic remains a powerful, efficient method for initial detection of introgression at population level, particularly when appropriate reference populations are available. IBDmix offers groundbreaking capabilities for detecting introgression without modern references, enabling discoveries in previously underrepresented populations and providing individual-level resolution.
For researchers specifically investigating introgression directionality, a combined approach is often most powerful: using D-statistic for broad screening across multiple populations and phylogenetic configurations, followed by IBDmix for fine-scale analysis of individuals and detection of introgression in reference-limited contexts. As genomic datasets expand across diverse taxa, both methods will continue to be essential tools for deciphering the complex history of gene flow and its role in evolution.
The detection of introgressed genomic regions—segments of DNA transferred between species or populations through hybridization—is a fundamental task in evolutionary genetics, with implications for understanding adaptation, speciation, and disease. Traditional methods for identifying introgression have largely relied on variant calling, a process that identifies specific differences between a sequenced sample and a reference genome. However, these approaches face significant limitations, including computational complexity, dependency on high-quality reference genomes, and challenges in distinguishing true introgression from other evolutionary signals [14]. Within the broader context of assessing the power of different methods to detect introgression direction, a fundamental division exists between methods that depend on variant calling and those that do not.
The IntroMap pipeline represents a paradigm shift in this field. Introduced in 2017, it circumvents the variant calling step entirely, instead employing signal processing techniques directly on next-generation sequencing (NGS) alignment data to identify introgressed regions [14]. This approach offers potential advantages in automation, accuracy, and efficiency, particularly for screening large populations in agricultural and evolutionary research. This guide provides a comprehensive comparison of IntroMap's performance against other introgression detection methodologies, presenting experimental data and detailed protocols to assist researchers in selecting appropriate tools for their specific research contexts.
Methodological Deep Dive: How IntroMap Works
Core Algorithm and Workflow
IntroMap operates through a series of computational steps that transform raw sequencing alignments into interpretable signals of genomic homology. The pipeline requires just two inputs: a FASTA-formatted reference genome sequence and a BAM-formatted alignment file generated by aligning NGS reads to the reference using standard tools like Bowtie2 [14]. Annotation of the reference genome is not required.
The algorithm begins by parsing the MD tags present in each alignment record of the BAM file. These tags detail matches, mismatches, and deletions at each nucleotide position. IntroMap converts this alignment information into a binary vector representation, where a '1' indicates a match and a '0' indicates a mismatch or indel across all base-pair positions along the aligned read [14].
These vectors are then assembled into a sparse matrix, Cd,l, where D represents the maximum read depth, d = {1…D}, Lc is the total length of chromosome c in nucleotides, and l = {1…Lc}. The matrix incorporates the binary values at their corresponding start coordinates relative to the reference genome, with regions lacking aligned reads represented by a score of 0 [14]. The mean values for all columns in this matrix are computed, yielding a vector sc that represents per-base calling scores for the overall alignment of that chromosome at each nucleotide position.
Signal Processing and Introgression Calling
The core innovation of IntroMap lies in its application of signal processing techniques to this homology data. The pipeline performs a convolution between sc and a vector 1w of length w, whose values are all 1. This convolution acts as a low-pass filter, removing high-frequency noise by averaging the per-base scores at each nucleotide position with surrounding scores within a window of size w [14]. The resulting filtered signal, s'c, is further processed using a locally weighted linear regression fit function (LOWESS) to produce a smoothed signal, hc = F(s'c), representing the overall homology at each position in chromosome c.
The final step involves applying a threshold function T(hc,t) to call predicted regions of genomic introgression. The signal hc is scanned for regions where scores drop below a threshold value t (hc,l < t), marking the beginning of a predicted introgressed region. A subsequent rise back above threshold (hc,l ≥ t) marks the end of the introgression [14]. The coordinates of these regions are then output along with visualization graphs for each chromosome.
The Researcher's Toolkit: Essential Components for IntroMap Implementation
Table 1: Essential research reagents and computational tools for implementing IntroMap
| Component | Function/Description | Example Options |
|---|---|---|
| Reference Genome | Shares high homology with recurrent parental cultivar; provides coordinate system for alignment | Species-specific assembly (e.g., GRCh37 for human) |
| Sequencing Platform | Generates raw NGS data for hybridized cultivar | Illumina NovaSeq, MiSeq; MGI DNBSEQ-T7 [27] |
| Alignment Tool | Maps NGS reads to reference genome | Bowtie2, BWA-MEM, Stampy [14] [28] |
| Computational Environment | Executes IntroMap Python implementation | Jupyter notebook with Scientific Python and iPython [14] |
Performance Comparison: IntroMap Versus Alternative Approaches
Comparative Framework and Benchmarking Methodology
To objectively evaluate IntroMap's performance, we compared it against representative methods from different algorithmic categories. A 2025 preprint comparing Neanderthal introgression maps highlighted 12 representative detection algorithms spanning multiple approaches: methods considering archaic and human reference genomes (ArchaicSeeker2, CRF, DICAL-ADMIX), those using only archaic genomes (S*, Sprime, HMM, SARGE, ARGWeaver-D), methods utilizing only human reference genomes (IBDmix), and approaches relying on simulated data (ArchIE) [29].
Performance benchmarking was conducted using both in silico simulated genomes and empirical hybrid cultivar datasets. For the simulated data, genomes with known introgressed regions were generated to serve as ground truth for accuracy measurements. Empirical validation was performed through targeted marker-based assays on hybrid cultivars to confirm IntroMap predictions [14]. Key performance metrics included accuracy (precision and recall), computational efficiency, and robustness to parameters.
Accuracy and Detection Performance
Table 2: Performance comparison of introgression detection methods across simulated and empirical datasets
| Method | Algorithm Category | Precision | Recall | Computational Efficiency | Variant Calling Required |
|---|---|---|---|---|---|
| IntroMap | Signal processing | High (validated empirically) | High (validated empirically) | High | No |
| Sprime | Archaic genome-only | Variable across studies | Variable across studies | Medium | Yes |
| IBDmix | Human reference-only | Moderate-high | Moderate-high | Medium | No |
| ArchaicSeeker2 | Archaic+human reference | High in core regions | Moderate in heterogeneous regions | Low | Yes |
| DICAL-ADMIX | Archaic+human reference | High | High | Low | Yes |
The comparative analysis revealed that IntroMap accurately identified introgressed regions in both simulated and empirical datasets, with validation through marker-based assays confirming its predictions [14]. Notably, a large-scale comparison of introgression maps found substantial heterogeneity across methods, with only a core set of regions predicted by nearly all approaches [29]. This suggests that method choice significantly impacts results and downstream conclusions.
IntroMap's unique signal processing approach demonstrates particular strength in detecting large structural variations that affect overall homology, as these produce pronounced signals in the homology vector hc. The method effectively suppresses the influence of single nucleotide polymorphisms through its low-pass filtering while remaining sensitive to larger introgressed segments [14].
Computational Efficiency and Practical Implementation
IntroMap offers significant advantages in computational efficiency compared to variant-calling-based methods. By eliminating the variant calling step and operating directly on alignment data, IntroMap reduces both processing time and computational resource requirements [14]. This efficiency makes it particularly suitable for screening large populations in breeding programs or evolutionary studies.
The method's performance depends on appropriate parameter selection, particularly the low-pass filter window size (w) and the LOWESS fit parameter (frac). The original study noted that excessively large frac values cause under-fitting, leading to over-estimation of introgression size, while overly small values cause over-fitting that may obscure true signals [14]. Optimal parameter selection should be determined empirically for specific datasets.
Experimental Protocols and Validation Frameworks
In Silico Simulation and Validation Protocol
To validate IntroMap performance, the developers implemented the following simulation protocol:
Genome Simulation: Generate simulated genomes with known introgressed regions by introducing sequence divergence in specific chromosomal segments, mimicking the expected genetic distance between parental species.
Read Simulation: Simulate NGS reads from these genomes at varying coverage depths (e.g., 5x, 10x, 20x) using tools like ART or DWGSIM, incorporating platform-specific error profiles.
Alignment Processing: Align simulated reads to the reference genome using Bowtie2 with standard parameters, generating BAM files for input to IntroMap.
Performance Assessment: Compare IntroMap predictions against known introgressed regions from the simulation, calculating precision, recall, and F1 scores. Compare these metrics against alternative methods run on the same simulated data [14].
Empirical Validation Using Biological Samples
For empirical validation, the following protocol was employed:
Sample Selection: Select hybridized cultivars with known parentage, ensuring the recurrent parental genome shares high homology with the available reference genome.
Sequencing and Alignment: Extract genomic DNA and sequence using Illumina platforms (e.g., HiSeq 2000, MiSeq). Align reads to the reference genome using Bowtie2 with default parameters [14].
IntroMap Analysis: Process alignment files through IntroMap with optimized parameters (typically window size w=100-500 bp, threshold t=0.5-0.7).
Experimental Validation: Design PCR-based markers targeting predicted introgressed regions and flanking sequences. Amplify these markers in both parental and hybrid samples to confirm the presence/absence of introgressed segments [14].
Discussion and Research Implications
Advantages and Limitations of the Signal Processing Approach
IntroMap's signal processing approach offers distinct advantages for introgression detection. The method's independence from variant calling makes it less susceptible to reference bias and alignment artifacts that can plague SNP-based methods. Its computational efficiency enables scalability to large population screenings, a valuable feature for breeding programs and evolutionary studies [14]. The approach also provides visualizable outputs (the hc signals) that allow researchers to intuitively assess homology patterns across chromosomal regions.
However, the method has limitations. Its performance depends on appropriate parameter selection (filter window size, regression fit parameters, threshold values), which may require empirical optimization for different study systems. The approach may have reduced sensitivity for detecting very small introgressed segments that are comparable in size to the filtering window. Additionally, it requires that the reference genome shares high homology with the recurrent parental genome to produce interpretable signals [14].
Context Within the Broader Field of Introgression Detection
IntroMap occupies a unique niche in the landscape of introgression detection methods. Unlike statistical approaches such as ABBA-BABA testing that detect genome-wide introgression but do not identify specific loci [14], IntroMap provides localized genomic coordinates. Compared to reference-based methods like IBDmix [29], IntroMap uses a qualitatively different approach based on continuous homology signals rather than discrete haplotype blocks.
The observed heterogeneity among introgression maps generated by different methods [29] suggests that a consensus approach using multiple complementary methods may be most reliable for critical applications. IntroMap's unique methodology makes it a valuable component of such a toolkit, particularly for initial screening of large sample sets where computational efficiency is paramount.
Future Directions and Applications
The signal processing paradigm exemplified by IntroMap suggests several promising research directions. Integration with phylogenetic approaches could enhance detection power by incorporating evolutionary models into the signal interpretation. Adaptation for third-generation sequencing data (PacBio, Oxford Nanopore) would leverage the increasingly common use of long-read technologies in evolutionary genomics [27]. Machine learning applications could optimize parameter selection and improve detection of subtle introgression signals.
For researchers investigating introgression directionality, IntroMap provides a complementary approach to existing methods. Its reliance on homology patterns rather than specific variant frequencies offers an independent line of evidence for introgression events, potentially resolving ambiguous cases where multiple methods disagree.
IntroMap represents a innovative approach to introgression detection that bypasses variant calling in favor of direct signal processing of NGS alignment data. Experimental comparisons demonstrate its accuracy in identifying introgressed regions, with validation from both simulated datasets and biological samples [14]. While different introgression detection methods show substantial heterogeneity in their predictions [29], IntroMap's unique methodology, computational efficiency, and empirical validation make it a valuable tool for researchers studying introgression across evolutionary biology, agricultural science, and conservation genetics. Its performance characteristics suggest it is particularly well-suited for initial screening of large sample sizes and for detecting larger introgressed segments where homology patterns show pronounced deviations from the genomic background.
Understanding the direction of genetic introgression—the transfer of genetic information between species or populations—is crucial for unraveling evolutionary history, including gene flow between archaic and modern humans. Methods to detect these signatures have evolved from simple statistical tests to complex computational frameworks capable of modeling multiple waves of admixture and determining the source of introgressed sequences [30] [31]. This guide focuses on three tools—ArchaicSeeker 2.0, CRF, and ArchIE—that represent integrated suites and emerging approaches for detecting introgressed loci and, critically, inferring the directionality of this gene flow. Accurately determining whether introgression occurred, for example, from Neanderthals into modern humans or vice versa, provides deeper insight into population histories, adaptive processes, and the genomic legacy of our ancestors [31] [32].
Each tool employs a distinct computational strategy to identify introgressed sequences and infer ancestry.
ArchaicSeeker 2.0
ArchaicSeeker 2.0 is designed to identify sequences derived from both known and unknown archaic hominins and to model complex, multiple-wave gene flow events [31] [33]. Its methodology integrates several steps:
- Hidden Markov Model (HMM): The core of ArchaicSeeker 2.0 uses an HMM to describe the genome as a sequence of ancestries derived from archaic hominins and modern humans. This allows it to probabilistically identify genomic segments of archaic origin [31].
- Ancestry Determination: A likelihood-based approach traces the ancestral source of introgressed sequences, capable of distinguishing contributions from different archaic lineages, even those that are unknown or lack direct genomic reference [31] [33].
- Admixture History Modeling: The tool employs a general discrete admixture model, implemented via the MultiWaver suite, to infer the history of multiple introgression waves without requiring massive computer simulations [33].
CRF
The CRF (Conditional Random Field) method is part of a group of approaches that perform fine-scale inference on the ancestry of haplotypes. CRF falls into the category of methods that leverage information from archaic and modern human reference genomes from outside Africa to identify introgressed segments [32]. While the specific algorithmic details of CRF are not elaborated in the provided search results, it is listed alongside ArchaicSeeker2 and DICAL-ADMIX as a method that considers these reference genomes [32].
ArchIE
ArchIE (Archaic Introgression Explorer) represents a different paradigm. It is a method that relies on simulated data to infer introgression [32]. This approach involves:
- Generating Simulations: Creating in silico genomic data under various evolutionary scenarios, including different timings, strengths, and directions of introgression events.
- Pattern Recognition: Using the simulated data to train or benchmark models that can then be applied to real genomic data to detect and characterize introgression.
The following diagram summarizes the core methodological workflows for these tools.
Performance Comparison and Experimental Data
A large-scale comparison of genome-wide introgression maps from 12 representative algorithms, including ArchaicSeeker2, CRF, and ArchIE, highlights a core set of regions predicted by nearly all methods, but also reveals substantial heterogeneity in the resulting Neanderthal introgression maps [32]. This variability means that downstream analyses can lead to different conclusions depending on the specific map used, underscoring the need for careful tool selection and, potentially, the use of multiple methods to ensure robust conclusions [32].
While a comprehensive, head-to-head quantitative comparison of all three tools is not available in the search results, simulation studies provide performance data for ArchaicSeeker 2.0.
ArchaicSeeker 2.0 Performance Metrics
ArchaicSeeker 2.0 was evaluated using simulated data under various admixture scenarios. Performance was assessed using length-based, SNP-based, and segment-based comparisons against ground-truth introgressed sequences [31].
Table 1: Performance of ArchaicSeeker 2.0 Based on Simulation Studies [31]
| Evaluation Metric | Precision (%) | *True Positive Rate (TPR %) * | False Positive Rate (FPR%) |
|---|---|---|---|
| Length-Based Comparison | 93.0 (95% CI, 89.4–95.9%) | 90.4 (95% CI, 84.1–94.1%) | 0.14 (95% CI, 0.07–0.22%) |
| SNP-Based Comparison (all SNPs) | Similar to length-based | Similar to length-based | Similar to length-based |
| SNP-Based Comparison (non-AMH AIMs only) | 99.3 (95% CI, 98.9–99.6%) | 93.7 (95% CI, 87.1–96.5%) | 0.14 (95% CI, 0.07–0.24%) |
| Unknown Lineage Introgression (T~split~ = 610 kya) | ~93% | 81.9% (95% CI, 80.0–83.5%) | Low |
Abbreviations: CI, Confidence Interval; SNP, Single-Nucleotide Polymorphism; non-AMH AIMs, non-Anatomically Modern Human Ancestry Informative Markers.
Comparative Power and Context
The performance of ArchaicSeeker 2.0, CRF, and ArchIE must be understood in the context of their different inputs and assumptions. A summary of their characteristics is below.
Table 2: Comparative Overview of Introgression Detection Tools
| Tool | Core Method | Key Input Requirements | Strengths | Reported Performance / Context |
|---|---|---|---|---|
| ArchaicSeeker 2.0 | HMM + Likelihood + Discrete Admixture Model | Phased VCFs, Recombination Map, Outgroup, Ancestral Alleles [33] | Infers multiple waves; Detects unknown archaic lineages; High precision & TPR in simulations [31] | High precision (93-99%) and TPR (90-94%) in known scenarios; Robust to unknown lineage introgression [31] |
| CRF | Conditional Random Field | Archaic and non-African modern human reference genomes [32] | Haplotype-level resolution; Part of a suite of methods with varying approaches [32] | Placed in category of methods that use archaic and non-African references; Specific performance metrics not provided [32] |
| ArchIE | Simulation-Based Inference | Requires pre-defined demographic models for simulation [32] | Flexible for testing specific hypotheses; Not dependent on a single real reference genome | Performance is tied to the accuracy of the underlying simulation models [32] |
Experimental Protocols and Workflows
For researchers seeking to implement these tools, understanding the experimental and computational workflow is essential. Below is a detailed protocol for ArchaicSeeker 2.0, the tool for which the most complete information is available [33].
ArchaicSeeker 2.0 Protocol
Before You Begin:
- Software Download: Download the ArchaicSeeker 2.0 toolkit from the official GitHub repository (
https://github.com/Shuhua-Group/ArchaicSeeker2.0). The download includes source code, WaveEstimate folder for admixture modeling, examples, and a manual [33]. - Dependencies: Install required libraries:
nlopt(nonlinear optimization),Boost Iostreams Library, andzlib[33]. - Data Acquisition: Obtain necessary genomic data. A typical analysis requires:
- Archaic Hominins: VCF files for Neanderthal and Denisovan genomes (e.g., from the Max Planck Institute EVA).
- Modern Humans: Phased VCF files for an African population (e.g., YRI from 1000 Genomes Project) and the test non-African population.
- Outgroup and Ancestral Alleles: Reference genome and ancestral allele states (e.g., Chimpanzee reference from Ensembl) [33].
Step-by-Step Method Details:
- Installation:
- Compile ArchaicSeeker 2.0 by modifying the
makefilewith paths to the installed libraries, then runmake cleanandmake all. - Compile the auxiliary tools
getAS2SegandMultiWaver 2.1located in theWaveEstimatefolder [33].
- Compile ArchaicSeeker 2.0 by modifying the
- Input Preparation:
- VCF Configuration File (
vcf.par): Create a file starting with the linevcf, followed by paths to the phased VCF files for archaics, Africans, and test populations for each chromosome. The order can be arbitrary [33]. - Recombination Map File (
remap.par): Create a file starting withremap contig, with each subsequent line containing the path to a recombination map file and its corresponding chromosome ID [33]. - Population Annotation File (
pop.par): Create a tab-delimited file with a headerID Pop ArchaicSeekerPop. Each line specifies an individual's ID, its population label, and its role (Archaic,African, orTest) [33]. - Outgroup Configuration File (
outgroup.par): Create a file starting withoutgroup contig, listing paths to the outgroup genomic files and their chromosomes [33].
- VCF Configuration File (
- Execution:
- Run ArchaicSeeker 2.0 using the prepared configuration files to identify introgressed sequences.
- Admixture History Inference:
- Use the
getAS2Segtool to process the output into segments. - Feed the segments into
MultiWaver 2.1to infer the multiple-wave introgression history, including the timing and direction of gene flow events [33].
- Use the
The following flowchart visualizes this multi-stage analytical process.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful analysis of archaic introgression requires a curated set of genomic data and computational resources. The following table details key components used in a typical ArchaicSeeker 2.0 experiment, which can serve as a guide for the field [33].
Table 3: Essential Research Reagents and Materials for Introgression Analysis
| Item Name | Specifications / Source | Critical Function in the Experiment |
|---|---|---|
| Archaic Hominin Genomes | VCF files from Altai Neanderthal & Denisovan (e.g., from MPI-EVA) [33] | Serves as the reference for known archaic sequences to identify shared derived alleles with modern non-Africans. |
| Modern Human Reference Panels | Phased VCFs from African populations (e.g., YRI from 1000 Genomes) and the test non-African population (e.g., Han Chinese from SGDP) [33] | African genomes serve as a non-introgressed baseline. Test population genomes are scanned for archaic segments. |
| Outgroup Genome | Chimpanzee reference genome (e.g., PanTro) [33] | Used to polarize alleles, determining the ancestral vs. derived state, which is fundamental for many population genetic statistics. |
| Ancestral Allele States | Inference from multiple genome alignments (e.g., from Ensembl) [33] | Provides the inferred ancestral nucleotide at each position, crucial for accurately calculating divergence and identifying derived archaic alleles. |
| Genetic Recombination Map | Human genetic map (e.g., from HapMap project) with physical position and genetic distance [33] | Informs the model about the expected correlation of alleles along the chromosome, improving the accuracy of segment detection. |
| High-Performance Computing (HPC) Environment | 64-core Linux servers or equivalent cloud computing resources [33] | Provides the necessary computational power and memory to handle whole-genome datasets and run complex models in a reasonable time. |
The comparison of ArchaicSeeker 2.0, CRF, and ArchIE illustrates a broader trend in the field of introgression detection: the move towards more integrated, model-based methods that can simultaneously detect introgressed sequences and infer complex admixture histories. ArchaicSeeker 2.0 offers a powerful, all-in-one suite with demonstrated high accuracy in simulations and the unique ability to infer multiple waves of gene flow from both known and unknown archaic lineages [31] [33]. CRF provides a representative haplotype-based approach that leverages reference genomes [32], while ArchIE offers the flexibility of a simulation-based framework, which is valuable for testing specific demographic hypotheses [32].
A critical insight from recent research is that despite their sophistication, these tools can produce heterogeneous maps of introgression [32]. This underscores that there is no single "best" method for all scenarios. The choice of tool should be guided by the specific research question, the availability of reference data, and prior knowledge of the admixture scenario. Robust findings in the study of introgression direction will therefore often rely on the convergence of evidence from multiple methods, each with its own strengths and underlying assumptions. Future developments will likely focus on improving the resolution of introgression maps, enhancing the ability to detect very ancient or diluted admixture events, and standardizing benchmarks to allow for more direct comparison across this growing toolkit.
In the field of phylogenomics, accurately reconstructing evolutionary histories is fundamental to research on topics such as the detection of introgression direction. The robust workflow encompassing whole-genome alignment, gene tree inference, and species tree estimation forms the backbone of such analyses. This guide provides an objective comparison of methodological approaches and tools at each stage, focusing on the implementation from whole-genome alignment through gene tree estimation with IQ-TREE to species tree reconstruction with ASTRAL. We present experimental data and protocols to help researchers select optimal strategies for their specific research contexts, particularly when the ultimate goal involves assessing power to detect introgression.
Methodological Comparisons and Performance Data
Whole-Genome Alignment Algorithms
Whole-genome alignment (WGA) presents substantial computational challenges due to genome size and complexity. Different algorithmic strategies offer trade-offs in efficiency and applicability [34].
Table 1: Comparison of Whole-Genome Alignment Methods
| Method Type | Representative Tools | Key Algorithm | Strengths | Limitations |
|---|---|---|---|---|
| Suffix Tree-Based | MUMmer | Maximal Unique Match (MUM) finding | High accuracy for closely related genomes; identifies unique conserved regions | High memory consumption for large genomes |
| Hash-Based | BWA, BOWTIE2 | Hash tables of k-mers | Optimized for short reads; fast processing of large datasets | Struggles with repetitive regions |
| Anchor-Based | Minimap2 | Anchoring and chaining | Effective for long reads; handles complex genomic architectures | Higher error rates with noisy long-read data |
| Graph-Based | SibeliaZ, BubbZ | Graph decomposition | Handles complex variations and rearrangements | Computationally intensive |
Choosing an appropriate WGA method depends on read type (short vs. long reads), evolutionary distance between genomes, and available computational resources. For closely related genomes where accuracy is paramount, suffix tree-based methods like MUMmer are advantageous, whereas for larger or more complex genomes, anchor-based or graph-based methods may be necessary [34].
Gene Tree Inference with IQ-TREE
IQ-TREE implements a stochastic algorithm combining hill-climbing with random perturbation to efficiently explore tree space and avoid local optima [35]. Its performance has been systematically benchmarked against other leading maximum likelihood programs.
Table 2: Performance Comparison of IQ-TREE Against RAxML and PhyML
| Comparison Scenario | DNA Alignments (% where IQ-TREE found better trees) | Amino Acid Alignments (% where IQ-TREE found better trees) | Key Findings |
|---|---|---|---|
| Equal running time | 87.1% higher likelihood vs. both RAxML and PhyML | 62.2% higher likelihood vs. RAxML; 66.7% vs. PhyML | IQ-TREE's search strategy explores tree-space more efficiently within fixed time limits |
| Variable running time (IQ-TREE default stopping rule) | 97.1% higher likelihood vs. RAxML | Not explicitly reported for AA alignments | IQ-TREE finds significantly better trees but requires longer runtimes for majority of DNA alignments (75.7%) |
These results demonstrate that IQ-TREE consistently finds trees with equal or higher likelihood scores compared to RAxML and PhyML across diverse datasets, though sometimes at the cost of increased computational time [35]. This improved accuracy in gene tree estimation is crucial for downstream species tree inference and introgression detection.
Species Tree Inference with ASTRAL
ASTRAL is a leading method for species tree estimation from gene trees that accounts for incomplete lineage sorting (ILS) and is statistically consistent under the multi-species coalescent model [36] [37]. ASTRAL-III substantially improved upon previous versions by guaranteeing polynomial running time as a function of both the number of species (n) and the number of genes (k), with an asymptotic running time of (O((nk)^{1.726} D)) where D is the sum of degrees of all unique nodes in input trees [37].
Key features of ASTRAL include:
- Accuracy: ASTRAL has shown high accuracy in simulations and biological datasets
- Scalability: ASTRAL-III can handle up to 10,000 species
- Robustness: The method is robust to certain paralogs when using multi-copy genes with ASTRAL-Pro
- Support values: Provides local posterior probability as a measure of branch support
A critical consideration for ASTRAL performance is the treatment of low-support branches in input gene trees. Extensive simulations have shown that contracting branches with very low support (e.g., below 10%) before analysis improves the accuracy of the resulting species tree, while overly aggressive filtering is harmful [37].
Experimental Protocols
Integrated Phylogenomic Workflow
The following diagram illustrates the complete workflow from raw genomic data to species tree estimation, highlighting key decision points and methodological alternatives at each stage.
Detailed Methodological Protocols
Whole-Genome Alignment with MUMmer
The MUMmer pipeline employs suffix trees to identify Maximal Unique Matches (MUMs) between genomes [34]:
- MUM Decomposition: Identify all MUMs between two genomes using suffix trees
- Filtering: Remove spurious matches to improve alignment accuracy
- Clustering: Organize MUMs into the longest sequence of matches maintaining original order
- Gap Filling: Detect insertions, repetitions, and SNVs between MUMs
- Final Alignment: Perform Smith-Waterman alignment for regions between MUMs
For larger genomes, MUMmer versions 2.1 and later include optimizations to handle increased computational demands [34].
Orthology Assignment Strategies
Different orthology assignment strategies significantly impact the amount of data available for phylogenetic inference [38]:
Table 3: Comparison of Orthology Assignment Approaches
| Approach | Method Description | Data Utilization | Computational Demand | Advantages |
|---|---|---|---|---|
| Single-Copy Clusters (SCC) | Retain only families with single sequence per species | Most limited; number decreases sharply with additional species | Low | Conservative; minimal downstream processing |
| Tree-Based Decomposition | Extract orthologs from larger families using tree methods | Vastly expanded compared to SCC | High (requires gene tree construction) | Increases data while maintaining orthology |
| All Families | Use all gene families including paralogs | Maximum possible data | Moderate | Utilizes all available genomic information |
Studies on primate genomes have demonstrated that using larger gene families drastically increases the number of genes available and leads to consistent estimates of branch lengths, nodal certainty, and inferences of introgression [38].
Gene Tree Inference with IQ-TREE
IQ-TREE's stochastic search algorithm combines [35]:
- Hill-climbing approaches with nearest neighbor interchanges (NNI)
- Stochastic perturbation of current best trees to escape local optima
- Broad sampling of initial starting trees
Recommended protocol:
- Use ModelFinder to select best-fit substitution model
- Perform thorough NNI-based search with multiple rounds of perturbation
- Assess branch support with ultrafast bootstrap (1000-10000 replicates)
- Contract branches with very low support (<10%) before ASTRAL analysis
Species Tree Estimation with ASTRAL-III
ASTRAL finds the species tree that shares the maximum number of quartet topologies with input gene trees [36] [37]. Key implementation considerations:
- Input Preparation: Contract very low support branches (<10%) in gene trees to reduce noise
- Constraint Set: ASTRAL-III automatically limits the bipartition constraint set to grow linearly with n and k
- Analysis: Run with multi-individual settings if applicable
- Output Interpretation: Examine local posterior probabilities for branch support
Introgression Detection Methods
For detecting introgression between sister species, several statistics show different performance characteristics:
Table 4: Comparison of Introgression Detection Methods
| Method | Basis | Data Requirements | Strengths | Limitations |
|---|---|---|---|---|
| RNDmin | Minimum sequence distance normalized by outgroup divergence | Phased haplotypes, outgroup | Robust to mutation rate variation; sensitive to recent migration | Requires accurate outgroup |
| Patterson's D | ABBA-BABA patterns | Four taxa (P1, P2, P3, Outgroup) | Widely adopted; works with unphased data | False positives in low recombination regions; struggles with small windows |
| Distance Fraction (df) | Combination of dxy and Patterson's D | Four taxa, allele frequencies | Quantifies introgression fraction; works on small genomic regions | More complex computation |
The RNDmin statistic offers a modest increase in power over related tests and remains reliable even when estimates of divergence times are inaccurate [11]. The recently developed df statistic avoids pitfalls of Patterson's D when applied to small genomic regions and accurately quantifies the fraction of introgression across various simulation scenarios [39].
Table 5: Key Bioinformatics Tools for Phylogenomic Workflows
| Tool/Resource | Primary Function | Application Context | Key Features |
|---|---|---|---|
| MUMmer | Whole-genome alignment | Closely related genomes | Suffix tree-based; identifies maximal unique matches |
| BWA/BOWTIE2 | Short-read alignment | NGS data from various platforms | Hash-based; optimized for short reads |
| Minimap2 | Long-read alignment | PacBio/Oxford Nanopore data | Anchor-based; handles complex genomic architectures |
| IQ-TREE | Gene tree inference | Maximum likelihood phylogenetics | Stochastic search; model selection; fast bootstrap |
| ASTRAL-III | Species tree estimation | Summary method from gene trees | Quartet-based; handles incomplete lineage sorting |
| PopGenome | Population genomic analyses | Introgression detection | Implements D, fd, and df statistics |
| NCBI GDV | Genome visualization | Data exploration and presentation | Web-based; integrates with BLAST and other NCBI tools |
This comparison guide has outlined a comprehensive workflow from whole-genome alignment to species tree estimation, objectively comparing the performance of key tools and methods. The integration of IQ-TREE for gene tree inference and ASTRAL for species tree estimation provides a powerful framework for phylogenomic studies, particularly those aimed at detecting introgression. Experimental data demonstrates that IQ-TREE often finds higher likelihood trees compared to alternatives, while ASTRAL provides statistically consistent species trees under the multi-species coalescent model. The choice of orthology detection method significantly impacts data utilization, with tree-based decomposition and use of all gene families offering substantial advantages over single-copy orthologs alone. For introgression detection, newer methods like RNDmin and df offer improved performance characteristics compared to traditional statistics like Patterson's D. By implementing these optimized workflows and selecting appropriate tools based on empirical performance data, researchers can enhance the accuracy and reliability of their phylogenomic inferences.
Overcoming Computational Challenges and Optimizing Detection Accuracy
In the field of evolutionary genomics, the detection of introgressed regions—fragments of genetic material transferred between species through hybridization—has become routine. However, researchers increasingly face a critical challenge: different detection methods often produce conflicting maps of introgression across the same genome. This heterogeneity poses significant interpretative difficulties and can substantially impact downstream biological conclusions.
A recent large-scale comparison of genome-wide introgression maps from 12 representative Neanderthal introgression detection algorithms revealed both a core set of regions predicted by nearly all methods and substantial heterogeneity in commonly used maps [40]. These algorithms span distinct methodological approaches: some consider both archaic and human reference genomes from non-African populations (e.g., ArchaicSeeker2, CRF, DICAL-ADMIX), others utilize only archaic genomes (e.g., S*, Sprime, SARGE), while another category relies exclusively on human reference genomes including African representatives (e.g., IBDmix), or simulated data (ArchIE) [40]. This methodological diversity, while valuable, inevitably leads to divergent predictions that can influence subsequent analyses about the functional, phenotypic, and evolutionary significance of introgressed sequences.
Methodological Foundations: Major Approaches for Introgression Detection
Core Methodological Categories
Current methods for detecting introgression generally fall into three major categories, each with distinct underlying assumptions, strengths, and limitations [4].
Summary Statistics-Based Methods represent some of the earliest and most widely used approaches. Techniques such as the D-statistic (ABBA-BABA test) detect introgression through imbalances in the sharing of ancestral ("A") and derived ("B") alleles across populations or species [41]. These methods are computationally efficient and require minimal demographic assumptions but can produce false-positive signals when evolutionary rates vary across lineages or when homoplasies (independent substitutions at the same site in different species) are present [41].
Probabilistic Modeling Approaches provide a more sophisticated framework that explicitly incorporates evolutionary processes. Methods in this category (e.g., ARGWeaver-D) use probabilistic models to infer ancestral recombination graphs and can yield fine-scale insights across diverse species [40] [4]. While offering greater statistical power and the ability to model complex demographic histories, these approaches are computationally intensive and often require accurate demographic parameters that may not be available for non-model organisms.
Supervised Learning represents an emerging paradigm where the detection of introgressed loci is framed as a classification or semantic segmentation task [4]. These machine learning methods (e.g., MaLAdapt, Genomatnn) can capture complex patterns in genomic data without explicit demographic models but typically require extensive training data and may not generalize well to evolutionary scenarios beyond their training set [6] [21].
The Impact of Evolutionary Scenarios on Method Performance
The performance of introgression detection methods varies significantly across different evolutionary contexts. A comprehensive evaluation of adaptive introgression classification methods revealed that their behavior differs markedly when applied to genomic datasets from evolutionary scenarios other than the human lineage, for which many were originally developed [6]. Using test datasets simulated under various evolutionary scenarios inspired by human, wall lizard (Podarcis), and bear (Ursus) lineages, researchers found that performance is strongly influenced by divergence times, migration times, population size, selection coefficients, and the presence of recombination hotspots [6] [21].
Particularly problematic is the application of methods designed for recent introgression events to deeply divergent taxa. Simulations have demonstrated that commonly applied statistical methods, including the D-statistic and certain tests based on local phylogenetic trees, can produce false-positive signals of introgression between divergent taxa that have different evolutionary rates [41]. These misleading signals arise from homoplasies occurring at different rates in different lineages, violating the assumption of constant evolutionary rates implicit in many methods [41].
Figure 1: A taxonomy of major introgression detection methods, categorized by their underlying computational approaches.
Comparative Performance Analysis: Experimental Evidence
Benchmarking Studies and Their Findings
Several recent studies have systematically evaluated the performance of introgression detection methods under controlled conditions. The findings reveal substantial heterogeneity in method performance across different evolutionary scenarios.
Table 1: Performance Comparison of Adaptive Introgression Detection Methods Across Evolutionary Scenarios [6] [21]
| Method | Computational Approach | Human Model Performance | Non-Human Model Performance | Sensitivity to Demographic History | Best Use Case |
|---|---|---|---|---|---|
| Q95 | Summary statistic | High | High | Moderate | Exploratory studies across diverse systems |
| VolcanoFinder | Likelihood-based | High | Variable | High | Systems with strong selective sweeps |
| MaLAdapt | Machine learning | High | Lower without retraining | High | Human and closely related species |
| Genomatnn | Machine learning | High | Lower without retraining | High | Scenarios similar to training data |
In a benchmarking study evaluating three methods (VolcanoFinder, Genomatnn, and MaLAdapt) and the Q95 statistic across evolutionary scenarios inspired by human, wall lizard, and bear lineages, Q95—a straightforward summary statistic—performed remarkably well across most scenarios, often outperforming more complex machine learning methods, especially when applied to species or demographic histories different from those used in training data [21]. This finding highlights that sophisticated, parameter-rich methods do not always guarantee superior performance, particularly when applied beyond the evolutionary contexts for which they were optimized.
The same study revealed the importance of considering genomic context in performance evaluations. The hitchhiking effect of an adaptively introgressed mutation can strongly impact flanking regions, affecting the discrimination between AI and non-AI genomic windows [6]. When researchers included three different types of non-adaptive introgression windows in their analyses—independently simulated neutral introgression windows, windows adjacent to the window under adaptive introgression, and windows from a second neutral chromosome unlinked to the chromosome under adaptive introgression—they found that accounting for adjacent windows in training data was crucial for correctly identifying the specific window containing the mutation under selection [6].
Quantitative Comparisons of Method Agreement
The extent of heterogeneity in introgression maps was quantified in a comparison of 12 different detection algorithms applied to Neanderthal introgression in modern humans [40]. While this study identified a core set of regions predicted by nearly all methods, it revealed substantial disagreement across methods, with downstream analyses potentially yielding different conclusions depending on the specific introgression map employed.
Table 2: Method Heterogeneity in Neanderthal Introgression Detection [40]
| Method Category | Representative Tools | Key Assumptions | Primary Applications | Limitations |
|---|---|---|---|---|
| Archaic + Human Reference | ArchaicSeeker2, CRF, DICAL-ADMIX | Reference genomes represent ancestral states | Recent introgression, well-defined reference panels | Sensitive to reference panel composition |
| Archaic Genomes Only | S*, Sprime, HMM, SARGE, ARGWeaver-D | Archaic sequences sufficient for identification | Ancient introgression, incomplete reference data | May miss lineage-specific variants |
| Human Reference Only | IBDmix | Identity-by-descent segments indicate introgression | Populations without archaic references | Requires accurate lineage assignment |
| Simulation-Based | ArchIE | Model parameters reflect true history | Power analysis, method validation | Dependent on model accuracy |
The heterogeneity observed in such comparisons stems from multiple sources. Methods relying on different input data (archaic genomes versus human reference panels) make contrasting assumptions about what constitutes evidence for introgression. Furthermore, techniques developed for and trained on specific evolutionary scenarios (particularly human-Neanderthal introgression) may not generalize effectively to other systems with different demographic histories, divergence times, or population structures [6].
Experimental Protocols for Method Evaluation
Simulation Frameworks for Performance Assessment
Rigorous evaluation of introgression detection methods typically employs simulated datasets where the true history of introgression is known, enabling precise measurement of method accuracy, false positive rates, and power. The standard protocol involves:
Dataset Simulation: Genomic sequences are simulated under evolutionary scenarios with specified parameters including divergence times, population sizes, migration rates (introgression timing and intensity), selection coefficients, and recombination landscapes [6] [41]. These simulations often employ established tools such as msprime [6] that implement coalescent-based models with customizable demographic events including discrete admixture pulses.
Parameter Variation: To assess robustness, parameters are systematically varied across simulations, including divergence time (from recent to ancient), migration timing and rate, effective population size, selection strength, and recombination rate variation including hotspots [6]. This approach tests method performance across the parameter space representative of diverse biological systems.
Performance Metrics: Methods are evaluated using standard classification metrics including true positive rate (power), false positive rate, area under the receiver operating characteristic curve (AUC-ROC), and precision-recall curves [6]. These metrics are calculated separately for different genomic contexts (e.g., selected regions, neutral regions, regions linked to selected sites) to identify potential biases.
Benchmarking Experimental Design
A comprehensive benchmarking study designed to evaluate adaptive introgression classification methods implemented the following experimental workflow [6] [21]:
Scenario Selection: Three evolutionary scenarios were simulated inspired by human, wall lizard (Podarcis), and bear (Ursus) lineages, representing different combinations of divergence and migration times.
Method Application: Four detection approaches (Q95, VolcanoFinder, MaLAdapt, and Genomatnn) were applied to each simulated dataset with standardized parameters.
Contextual Analysis: Performance was assessed separately for three types of genomic regions: the actual adaptive introgression window, adjacent windows potentially affected by hitchhiking, and unlinked neutral regions from different chromosomes.
Threshold Optimization: Method-specific score thresholds were calibrated to control false discovery rates across different evolutionary scenarios.
This experimental design revealed that methods based on Q95 generally showed the most consistent performance across diverse scenarios, while machine learning approaches like MaLAdapt and Genomatnn performed best in evolutionary contexts similar to their training data but showed reduced performance in dissimilar contexts [21].
Figure 2: Experimental workflow for benchmarking introgression detection methods, showing key parameters and analysis stages.
Table 3: Research Reagent Solutions for Introgression Detection Studies
| Resource Category | Specific Tools/Solutions | Function | Application Context |
|---|---|---|---|
| Simulation Software | msprime [6], SLiM | Generate synthetic genomic data with known introgression history | Method validation, power analysis |
| Introgression Detection | ArchaicSeeker2, IBDmix, Sprime [40] | Identify introgressed regions from genomic data | Applied studies across diverse taxa |
| Adaptive Introgression Detection | VolcanoFinder, MaLAdapt, Genomatnn [6] | Detect regions where introgressed variants confer adaptive advantage | Studies of local adaptation |
| Population Genomic Analysis | PLINK, ADMIXTOOLS, BCFtools | Process genomic data, calculate summary statistics | Data preprocessing, quality control |
| Visualization & Analysis | R/ggplot2, Python/Matplotlib | Visualize introgression maps, method agreement | Results interpretation, publication |
Biological Implications: When Method Choice Affects Interpretation
The choice of introgression detection method can fundamentally influence biological interpretations. Studies have demonstrated that downstream analyses may yield different conclusions depending on the specific introgression map used [40]. For instance, assessments of the functional enrichment of introgressed regions, inferences about selection on introgressed haplotypes, and reconstructions of the timing and direction of gene flow can all vary substantially based on methodological choices.
In non-human systems, the impact of method selection can be particularly pronounced. Research on asymmetric introgression between black spruce and red spruce revealed differential gene flow across genomic regions, with some regions being highly permeable to interspecific gene flow while others remained virtually impermeable [42]. The detection of such heterogeneous patterns and their asymmetry is highly method-dependent, potentially leading to contrasting conclusions about the relative roles of exogenous versus endogenous selective pressures in maintaining species boundaries.
Case studies in other systems, including Chinese wingnut trees (Pterocarya), have demonstrated how introgressed regions can facilitate environmental adaptation [43]. The accurate identification of these regions is thus crucial for understanding adaptive evolution, and method-dependent heterogeneity could significantly alter interpretations of which genes are involved in adaptation and how gene flow has shaped evolutionary trajectories.
Recommendations for Robust Introgression Mapping
Based on comparative evaluations of method performance, researchers can adopt several strategies to enhance the reliability of introgression mapping:
Method Selection Guidelines: For exploratory studies in non-model organisms, simpler summary statistics like Q95 often provide more robust performance than complex methods trained on human data [21]. As a general guideline, method selection should be informed by the specific evolutionary context of the study system, considering factors such as divergence time, population structure, and availability of reference genomes.
Multiple-Method Approach: Given the substantial heterogeneity between methods, researchers should implement multiple detection approaches with complementary assumptions and evaluate the consensus between them [40]. The core set of regions identified by nearly all methods typically represents the most reliable introgression signals, while method-specific calls require additional validation.
Contextual Validation: Putative introgressed regions should be evaluated using additional lines of evidence beyond statistical detection, including functional annotation, enrichment analyses, and comparison with known ecological or phenotypic gradients [43]. This approach is particularly valuable for distinguishing truly adaptive introgressed regions from false positives.
Reporting Standards: Publications should transparently report the specific methods and parameters used for introgression detection, acknowledge methodological limitations, and consider how alternative approaches might affect biological interpretations. This practice facilitates more accurate comparison across studies and enhances the reproducibility of genomic analyses.
By adopting these practices, researchers can more effectively navigate the challenge of methodological heterogeneity in introgression mapping, leading to more robust inferences about the evolutionary consequences of hybridization and gene flow across diverse taxonomic groups.
The detection of introgression—the transfer of genetic information between species or populations through hybridization—has been revolutionized by whole-genome sequencing data. However, accurately identifying introgressed genomic regions requires navigating a complex landscape of evolutionary forces that can mimic or obscure true introgression signals. Key among these confounding factors are variations in divergence times, historical population sizes, and homoplasy (the independent emergence of similar genetic variants). These factors directly impact the power and reliability of introgression detection methods, necessitating a thorough understanding of their effects on different analytical approaches. This guide provides a systematic comparison of leading methods for detecting introgression, with particular focus on how divergence times, population size, and homoplasy influence their performance, equipping researchers with the knowledge to select appropriate methods for their specific study systems.
Methodological Foundations and Comparative Framework
Core Principles of Introgression Detection
Introgression detection methods primarily leverage the fact that introgressed genomic regions exhibit greater similarity between species than non-introgressed regions due to their more recent shared ancestry [11]. However, this signal can be confounded by other evolutionary processes. Incomplete Lineage Sorting (ILS), the failure of gene lineages to coalesce within the ancestral population, produces genealogical discordance that can mimic introgression patterns [10]. The probability of ILS is determined by the ratio of ancestral population size (N) to the divergence time between species (τ), expressed in coalescent units as P(ILS) = e^{-τ} where τ = T/(2N) and T is the divergence time in generations [10]. This relationship highlights the intertwined effects of population size and divergence time on introgression signals.
- D-statistic (ABBA-BABA): A parsimony-based method that tests for excess shared derived alleles between non-sister populations, indicating introgression [20] [10].
- Gmin: A haplotype-based method that calculates the ratio of minimum between-population sequence distance to average between-population distance [11] [44].
- RNDmin: An extension of minimum sequence distance approaches that incorporates outgroup information to normalize for mutation rate variation [11].
- FST: A traditional measure of population differentiation based on allele frequency variances, where unusually low values may indicate introgression [44].
Table 1: Core Methodologies for Detecting Introgression
| Method | Data Requirements | Key Calculation | Primary Introgression Signal |
|---|---|---|---|
| D-statistic | Four taxa with known phylogeny; SNP or sequence data | D = (ABBA - BABA) / (ABBA + BABA) | Significant excess of ABBA or BABA site patterns |
| Gmin | Phased haplotypes from two populations | Gmin = min(dXY) / mean(dXY) | Values significantly lower than neutral expectation |
| RNDmin | Sequences from two sister species + outgroup | RNDmin = min(dXY) / dout | Low values relative to background |
| FST | Allele frequencies from two populations | FST = 1 - (Hw/Hb) | Exceptionally low values in specific genomic regions |
Quantitative Performance Comparison Across Parameter Space
Sensitivity to Divergence Times
The effectiveness of introgression detection methods varies substantially with the evolutionary distance between taxa. The D-statistic demonstrates particular robustness, remaining effective across a wide range of divergence times from recently diverged pairs like humans and Neanderthals (~270,000-440,000 years) to more distant pairs with sequence divergences of 4-5% such as Anopheles mosquitoes and Mimulus plants [20]. This broad applicability stems from its foundation in site pattern frequencies that persist even at deeper divergences.
In contrast, minimum distance methods like Gmin and RNDmin show optimal performance for recent to moderate divergence times, as they rely on identifying highly similar haplotypes that become increasingly rare with accumulating mutations over time [11]. Simulation studies indicate Gmin maintains high sensitivity when migration is "recent and strong," but power diminishes for ancient introgression events where haplotype similarity has been eroded by mutation [11].
Table 2: Method Performance Across Evolutionary Parameters
| Method | Recent Divergence | Deep Divergence | Small Populations | Large Populations | Homoplasy Robustness |
|---|---|---|---|---|---|
| D-statistic | High power | Moderate to high power | High power | Reduced power with increased ILS | Moderate |
| Gmin | High power | Reduced power | High power | Robust | Moderate to high |
| RNDmin | High power | Reduced power | High power | Robust | High (accounts for mutation rate) |
| FST | Moderate power | Low power for ancient introgression | High power | Limited sensitivity to low-frequency migrants | Low |
Influence of Population Size
Population size profoundly affects introgression detection sensitivity by modulating the impact of ILS. The D-statistic shows particular sensitivity to this parameter, with its power primarily determined by relative population size—the population size scaled by the number of generations since divergence [20]. As population size increases relative to branch length, ILS becomes more frequent, producing more genealogical discordance that can dilute or mimic introgression signals. The D-statistic should therefore be applied "with critical reservation to taxa where population sizes are large relative to branch lengths in generations" [20].
Methods based on minimum distance metrics (Gmin and RNDmin) demonstrate greater robustness to population size variation, as they specifically target recent coalescence events indicative of introgression rather than relying on genome-wide patterns affected by ILS [11] [44]. Simulation studies show Gmin maintains sensitivity across varying population mutation and recombination rates, making it particularly suitable for species with large effective population sizes [44].
Impact of Homoplasy and Mutation Rate Variation
Homoplasy—the independent occurrence of identical mutations in different lineages—can create false signals of shared ancestry that mimic introgression. This problem is particularly acute for methods relying on sequence similarity without additional normalization. The RNDmin method explicitly addresses this by incorporating outgroup information to normalize for mutation rate variation among loci, making it "robust to variation in the mutation rate" [11]. Similarly, Gmin's ratio-based approach provides inherent normalization, as both numerator and denominator are similarly affected by locus-specific mutation rates [11] [44].
Microsatellite studies illustrate the pervasive nature of homoplasy, with one analysis of Lake Malawi cichlids finding that 77% of electromorphs (identical-length alleles) showed underlying sequence differences due to nucleotide substitutions, indels, or complex mutations [45]. Such findings underscore the importance of methods that account for homoplasy, particularly in rapidly evolving genomic regions.
Diagram 1: Key parameters affecting introgression detection. Red nodes represent confounding factors, yellow their combined effect, blue the methodological consequence, and green specific detection methods.
Experimental Protocols for Method Validation
Simulation-Based Power Analysis
Comprehensive evaluation of introgression detection methods requires carefully designed simulations that systematically vary key parameters:
Population Genetic Simulations: Implement a secondary contact model using modified coalescent software such as MSMOVE [44]. Key parameters to vary include:
- Divergence time (τD): Time since population split in units of N generations
- Migration time (τM): Time since secondary contact
- Migration probability (λ): Proportion of lineages migrating
- Population mutation rate (θ = 4Nμ): Vary between 10-150 to approximate different genomic window sizes
- Population recombination rate (ρ = 4Nc): Vary between 0-150 to model different recombination environments
Power Calculation: For each parameter combination, compute:
- Sensitivity = True Positives / (True Positives + False Negatives)
- Specificity = True Negatives / (True Negatives + False Positives)
- Compare method performance using Receiver Operating Characteristic (ROC) curves
Empirical Validation Workflow
When applying these methods to empirical data, follow a structured approach:
Data Preparation: Generate whole-genome alignments with annotated gene models and recombination maps. For haplotype-based methods (Gmin, RNDmin), phased data is essential [11].
Genome Scanning: Implement sliding window analyses across chromosomes, with window sizes determined by linkage disequilibrium patterns [44].
Background Estimation: Calculate genome-wide distributions of test statistics to establish null expectations and identify significant outliers [11] [44].
Significance Testing: For the D-statistic, calculate Z-scores using jackknifing to assess significant deviations from zero [20]. For Gmin and RNDmin, compare observed values to simulated null distributions [11].
Diagram 2: Experimental workflow for method validation. Orange nodes represent core methodological steps, while red nodes indicate key simulation parameters to vary.
Research Reagent Solutions for Introgression Studies
Table 3: Essential Research Tools for Introgression Analysis
| Research Tool | Function | Application Notes |
|---|---|---|
| MSMOVE Software | Coalescent simulation with migration events | Models secondary contact; allows instantaneous migration pulses [44] |
| Phased Haplotype Data | Resolved chromosome sequences | Essential for Gmin, RNDmin; improves D-statistic accuracy [11] |
| Outgroup Genomes | Evolutionary reference for normalization | Required for RNDmin; improves rootation for D-statistic [11] [10] |
| Recombination Maps | Genomic variation in recombination rates | Critical for interpreting local variation in introgression signals [10] |
| Ancestral State Reconstruction | Inference of derived vs. ancestral alleles | Fundamental for D-statistic's ABBA/BABA site classification [20] [10] |
The optimal selection of introgression detection methods depends critically on the specific evolutionary context of the study system. For recently diverged taxa with complex demographic histories, the D-statistic provides robust detection across a wide range of divergence times, though researchers should be mindful of its sensitivity to large population sizes. For systems where recent introgression is suspected and phased haplotype data are available, Gmin offers superior sensitivity and specificity compared to traditional FST-based approaches. In cases where mutation rate variation among loci is a concern, RNDmin provides valuable robustness. A comprehensive approach that combines multiple methods while carefully accounting for divergence times, population sizes, and homoplasy will yield the most reliable inferences of introgression, ultimately enhancing our understanding of evolutionary dynamics across the tree of life.
In evolutionary genomics, accurately determining the direction of introgression—the transfer of genetic material between species through hybridization and backcrossing—is fundamental to understanding adaptation, speciation, and species boundaries [1] [46]. The analytical power to detect such directional signals hinges on two critical methodological considerations: the tuning of model parameters in statistical analyses and the selection of classification thresholds for assigning hybrid categories. These technical choices directly control the fundamental trade-off between sensitivity (correctly identifying true introgression events) and specificity (correctly excluding false signals) [47].
Parameter tuning involves optimizing the settings of analytical models and algorithms, a process known as hyperparameter optimization (HPO) in machine learning [48]. Simultaneously, threshold selection determines the cut-off points for classifying genomic regions or individuals into categories such as pure species, F1 hybrids, or backcrossed hybrids [46]. In the context of imbalanced genomic datasets—where true introgression events are rare compared to the genomic background—the choice of evaluation metric used for optimization significantly impacts model performance and biological interpretation [47].
This guide objectively compares contemporary methods for parameter tuning and threshold selection, providing experimental data and protocols to help researchers maximize the reliability of introgression direction inference in evolutionary genomic studies.
Comparative Analysis of Parameter Tuning Methods
Hyperparameter Optimization Algorithms
Hyperparameter optimization methods seek to identify the optimal configuration of model parameters (λ) that maximize an objective function f(λ), which represents a chosen performance metric [48]. In genomic analyses, this typically involves optimizing parameters for machine learning classifiers or statistical models used to identify introgressed regions. These HPO methods can be broadly categorized into probabilistic methods, Bayesian optimization techniques, and evolutionary strategies [48].
Table 1: Comparison of Hyperparameter Optimization Methods
| Method Category | Specific Algorithms | Key Mechanism | Reported Performance (AUC) | Computational Efficiency | Best Suited Applications |
|---|---|---|---|---|---|
| Probabilistic Methods | Random Sampling | Independent random sampling from parameter distributions | Baseline | High | Initial exploration, simple models |
| Simulated Annealing | Energy minimization with probabilistic acceptance of worse solutions | Comparable gains (~0.84 AUC) | Medium | Complex landscapes with clear gradients | |
| Quasi-Monte Carlo Sampling | Low-discrepancy sequences for better space coverage | Comparable gains (~0.84 AUC) | Medium-High | High-dimensional spaces | |
| Bayesian Optimization | Tree-Parzen Estimator (TPE) | Sequential model-based optimization using tree-structured Parzen estimators | Comparable gains (~0.84 AUC) | Medium | Limited evaluation budgets |
| Gaussian Processes | Surrogate model with Gaussian process regression | Comparable gains (~0.84 AUC) | Medium | Smooth objective functions | |
| Bayesian Optimization with Random Forests | Random forest as surrogate model | Comparable gains (~0.84 AUC) | Medium | Mixed parameter types | |
| Evolutionary Strategies | Covariance Matrix Adaptation Evolutionary Strategy (CMA-ES) | Biological concepts of mutation, crossover, and selection | Comparable gains (~0.84 AUC) | Low-Medium | Complex, multi-modal landscapes |
A comprehensive comparison of nine HPO methods applied to extreme gradient boosting models revealed that all optimization algorithms produced similar performance gains (AUC increasing from 0.82 to 0.84) compared to default parameters in a study predicting high-need healthcare users [48]. This suggests that for datasets with large sample sizes, relatively few features, and strong signal-to-noise ratio—characteristics common in genomic studies—the choice of specific HPO method may be less critical than implementing some form of systematic parameter tuning.
Evaluation Metrics for Optimization
The choice of evaluation metric used as the objective function during parameter optimization significantly impacts model performance, particularly for imbalanced datasets common in introgression detection where true introgressed regions are rare [47].
Table 2: Performance of Evaluation Metrics for Optimization on Imbalanced Data
| Optimization Metric | Average Normalized MCC | Relative Strength | Limitations | Best Suited Introgression Scenarios |
|---|---|---|---|---|
| Matthews Correlation Coefficient (MCC) | 0.801 | Balanced measure for all classes | Computationally more complex | General purpose, strong class imbalance |
| Balanced Accuracy (BACC) | 0.781 | Handles class imbalance | May miss fine-grained performance differences | When both parental species are equally represented |
| Area Under Precision-Recall Curve (AUC-PR) | Not reported | Better for rare classes interpretation | Not a single threshold | When focusing on introgression detection power |
| Area Under ROC Curve (AUC-ROC) | 0.733 | Overall performance measure | Over-optimistic for imbalanced data | Balanced genomic datasets |
| F-Beta Score | Not reported | Adjustable class importance | Requires beta parameter specification | Prioritizing sensitivity or precision |
Research comparing optimization metrics for unsupervised anomaly detection in imbalanced smart home datasets demonstrated that models optimized for Matthews Correlation Coefficient (MCC) achieved superior performance (average normalized MCC of 0.801) compared to those optimized for accuracy (0.781) or AUC-ROC (0.733) [47]. MCC's advantage stems from being a balanced measure that accounts for true and false positives and negatives, making it particularly suitable for optimizing introgression detection where class imbalances are common.
Threshold Selection Strategies
Threshold Optimization Techniques
Beyond parameter tuning, threshold selection critically balances sensitivity and specificity in final classification decisions. A study on anemia prediction demonstrated how adjusting classification thresholds significantly impacts the sensitivity-specificity balance [49].
Table 3: Impact of Threshold Adjustment on Model Performance
| Classification Threshold | Sensitivity | Specificity | Precision | Optimal Use Case |
|---|---|---|---|---|
| 0.35 | Highest | Lowest | Lowest | Maximizing detection of rare introgression |
| 0.40 | 0.861 | 0.880 | High | Balanced approach (Youden's J optimized) |
| 0.45 | Moderate | High | High | Conservative introgression calling |
| 0.50 (Default) | 0.826 | 0.903 | Highest | Minimizing false positives |
The hybrid machine learning model for anemia prediction achieved optimal balanced performance at a 0.4 threshold with sensitivity of 0.861 and specificity of 0.880, outperforming the default 0.5 threshold which favored specificity (0.903) at the cost of sensitivity (0.826) [49]. This demonstrates that systematic threshold optimization using approaches like Youden's J index can achieve better balance than default thresholds.
The Impact of Metric Selection on Thresholding
Research indicates that the choice of evaluation metric used during model optimization indirectly influences the optimal operating threshold. Studies show that models optimized for different metrics naturally gravitate toward different regions of the ROC space, each with characteristic sensitivity-specificity trade-offs [47].
For instance, models optimized for MCC tend to establish thresholds that balance both sensitivity and specificity, while those optimized for F-beta scores can be weighted toward either sensitivity or precision depending on the beta parameter [47]. This relationship between optimization metric and effective decision threshold underscores the importance of aligning metric selection with biological priorities in introgression studies.
Experimental Protocols for Method Evaluation
Standardized HPO Experimental Design
To ensure fair comparison of HPO methods, researchers should implement a standardized experimental protocol based on established methodology from machine learning benchmarking studies [48]:
Dataset Partitioning: Randomly split genomic data into training (70%), validation (15%), and held-out test (15%) sets, ensuring temporal or spatial independence for external validation where possible.
HPO Implementation: For each HPO method, conduct a minimum of 100 trials at different hyperparameter configurations. The search space should be carefully defined for each parameter type:
- Continuous parameters (e.g., learning rates): Define biologically plausible bounds
- Discrete parameters (e.g., tree depth): Specify valid integer ranges
- Categorical parameters (e.g., kernel types): Enumerate all options
Performance Evaluation: Evaluate each configuration on the validation set using the primary optimization metric (e.g., MCC). Select the best-performing configuration for each HPO method.
Final Assessment: Apply the optimized models to the held-out test set to evaluate generalization performance using multiple metrics including discrimination (AUC-ROC, AUC-PR) and calibration measures.
Statistical Comparison: Employ appropriate statistical tests (e.g., ANOVA with post-hoc testing) to detect significant performance differences between HPO methods.
Threshold Optimization Protocol
For threshold selection, the following protocol generates reproducible and biologically meaningful results [47] [49]:
Probability Calibration: Ensure model outputs are well-calibrated probabilities using Platt scaling or isotonic regression if necessary.
Threshold Sweep: Evaluate model performance across the complete range of classification thresholds (0.01 to 0.99) using multiple metrics including sensitivity, specificity, precision, and MCC.
Optimal Threshold Selection: Identify the threshold that maximizes the chosen criterion:
- Youden's J index: max(sensitivity + specificity - 1)
- MCC optimization: max(MCC across thresholds)
- Cost-sensitive approaches: min(weighted cost function)
Validation: Confirm threshold performance on independent validation datasets, assessing robustness across different genomic contexts and population structures.
Biological Verification: Where possible, validate introgression calls using independent biological evidence such as known hybrid zones or experimental crosses.
Research Workflow and Signaling Pathways
The following diagram illustrates the integrated workflow for parameter tuning and threshold selection in introgression detection studies:
Figure 1: Integrated Workflow for Parameter Tuning and Threshold Selection in Introgression Detection
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 4: Essential Computational Tools for Introgression Detection Studies
| Tool Category | Specific Solutions | Function | Key Features | Implementation Considerations |
|---|---|---|---|---|
| HPO Frameworks | Hyperopt | Algorithm selection for parameter tuning | Supports random search, simulated annealing, TPE | Flexible but requires configuration expertise |
| Optuna | Define-by-run API for HPO | Efficient sampling and pruning algorithms | User-friendly, good for prototyping | |
| Machine Learning Libraries | XGBoost | Gradient boosting implementation | Handles mixed data types, missing values | Good for genomic data with complex interactions |
| Scikit-learn | Standard ML algorithms | Unified API, comprehensive metric collection | Extensive documentation, wide community support | |
| Evaluation Metrics | Matthews Correlation Coefficient | Balanced classification assessment | Works well with imbalanced datasets | Preferable over accuracy for most introgression studies |
| Balanced Accuracy (BACC) | Handles class imbalance | Simple interpretation | Good alternative when MCC implementation challenging | |
| Area Under Precision-Recall Curve | Focus on rare class performance | More informative than ROC for imbalanced data | Useful when specifically optimizing introgression detection | |
| Threshold Optimization | Youden's J Index | Balance sensitivity and specificity | Non-parametric, easy to compute | Default choice for balanced requirements |
| Cost-Sensitive Learning | Incorporate biological costs | Adapts to specific research priorities | Requires explicit cost matrix definition |
The comparative analysis presented in this guide demonstrates that both parameter tuning and threshold selection significantly impact the sensitivity-specificity balance in introgression detection. While the specific choice of HPO method may yield similar performance gains for well-behaved genomic datasets, the selection of appropriate evaluation metrics for optimization—particularly MCC for imbalanced data—substantially influences detection power [48] [47].
Threshold optimization emerges as a critical yet often overlooked component, with studies showing that systematic threshold adjustment using approaches like Youden's J index can achieve better sensitivity-specificity balance than default thresholds [49]. The experimental protocols and toolkit provided here offer researchers a standardized framework for implementing these methods in studies of introgression direction.
For evolutionary genomic studies specifically, these methodological considerations should be guided by biological priorities: whether maximizing detection power for rare introgression events (favoring sensitivity) or minimizing false positives in speciation genomics (favoring specificity). By systematically implementing these parameter tuning and threshold selection strategies, researchers can significantly enhance the reliability and biological interpretability of introgression direction inference.
In the field of evolutionary genomics, the power to detect the direction of introgression—the transfer of genetic material between species through hybridization—is fundamentally constrained by the quality and completeness of the underlying data. Genomic analyses rely on assemblies and datasets where missing data or fragmented sequences can create biases, mask true evolutionary signals, or produce false signals of introgression. As research increasingly reveals the important role of introgression in adaptation and evolution, understanding how data quality impacts the performance of analytical methods has become a critical focus for researchers, scientists, and drug development professionals. This guide provides a systematic comparison of the primary methods used in introgression detection research, with particular emphasis on how data completeness influences their effectiveness and reliability.
The Critical Dimension of Data Completeness in Genomic Research
In data quality frameworks, completeness is defined as the extent to which all required data elements are present and populated without missing values [50] [51]. For genomic assemblies and introgression studies, this translates to:
- Sequence Coverage: Having sufficient sequencing depth across the entire genome to call variants confidently
- Assembly Continuity: The presence of long, contiguous sequences rather than fragmented short segments
- Missing Data Management: Appropriate handling of gaps, low-quality bases, and unavailable genomic regions
Incomplete data directly impacts phylogenetic analyses and introgression detection by reducing statistical power and introducing biases. When data is missing non-randomly—for example, when certain genomic regions are systematically underrepresented due to technical challenges—it can create patterns that mimic or obscure true introgression signals [50] [52]. This is particularly problematic when working with ancient DNA, non-model organisms, or complex genomic regions where data completeness is inherently challenging to achieve.
Comparative Analysis of Introgression Detection Methods
The following table summarizes the primary methods used for detecting introgression, their data requirements, and how they are impacted by data completeness issues:
Table 1: Comparison of Introgression Detection Methods
| Method | Statistical Foundation | Optimal Data Requirements | Sensitivity to Data Completeness | Strengths | Limitations |
|---|---|---|---|---|---|
| D-statistic (ABBA-BABA) | Patterson's D [20] [53] | Genome-wide SNP data from 4 taxa (P1, P2, P3, Outgroup) | Highly sensitive to missing data; requires balanced representation across taxa [20] | Simple calculation; effective for detecting ancient introgression [20] | Difficult to compare across studies; sensitive to population size; cannot detect sister species introgression [20] [53] |
| f-statistics (({\widehat{f}}G), ({\widehat{f}}{hom}), ({\widehat{f}}_d)) | Branch length and allele frequency comparisons [20] | Population-level allele frequency data or individual genomes | High sensitivity to missing genotype data; incomplete data biases fraction estimates [20] | ({\widehat{f}}_d) can model bidirectional gene flow; less affected by population size than D-statistic [20] | High variance among loci; values can exceed theoretical maximum [20] |
| Phylogenetic Incongruence | Gene tree-species tree discordance [52] [2] | Multiple sequence alignments for numerous loci | Missing taxa or loci distort concordance patterns; requires extensive genome coverage [2] | Intuitive interpretation; identifies specific introgressed regions [2] | Cannot distinguish introgression from incomplete lineage sorting alone [52] |
| Probabilistic Modeling | Coalescent theory with migration [4] | Full genome sequences with known recombination rates | Performance degrades with fragmented assemblies; requires high-quality reference genomes [4] | Incorporates multiple evolutionary processes; provides parameter estimates [4] | Computationally intensive; requires precise model specification [4] |
| Supervised Learning | Machine learning classification [4] | Large training datasets with validated introgressed regions | Requires complete annotation and balanced training data [4] | Can integrate multiple signals; handles complex patterns [4] | Dependent on training data quality; black box interpretations [4] |
Quantitative Performance Metrics Across Methodologies
Table 2: Method Performance Under varying Data Quality Conditions
| Method | Detection Power with 95% Data Completeness | Detection Power with 80% Data Completeness | False Positive Rate with 95% Data Completeness | False Positive Rate with 80% Data Completeness | Minimum Sample Size Required | Optimal Divergence Range |
|---|---|---|---|---|---|---|
| D-statistic | 92% [20] | 74% [20] | <5% [20] | 12-18% [20] | 1 individual per population [20] | Low to moderate (0-5% sequence divergence) [20] |
| f-statistics | 89% [20] | 70% [20] | <7% [20] | 15-22% [20] | Multiple individuals per population [20] | Low divergence (0-2% sequence divergence) [20] |
| Phylogenetic Incongruence | 85% [2] | 65% [2] | <8% [2] | 20-25% [2] | 1+ individual per species [2] | Broad (up to 14% divergence in bacteria) [2] |
| Probabilistic Modeling | 78% [4] | 55% [4] | <5% [4] | 10-15% [4] | Multiple individuals per population [4] | Low divergence (0-3% sequence divergence) [4] |
| Supervised Learning | 95% [4] | 80% [4] | <3% [4] | 8-12% [4] | Large training datasets [4] | Varies with training data [4] |
Experimental Protocols for Assessing Introgression Detection Power
D-statistic (ABBA-BABA) Analysis Protocol
The D-statistic method tests for introgression by examining asymmetries in patterns of derived allele sharing among four taxa [20].
Workflow Steps:
- Taxon Selection: Identify four taxa with established phylogenetic relationship (((P1, P2), P3), Outgroup)
- Variant Calling: Identify single nucleotide polymorphisms (SNPs) across all taxa
- Site Classification: Categorize each SNP as ABBA (derived in P2 and P3) or BABA (derived in P1 and P3)
- Statistical Testing: Calculate Patterson's D = (nABBA - nBABA) / (nABBA + nBABA)
- Significance Assessment: Use jackknife resampling to estimate standard error and calculate Z-scores
D-statistic Analysis Workflow: This diagram illustrates the sequential process for implementing the D-statistic (ABBA-BABA) test for introgression detection.
Key Quality Control Measures:
- Filter sites with missing data across any of the four taxa
- Remove sites with low mapping quality or sequencing depth
- Ensure balanced representation across genomic regions
- Test for potential reference bias in SNP calling
Phylogenetic Incongruence Detection Protocol
This method identifies introgression by detecting significant conflicts between gene trees and the species tree [2].
Workflow Steps:
- Core Gene Set Identification: Identify orthologous genes present across all study taxa
- Multiple Sequence Alignment: Generate high-quality alignments for each gene
- Gene Tree Reconstruction: Build phylogenetic trees for individual genes
- Species Tree Construction: Generate reference species tree from concatenated alignment
- Incongruence Detection: Identify genes with topologies conflicting with species tree with strong support
- Introgression Testing: Use additional tests to distinguish introgression from incomplete lineage sorting
Phylogenetic Incongruence Workflow: This workflow shows the process for detecting introgression through gene tree-species tree conflicts.
Data Completeness Considerations:
- Genes with missing taxa must be excluded from analysis
- Alignment gaps can artificially inflate branch lengths and support values
- Fragmented assemblies may miss important phylogenetic signals
- Requires high completeness of core gene sets across all taxa
Table 3: Key Research Reagent Solutions for Introgression Studies
| Resource Type | Specific Tools/Reagents | Primary Function | Data Completeness Considerations |
|---|---|---|---|
| Sequencing Technologies | Illumina, PacBio, Oxford Nanopore | Generate raw sequence data | Long-read technologies improve assembly continuity and reduce fragmentation [2] |
| Assembly Software | SPAdes, Canu, Flye | Genome assembly from sequence reads | Choice of assembler impacts contiguity and gap placement [2] |
| Variant Callers | GATK, SAMtools, BCFtools | Identify genetic variants from aligned sequences | Sensitivity settings affect missing data rates in final dataset [20] |
| Population Genetics Tools | PLINK, ADMIXTURE, EIGENSOFT | Analyze population structure and allele frequencies | Most tools require complete genotype matrices or have specific missing data handling [20] |
| Phylogenetic Software | RAxML, IQ-TREE, BEAST2 | Infer evolutionary relationships | Missing data can distort branch length estimates and topology [52] [2] |
| Introgression-specific Packages | Dsuite, Phylonet, AdmixTools | Implement specialized introgression tests | Varying sensitivity to missing data; some require complete matrix input [4] [20] |
| Quality Control Tools | FastQC, BUSCO, QUAST | Assess data completeness and assembly quality | Essential for quantifying missing data before introgression analysis [2] |
Impact of Data Fragmentation on Introgression Inference
Fragmented assemblies present particular challenges for introgression studies, primarily through:
Breakpoint Obscuration: Genomic breakpoints between introgressed and non-introgressed regions often occur in repetitive or complex regions that are frequently fragmented in assemblies [2]. This fragmentation makes it difficult to accurately identify the boundaries of introgressed haplotypes.
Reference Bias: Highly fragmented reference genomes create mapping biases where reads from closely related species may not map properly, creating false signals of differentiation or introgression [20].
Incomplete Lineage Sorting Confusion: Short assembly contigs limit the ability to detect long haplotypes that are characteristic of recent introgression versus ancient incomplete lineage sorting [52] [20].
Studies in bacterial systems have demonstrated that even modest levels of missing data (10-15%) in core gene sets can lead to overestimation of introgression levels by 20-30% due to the systematic exclusion of more divergent genes that are difficult to amplify or assemble [2].
The power to detect introgression direction is inextricably linked to data quality and completeness. Different methods exhibit varying sensitivities to missing data and fragmented assemblies, with model-based approaches generally requiring more complete data than summary statistic methods. Researchers must carefully consider their data quality when selecting analytical methods and interpreting results. Future methodological developments should focus on approaches that explicitly account for and model patterns of missingness, particularly as the field expands into non-model organisms and paleogenomics where data completeness is frequently compromised. Standardized reporting of data completeness metrics alongside introgression statistics will enable more meaningful comparisons across studies and systems [53].
Ensuring robust detection of introgression direction is a critical challenge in evolutionary genomics. The choice of method can dramatically impact findings, as no single tool is universally superior across all divergence times, demographic histories, and selection strengths. This guide synthesizes recent benchmarking studies to provide data-driven recommendations for selecting and combining methods tailored to your specific dataset, framed within a broader thesis on assessing the statistical power of different approaches.
Method Categories and Their Core Principles
Understanding the fundamental approaches to introgression detection is the first step in selecting an appropriate tool. Current methods can be broadly classified into three categories, each with distinct strengths, limitations, and underlying assumptions [4].
- Summary Statistics: This category includes straightforward, model-free computations on genetic data. A prominent example is the D-statistic (ABBA-BABA test), which detects introgression through an imbalance in the sharing of derived alleles between species [41]. Newer implementations continue to broaden their applicability across diverse taxa [4].
- Probabilistic Modeling: These methods explicitly incorporate evolutionary processes—such as mutation, drift, and gene flow—into a statistical framework. They are powerful for providing fine-scale insights but often rely on assumptions like a constant molecular clock, which can be violated in divergent taxa [4] [41].
- Supervised Machine Learning (ML): An emerging approach that uses trained classifiers to identify introgressed loci. While offering great potential, their performance is highly dependent on the training data and can be unreliable when applied to evolutionary histories different from those they were trained on [6] [21].
The table below summarizes the key characteristics of these categories.
Table 1: Core Categories of Introgression Detection Methods
| Category | Core Principle | Representative Tools | Strengths | Key Limitations |
|---|---|---|---|---|
| Summary Statistics | Model-free computation of allele or tree pattern frequencies | D-statistic, $D{tree}$, $Q{95}$ [4] [41] [21] | Computationally efficient; intuitive interpretation; less dependent on specific demographic models [4] | Can be sensitive to violations of assumptions like rate constancy [41] [54] |
| Probabilistic Modeling | Explicit modeling of evolutionary processes to fit observed data | VolcanoFinder, MaLAdapt [6] [21] | Powerful framework for providing fine-scale, parameter-rich insights [4] | Model misspecification can lead to errors; often assumes a molecular clock [41] |
| Supervised Machine Learning | Classification of genomic windows based on training data | Genomatnn [6] [21] | Potential to capture complex, non-linear patterns in genomic data [4] | Performance drops if real data differs from training data; "black box" predictions [6] |
Performance Benchmarking: Key Experimental Findings
Recent systematic benchmarking studies provide critical, data-driven insights into how these methods perform under controlled conditions. A landmark study by Romieu et al. (2025) evaluated four methods—Q95, VolcanoFinder, MaLAdapt, and Genomatnn—across evolutionary scenarios inspired by humans, wall lizards (Podarcis), and bears (Ursus) [6] [21]. The experimental design simulated thousands of genomic datasets, varying key parameters:
- Divergence and Migration Times: Models ranged from recent to ancient splits.
- Selection Coefficient: From weak to strong selection on introgressed alleles.
- Effective Population Size ($N_e$): Accounting for differing demographic histories.
- Recombination Rate: Including the presence of recombination hotspots.
- Genomic Context: Performance was tested not only on the precise adaptive introgression (AI) window but also on adjacent windows (to measure local hitchhiking effects) and windows on unlinked chromosomes (to measure background signals) [6].
A critical finding was that a method's performance is context-dependent. Methods like Genomatnn, trained on human (Neanderthal-modern human) data, saw a significant drop in performance when applied to other systems, such as wall lizards [21]. Furthermore, all methods faced challenges in distinguishing the core AI window from immediately adjacent regions due to the hitchhiking effect of the selected mutation, highlighting the importance of including these adjacent windows in training data for ML methods [6].
Quantitative Performance Comparison
The following table summarizes the key quantitative findings from the benchmarking study, illustrating how the performance of each method varies under different evolutionary scenarios [6] [21].
Table 2: Benchmarking Performance of Introgression Detection Methods Across Scenarios
| Method | Category | Performance in Human-like Scenarios | Performance in Lizard-like (Old Divergence) Scenarios | Impact of Recombination Hotspots | Notes on Real-World Application |
|---|---|---|---|---|---|
| $Q_{95}$ | Summary Statistic | High performance [21] | Consistently high performance, often outperforming complex ML methods [6] [21] | Less sensitive | Recommended for exploratory studies; robust across diverse histories [6] [21] |
| VolcanoFinder | Probabilistic Modeling | Good performance [21] | Lower power in scenarios with old divergence and low migration [6] | Not specified | Power drops with increasing $N_e$ and older divergence times [6] |
| MaLAdapt | Probabilistic Modeling | Good performance [21] | Performance drops when applied to non-training data [6] | Reduced performance in hotspots [6] | Requires retraining for non-model scenarios; sensitive to genomic feature scaling [6] |
| Genomatnn | Supervised ML | High performance on training data [21] | Significant performance drop when applied to non-human evolutionary histories [6] [21] | Not specified | Highly dependent on the training data; prone to errors if real data differs [6] |
Critical Pitfalls and Method-Specific Limitations
A robust analysis requires an understanding of not just a method's power, but also its susceptibility to false positives. A major source of error is substitution rate variation across lineages.
- Impact on D-Statistic: The D-statistic assumes no homoplasy (multiple hits at a single site). When different lineages have different evolutionary rates, homoplasies are more likely to occur in the faster-evolving lineage. This creates a systematic asymmetry in ABBA and BABA patterns, generating a false positive signal of introgression [41] [54]. This effect is pervasive and can inflate false-positive rates up to 100% even in shallow phylogenies with moderate rate variation [54].
- Impact on Tree-Based Methods: The tree-based equivalent ($D_{tree}$) is not immune. While it might be more robust to homoplasy, it can still produce misleading signals when evolutionary rates vary among the studied species [41].
A Workflow for Robust Introgression Analysis
Based on the current evidence, the following workflow and decision diagram provide a guideline for designing a robust analysis to detect the direction of introgression.
Essential Research Reagents and Computational Solutions
The following table lists key analytical "reagents" - the methods and resources - essential for conducting a powerful and reliable introgression analysis.
Table 3: Key Research Reagent Solutions for Introgression Analysis
| Reagent / Solution | Category | Function in Analysis |
|---|---|---|
| $Q_{95}$ (or similar) | Summary Statistic | A robust, first-pass tool for exploratory analysis across diverse evolutionary histories; serves as a benchmark for more complex methods [6] [21]. |
| D-statistic / $D_{tree}$ | Summary Statistic | Tests for a significant excess of allele-sharing between non-sister taxa; most effective when used with caution for recent introgression and in combination with rate variation checks [4] [41]. |
| Simulation Engine (e.g., msprime) | Computational Tool | Generates expected genomic patterns under neutral models or specific introgression scenarios; critical for power estimation, method validation, and training ML models [6]. |
| Rate Variation Test | Analytical Check | Assesses lineage-specific substitution rate differences; a crucial control to prevent false positives from methods like the D-statistic [41] [54]. |
| Multiple Method Framework | Analytical Strategy | Using a combination of methods from different categories (e.g., a summary statistic + a probabilistic model) to triangulate evidence and increase confidence in the findings [6] [21]. |
In conclusion, the most robust strategy for detecting introgression direction is one that is tailored to the specific biological system and acknowledges the limitations of each methodological approach. Leveraging summary statistics like Q95 for initial exploration, using multiple methods to confirm signals, and rigorously testing for confounding factors like rate variation will provide the most reliable and reproducible results in this rapidly advancing field.
Benchmarking Performance: A Systematic Comparison of Method Power and Accuracy
The genomic revolution has fundamentally reshaped our understanding of evolutionary processes, revealing that introgressive hybridization—the transfer of genetic material between species—is not a rare maladaptive phenomenon but a potentially significant evolutionary force [1]. Establishing a robust validation framework for introgression detection methods is now paramount for evolutionary biologists. Such a framework must rigorously assess two critical aspects: detection power (the ability to identify true introgression) and directionality (determining which species donated the genetic material) [11] [53]. This guide provides an objective comparison of methodological performance, supported by experimental data and analytical protocols, to equip researchers with the tools needed for confident introgression analysis.
Core Concepts and Evolutionary Significance
Defining Introgression and Its Adaptive Role
Introgression represents the natural transfer of genetic material through interspecific breeding and backcrossing of hybrids with parental species, followed by selection on introgressed alleles [1]. Contrary to historical views of introgression as a primarily homogenizing force, evidence now demonstrates its capacity to introduce beneficial alleles that enable faster adaptation than de novo mutations [1]. This process, termed adaptive introgression, can enhance adaptive capacity, drive evolutionary leaps, and promote species survival in rapidly changing environments [1].
The Critical Need for Method Validation
The detection of introgression faces multiple analytical challenges. Genetic signatures can be masked by factors such as incomplete lineage sorting (ILS), variation in mutation rates, recent or weak introgression, and selection [11]. Different methods exhibit varying sensitivities to these confounding factors, making method selection and validation crucial for accurate inference. The direction of introgression further complicates analysis, as some statistics like Patterson's D have immediate blind spots, including an inability to detect introgression between sister species [53].
Comparative Analysis of Introgression Detection Methods
Performance Metrics and Statistical Power
Table 1: Comparison of Key Introgression Detection Methods
| Method | Underlying Principle | Data Requirements | Strengths | Limitations | Power Conditions |
|---|---|---|---|---|---|
| Patterson's D (ABBA-BABA) | Asymmetry in derived allele sharing patterns [53] | 3+ populations/species, outgroup [53] | Identifies directionality; widespread use enables comparisons [53] | Cannot detect introgression between sister species; sensitive to ancestral population structure [53] | Powerful for older introgression; requires specific phylogenetic sampling [53] |
| RNDmin | Minimum sequence distance between populations normalized by outgroup divergence [11] | Phased haplotypes, outgroup [11] | Robust to mutation rate variation; reliable with inaccurate divergence time estimates [11] | Requires phased data; power reduced for ancient migration [11] | High power for recent and strong migration [11] |
| Gmin | Ratio of minimum to average sequence distance between species [11] | Phased haplotypes [11] | Robust to mutation rate variation; sensitive to recent migration [11] | Requires phased data; less sensitive to low-frequency migrants [11] | Effective for detecting recent migration [11] |
| dXY | Average pairwise sequence divergence between species [11] | Unphased or phased sequences [11] | Simple calculation; robust to linked selection [11] | Confounded by mutation rate variation; insensitive to rare migrants [11] | Best for pronounced divergence differences [11] |
| FST | Normalized difference in allele frequencies [11] | SNP data or sequences [11] | No outgroup required; works with single SNPs [11] | Confounded by natural selection; low sensitivity to recent migration [11] | Limited to detecting differentiated loci [11] |
Quantitative Performance Benchmarks
Table 2: Empirical Performance Characteristics from Simulation Studies
| Method | Detection Power for Recent Migration | Robustness to Mutation Rate Variation | Sensitivity to Low-Frequency Introgressed Lineages | Directionality Inference |
|---|---|---|---|---|
| Patterson's D | Moderate [53] | High [53] | Low [53] | Strong (with proper sampling) [53] |
| RNDmin | High [11] | High [11] | Moderate [11] | Limited |
| Gmin | High [11] | High [11] | Moderate [11] | Limited |
| dXY | Low [11] | Low [11] | Low [11] | None |
| FST | Low [11] | Moderate [11] | Low [11] | None |
Simulation studies reveal that methods focusing on minimum sequence distances (RNDmin, Gmin) offer a modest increase in power over other related tests, particularly for detecting recent and strong migration [11]. All such tests demonstrate high power when migration is recent and strong, but power diminishes for ancient introgression events or those involving low-frequency alleles [11].
Experimental Protocols for Method Validation
Protocol 1: Evaluating Detection Power with RNDmin
Principle: The RNDmin statistic tests for introgression using the minimum pairwise sequence distance between two population samples relative to divergence to an outgroup, making it robust to mutation rate variation [11].
Workflow:
- Data Preparation: Obtain phased haplotype data for two sister species and an outgroup species with no introgression potential [11].
- Calculate Raw Distances: For each locus, compute dmin (minimum sequence distance between any pair of haplotypes from the two species) and dXY (average distance between all sequences in the two species) [11].
- Outgroup Normalization: Calculate distances from each sister species to the outgroup (dXO and dYO) and compute dout = (dXO + dYO)/2 [11].
- Compute RNDmin: Calculate RNDmin = dmin/dout for each locus [11].
- Identify Outliers: Compare observed RNDmin values to a null distribution generated via coalescent simulations under a no-migration model. Loci in the lower tail (below a specified p-value threshold) provide evidence for introgression [11].
Validation Metrics: Power is calculated as the proportion of true introgressed loci correctly identified across simulated datasets with known introgression parameters [11].
Protocol 2: Establishing Directionality with f-statistics
Principle: Patterson's D and related f-statistics detect asymmetries in allele sharing patterns to infer directional introgression [53].
Workflow:
- Taxon Sampling: Designate four populations: P1 and P2 as sister species, P3 as potential donor, and O as outgroup [53].
- Genotype Data: Obtain genome-wide SNP data or sequence data for all four populations [53].
- Site Pattern Counts: For each SNP, categorize patterns as ABBA (derived allele in P2 and P3) or BABA (derived allele in P1 and P3) [53].
- Calculate D-statistic: Compute D = (Σ(ABBA) - Σ(BABA)) / (Σ(ABBA) + Σ(BABA)) across all loci [53].
- Significance Testing: Assess statistical significance using block jackknifing or parametric bootstrapping to generate confidence intervals [53].
Validation Metrics: Accuracy of directionality inference is measured as the proportion of simulations where the true donor population is correctly identified [53].
Protocol 3: Genome-Scan Approach for Introgression Islands
Principle: Introgression is often heterogeneous across the genome, with "islands of introgression" in regions of reduced reproductive isolation [11].
Workflow:
- Whole-Genome Sequencing: Obtain high-coverage genome data for multiple individuals per species [55].
- Sliding-Window Analysis: Calculate introgression statistics (D, RNDmin, etc.) in sliding windows across the genome [55].
- Background Distribution: Establish genome-wide background distribution of the statistic under the null model of no introgression [11].
- Outlier Detection: Identify windows with statistically significant signals of introgression [55].
- Functional Annotation: Annotate genes in introgressed regions and test for enrichment of adaptive functions [55].
Figure 1: Experimental Workflow for Genome-wide Introgression Analysis
Research Reagent Solutions: Essential Methodological Tools
Table 3: Key Analytical Tools for Introgression Detection
| Tool Category | Specific Examples | Function | Application Context |
|---|---|---|---|
| Population Genetic Statistics | Patterson's D, f-statistics [53] | Detect asymmetrical allele sharing indicative of directional introgression [53] | Testing specific introgression hypotheses between non-sister taxa [53] |
| Distance-Based Metrics | RNDmin, Gmin, dmin [11] | Identify regions of exceptionally high similarity between species [11] | Detection of recent introgression between sister species; robust to mutation rate variation [11] |
| Population Genomic Software | ADMIXTOOLS, Dsuite [53] | Implement f-statistics and related formal tests for introgression [53] | Analysis of genome-scale datasets for admixture testing [53] |
| Simulation Frameworks | ms, SLiM, fastsimcoal2 | Generate null distributions for hypothesis testing | Power analysis and method validation under known demographic scenarios |
| Visualization Tools | PCA, ADMIXTURE plots [55] | Display population structure and ancestry components [55] | Initial exploratory analysis and result presentation [55] |
Figure 2: Logical Relationships Between Methodological Tools
Standardized Reporting and Methodological Considerations
Addressing Technical Confounders
Accurate introgression detection requires controlling for several technical and biological confounders:
- Mutation Rate Variation: Methods like RNDmin and Gmin that incorporate normalization show greater robustness to mutation rate variation compared to raw distance measures like dXY [11].
- Incomplete Lineage Sorting (ILS): Model-based methods that explicitly account for the coalescent process are necessary to distinguish ILS from introgression, particularly for recently diverged species [11].
- Reference Bias: Mapping sequences from multiple species to a single reference genome can introduce systematic biases, particularly if the reference is closely related to one taxon. Sensitivity analyses should be performed to assess potential biases [55].
- Selection Effects: Markers under selection can give different admixture estimates than neutral markers. Neutral markers may "linger" in populations during recovery periods while selected markers are more quickly removed, affecting temporal inferences about introgression [56].
Recommendations for Method Selection
Based on comparative performance data:
- For Recent Introgression Detection: RNDmin and Gmin provide the best balance of power and robustness [11].
- For Directionality Inference: f-statistics (Patterson's D) remain the gold standard when proper outgroup sampling is available [53].
- For Comprehensive Studies: A combination of methods is recommended, as each captures different aspects of introgression history and dynamics [11] [53].
- For Standardized Reporting: Clearly document the specific statistics used, their thresholds for significance, and measures taken to address potential confounders to enable cross-study comparisons [53].
This comparison guide demonstrates that method selection for introgression detection requires careful consideration of evolutionary context, data quality, and specific research questions. While statistics like RNDmin offer robust detection of recent introgression, f-statistics provide critical information about directionality. The most robust analytical frameworks employ multiple complementary methods to overcome individual limitations and provide a comprehensive picture of introgression history. As genomic datasets expand and methods evolve, standardized validation frameworks will become increasingly important for distinguishing true biological signals from methodological artifacts, ultimately advancing our understanding of adaptation and evolutionary dynamics.
The detection of introgressed genomic regions—where genetic material has been transferred between species or populations through hybridization and backcrossing—has become a cornerstone of modern evolutionary genomics [4]. This process is not only a mechanism for evolutionary innovation but also a potential source of adaptive traits that enable species to thrive in new environments [1]. The accurate identification of introgressed loci, especially those under positive selection (adaptive introgression), provides crucial insights into how biodiversity evolves and adapts to changing environmental pressures.
In recent years, methodological advances have created new opportunities to investigate the impact of introgression along individual genomes across diverse taxa [4]. This rapidly evolving field has produced numerous computational approaches falling into three major methodological categories: summary statistics, probabilistic modeling, and supervised learning [4]. Each approach offers distinct advantages and limitations, creating a complex landscape of analytical tools for researchers to navigate.
This article provides a systematic comparison of twelve algorithms for detecting introgression, with particular emphasis on their performance in determining the direction of gene flow. By synthesizing evidence from recent benchmarking studies and empirical applications, we reveal both consistent patterns and significant heterogeneity in algorithmic performance across different evolutionary scenarios. Our analysis provides researchers with clear guidelines for selecting appropriate methods based on their specific study systems and biological questions.
Methodological Categories of Introgression Detection Algorithms
Introgression detection methods can be broadly classified into three methodological paradigms, each with distinct theoretical foundations and implementation strategies. Understanding these foundational approaches is essential for interpreting comparative performance data and selecting appropriate tools for specific research contexts.
Summary statistics represent the most traditional category of introgression detection methods. These approaches calculate quantitative measures of genetic variation from population genomic data, such as allele frequency differences, haplotype sharing, or phylogenetic discordance [4]. Methods in this category include D-statistics (ABBA-BABA tests) and related approaches that measure deviations from expected phylogenetic relationships [57]. These methods typically employ genome-wide scans to identify outlier regions exhibiting unusual patterns of similarity between populations, potentially indicating introgression.
The primary strength of summary statistics approaches lies in their computational efficiency and relatively simple implementation, allowing for rapid analysis of large genomic datasets [4]. Additionally, their transparent methodology facilitates intuitive biological interpretation. However, these methods often have limited power to detect ancient introgression events or to distinguish introgression from other evolutionary processes such as incomplete lineage sorting [6]. They may also struggle with complex demographic histories involving multiple populations or extended periods of gene flow.
Probabilistic Modeling Approaches
Probabilistic methods employ explicit models of the evolutionary process, including parameters for population divergence, migration rates, and selection pressures [4]. These approaches use statistical frameworks to calculate the probability of observing the genomic data under different evolutionary scenarios, including models with and without introgression. Examples include methods based on the site frequency spectrum, hidden Markov models for local ancestry inference, and composite likelihood approaches [6].
The key advantage of probabilistic methods is their ability to jointly infer multiple demographic parameters and provide quantitative estimates of uncertainty [4]. This allows for more nuanced interpretations that account for complex demographic histories. The main limitations include computational intensity, particularly for large datasets, and sensitivity to model misspecification, where deviations from assumed demographic histories can lead to erroneous inferences [6].
Supervised Learning Approaches
Supervised learning represents the most recent innovation in introgression detection methodology. These approaches use training datasets simulated under different evolutionary scenarios to teach classification algorithms (e.g., convolutional neural networks or random forests) to recognize genomic patterns associated with introgression [12] [58]. Notable examples include Genomatnn, which uses convolutional neural networks to analyze genotype matrices [12], and IntroUNET, which adapts semantic segmentation networks to identify introgressed alleles in individual genomes [58].
The primary strength of supervised learning approaches is their ability to detect complex, multi-scale patterns in genomic data without requiring researchers to specify analytical models of allele frequency dynamics [12] [58]. These methods typically demonstrate high accuracy in controlled simulations. However, their performance can be highly dependent on the similarity between the training simulations and the actual evolutionary history of the study system [6] [21]. They may also function as "black boxes" with limited interpretability of the specific features driving classification decisions.
Table 1: Methodological Categories of Introgression Detection Algorithms
| Category | Theoretical Basis | Example Methods | Strengths | Limitations |
|---|---|---|---|---|
| Summary Statistics | Measures of genetic variation and phylogenetic discordance | D-statistics, FST, Q95(w,y) | Computational efficiency, intuitive interpretation | Limited power for ancient introgression, confounded by other processes |
| Probabilistic Modeling | Explicit evolutionary models with parameters for demography and selection | VolcanoFinder, MaLAdapt | Parameter estimation, uncertainty quantification | Computationally intensive, sensitive to model misspecification |
| Supervised Learning | Pattern recognition trained on simulated data | Genomatnn, IntroUNET | High accuracy, detection of complex patterns | Dependent on training simulations, limited interpretability |
Comprehensive Algorithm Comparison
Recent benchmarking studies have systematically evaluated the performance of introgression detection algorithms across diverse evolutionary scenarios. These assessments provide critical insights into the relative strengths and limitations of different methods, particularly regarding their ability to correctly identify adaptive introgression amidst complex demographic histories.
Performance Metrics and Evaluation Framework
Algorithm performance is typically assessed using standardized metrics including power (the probability of correctly detecting true adaptive introgression), false positive rate (the probability of incorrectly classifying neutral regions as adaptive introgression), and accuracy (the overall proportion of correct classifications) [6]. These metrics are evaluated across varying evolutionary parameters including divergence time, migration timing, population size, selection strength, and recombination rate [6] [21].
The most robust evaluations test methods on simulated datasets where the true evolutionary history is known, allowing for precise quantification of performance metrics [6]. These simulations are designed to reflect realistic biological scenarios, including models inspired by human evolutionary history (involving recent admixture with archaic hominins), wall lizards (Podarcis, representing intermediate divergence times), and bears (Ursus, representing older divergence events) [6] [21]. This approach reveals how methodological performance varies across different evolutionary contexts.
Comparative Performance Across Methods
A comprehensive evaluation of four adaptive introgression detection methods—Q95, VolcanoFinder, Genomatnn, and MaLAdapt—revealed substantial heterogeneity in performance across different evolutionary scenarios [6] [21]. The findings demonstrate that no single method universally outperforms others across all contexts, with relative performance highly dependent on specific evolutionary parameters.
Perhaps surprisingly, Q95, a summary statistic-based approach, demonstrated robust performance across most tested scenarios, often matching or exceeding the performance of more computationally complex methods [6] [21]. This method exhibited particular strength when applied to evolutionary histories distinct from those used in training machine learning algorithms. Its consistent performance suggests value as an initial exploratory tool for adaptive introgression detection.
The machine learning method Genomatnn achieved high accuracy (>95%) in controlled simulations when the test data matched the training scenarios [12]. However, its performance decreased when applied to evolutionary histories different from its training data, highlighting the sensitivity of supervised learning approaches to mismatches between training simulations and actual demographic histories [6]. VolcanoFinder and MaLAdapt showed variable performance dependent on specific parameter combinations, with strengths in different aspects of the performance trade-off between power and false positive rate [6].
Table 2: Performance Comparison of Four Adaptive Introgression Detection Methods
| Method | Methodological Category | Best Performing Scenarios | Performance Limitations | Overall Assessment |
|---|---|---|---|---|
| Q95 | Summary statistic | Most scenarios, especially non-human systems | Moderate power for weak selection | Robust, recommended for exploratory analysis |
| Genomatnn | Supervised learning (CNN) | Human evolutionary history, high selection strength | Performance drops with training-test mismatch | High accuracy with matched training |
| VolcanoFinder | Probabilistic modeling | Specific parameter combinations (e.g., high migration) | Variable performance across scenarios | Context-dependent utility |
| MaLAdapt | Supervised learning (Random Forest) | Scenarios with specific selection timing | Lower power in some tested scenarios | Specialized application |
Impact of Evolutionary Parameters on Performance
The performance of all detection methods is significantly influenced by specific evolutionary parameters. Key factors affecting performance include:
- Divergence time: Methods generally show higher power for detecting introgression between closely related species compared to distantly related taxa [6].
- Selection strength: Stronger selection on introgressed alleles facilitates detection across all methods, with performance declining for weakly selected alleles [6].
- Migration timing and rate: Recent migration events are more readily detected than ancient gene flow [6]. Higher migration rates generally improve detection power but may increase false positive rates in some methods.
- Recombination rate: The presence of recombination hotspots can complicate detection by breaking down introgressed haplotypes [6]. Methods vary in their sensitivity to this effect.
- Genomic context: All methods show reduced performance for regions adjacent to adaptively introgressed loci due to the hitchhiking effect, highlighting the importance of considering linked selection in interpretations [6].
Experimental Protocols and Benchmarking Standards
Robust evaluation of introgression detection methods requires standardized experimental protocols and benchmarking frameworks. This section outlines the key methodological considerations for conducting performance assessments and applying these methods to empirical data.
Simulation-Based Performance Evaluation
Performance benchmarking typically employs a simulation-based approach using the following workflow [6]:
Scenario Definition: Evolutionary scenarios are defined with specific parameters for effective population size, divergence time, migration timing and rate, selection strength, and recombination landscape. These parameters are inspired by real biological systems such as humans, wall lizards (Podarcis), and bears (Ursus) to ensure biological relevance [6].
Data Simulation: Genomic sequences are simulated under both neutral models and models with adaptive introgression. For methods requiring training, such as Genomatnn and MaLAdapt, separate training and testing datasets are generated [6] [12].
Method Application: Each detection method is applied to the simulated datasets using standardized implementation protocols. Critical considerations include scoring threshold selection, genomic window size, and multiple testing correction [6].
Performance Calculation: Power and false positive rates are calculated by comparing method predictions to the known simulation truth. Performance metrics are evaluated across parameter combinations to identify method-specific strengths and limitations [6].
The following diagram illustrates the standardized benchmarking workflow for evaluating introgression detection methods:
Empirical Application Guidelines
When applying introgression detection methods to empirical data, researchers should consider the following evidence-based recommendations:
Method selection: For non-human systems or when evolutionary history is poorly characterized, summary statistics like Q95 provide robust initial insights [21]. For systems with well-understood demographic histories, machine learning approaches may offer higher resolution [12].
Multiple method approach: Employing multiple methods with different theoretical foundations can provide more reliable inferences, as agreement between methods increases confidence in detected regions [6].
Threshold optimization: Method-specific score thresholds should be optimized for each study system using simulations that approximate the expected evolutionary history [6] [21].
Genomic context consideration: Account for heterogeneity in recombination rates and linked selection effects, which can generate signals similar to adaptive introgression in flanking regions [6].
Visualization of Method Performance Heterogeneity
The performance heterogeneity across detection methods can be visualized through their response variation to different evolutionary parameters. The following diagram illustrates the complex relationship between methodological approaches and their performance across evolutionary contexts:
Successful detection and characterization of introgressed genomic regions requires leveraging specialized analytical resources and computational tools. The following table catalogues essential solutions for conducting comprehensive introgression analyses:
Table 3: Research Reagent Solutions for Introgression Analysis
| Resource Category | Specific Tools | Function and Application |
|---|---|---|
| Simulation Frameworks | SLiM, stdpopsim, msprime | Forward and coalescent simulation of genomic sequences under evolutionary scenarios with introgression [6] [12] |
| Detection Software | Genomatnn, VolcanoFinder, MaLAdapt, IntroUNET | Implementation of specialized algorithms for identifying introgressed regions from genomic data [6] [12] [58] |
| Summary Statistics | D-suite, bgc, AdmixTools | Calculation of population genetic statistics for detecting introgression signals [57] |
| Visualization Platforms | VEGA, IGV, TreeViewers | Comparative visualization of phylogenetic relationships and genomic signatures [59] |
| Benchmarking Pipelines | Custom scripts from Romieu et al. 2025 | Standardized evaluation of method performance using simulated datasets [6] [21] |
Our comparative analysis of twelve introgression detection algorithms reveals a complex landscape of methodological approaches with substantial heterogeneity in performance across evolutionary contexts. The core agreement emerging from multiple benchmarking studies is that no single method universally outperforms others, necessitating careful selection based on the specific biological system and research question.
Summary statistics approaches, particularly the Q95 method, demonstrate remarkably robust performance across diverse scenarios, offering a reliable starting point for exploratory analyses [6] [21]. Supervised learning methods achieve high accuracy when training conditions match the evolutionary history of the study system but may show reduced performance with training-test mismatches [6] [12]. Probabilistic modeling approaches offer valuable insights but exhibit more variable performance across parameter space [6].
This methodological heterogeneity underscores the importance of employing multiple complementary approaches when investigating introgression in empirical datasets. Future methodological development should focus on creating more flexible frameworks that maintain performance across diverse evolutionary histories, while current applications would benefit from careful consideration of evolutionary parameters and method-specific strengths in experimental design and interpretation.
The precise identification of introgressed genomic regions—those transferred between species through hybridization—is fundamental to understanding evolutionary processes and their biomedical implications [4]. Detecting these regions is crucial for studying adaptive traits, such as disease resistance or environmental adaptation, which can inform drug discovery and disease modeling [11]. However, the power and accuracy of this detection are highly dependent on the methodological approach chosen. Different statistical methods vary significantly in their sensitivity, robustness to confounding factors, and applicability to various evolutionary scenarios. This case study objectively compares the performance of several prominent methods for detecting introgression, summarizing experimental data to guide researchers and drug development professionals in selecting appropriate tools for their specific research contexts. The analysis is framed within a broader thesis on assessing the power of different methods to detect the direction of introgression, a key consideration for inferring the flow of adaptive alleles.
Methodologies for Detecting Introgression: Principles and Protocols
The development of methods to identify introgression has evolved with next-generation sequencing, yielding approaches that can be broadly categorized into summary statistics-based methods and probabilistic models [4]. This section details the experimental protocols and workflows for key methods featured in this comparison.
These methods use calculated statistics from genetic data to identify regions with unusual patterns of similarity indicative of introgression.
The RNDmin Protocol: This method is designed to test for introgression by leveraging the minimum pairwise sequence distance between two population samples relative to divergence from an outgroup [11].
- Data Requirements: Phased haplotypes from two sister species and an outgroup assumed to have no introgression.
- Core Calculation: The statistic is computed as RNDmin = dmin / dout, where dmin is the minimum sequence distance between any pair of haplotypes from the two sister species, and dout is the average distance from each sister species to the outgroup (dout = (dXO + d_YO)/2) [11].
- Interpretation: Exceptionally low values of RNDmin indicate regions with higher similarity between species than expected, suggesting recent introgression. The method is robust to variation in mutation rates and inaccurate species divergence times [11].
The Gmin Protocol: Developed to improve sensitivity to recent migration while accounting for mutation rate variation.
- Data Requirements: Phased haplotypes from two species.
- Core Calculation: The statistic is Gmin = dmin / dXY, where dXY is the average sequence distance between all sequences in the two species [11].
- Interpretation: Low Gmin values signal introgression. The normalization by dXY makes it robust to variable rates of evolution among loci [11].
The dmin Protocol: This approach focuses on identifying highly similar haplotypes between species.
- Data Requirements: Phased haplotypes from two taxa.
- Core Calculation: dmin = min{x∈X, y∈Y}{d_x,y}, the minimum number of sequence differences among all pairings of haplotypes from the two species [11].
- Interpretation: Highly similar haplotypes (low dmin) that coalesce more recently than the species divergence are considered evidence of introgression. The null distribution under no migration is typically generated via coalescent simulations [11].
The dXY and FST Protocols:
- dXY: Calculated as the average number of sequence differences between all sequences in two species. Low values suggest introgression but are confounded by low mutation rates [11].
- FST: A classic measure of population differentiation based on allele frequencies. Exceptionally low FST values relative to the genomic background can indicate gene flow [11].
Workflow and Logical Relationships in Introgression Detection
The following diagram illustrates the logical workflow and key decision points for selecting and applying different introgression detection methods.
Comparative Performance Analysis
The choice of method significantly impacts the ability to detect introgressed regions accurately and can lead to different biological conclusions. The following table summarizes the quantitative performance and characteristics of the key methods discussed.
Table 1: Comparative Performance of Introgression Detection Methods
| Method | Power Under Recent/Strong Migration | Robustness to Mutation Rate Variation | Sensitivity to Low-Frequency Migrants | Data Requirements | Key Strengths | Key Limitations |
|---|---|---|---|---|---|---|
| RNDmin | High [11] | High (explicitly accounts for it) [11] | Moderate [11] | Phased haplotypes, outgroup [11] | Robust to inaccurate divergence times; offers modest power increase over related tests [11] | Requires an outgroup; power is modest over other tests [11] |
| Gmin | High for recent migration [11] | High (normalized by dXY) [11] | Moderate [11] | Phased haplotypes [11] | Sensitive to recent migration; robust to variable evolutionary rates [11] | Less sensitive to low-frequency migrants compared to dmin [11] |
| dmin | High when assumptions are met [11] | Low (can be confounded by low mutation rates) [11] | High (designed for rare lineages) [11] | Phased haplotypes [11] | High power to detect even rare introgressed lineages [11] | Assumptions often violated; confounded by mutation rate variation [11] |
| dXY | Moderate [11] | Low (low values mimic introgression) [11] | Low (uses averages) [11] | Unphased or phased sequences [11] | Simple calculation; does not require phased data [11] | Not sensitive to low-frequency migrants; confounded by selection and mutation rate [11] |
| FST | Moderate [11] | N/A (based on allele frequencies) | Low [11] | Allele frequency data [11] | Can be calculated from SNPs; does not require full sequence or phased data [11] | Confounded by natural selection; not sensitive to low-frequency migrants [11] |
The application of RNDmin to population genomic data from the African mosquitoes Anopheles quadriannulatus and A. arabiensis exemplifies how method choice impacts conclusions. This approach identified three novel candidate regions for introgression, including one on the X chromosome outside a known inversion, suggesting that significant but rare allele sharing occurs between species that diverged over 1 million years ago [11]. A less powerful method might have missed these signals, leading to an underestimation of gene flow and an incomplete understanding of the evolutionary history between these species. Furthermore, methods like dmin and Gmin, which are sensitive to recent migration and rare lineages, are crucial for identifying nascent introgression events that may have adaptive potential [11]. Conversely, relying solely on FST or dXY could lead to false positives if regions with low neutral mutation rates are mistaken for introgressed loci [11].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful detection of introgression relies on a combination of biological materials, computational tools, and data resources. The following table details key components of the research toolkit for this field.
Table 2: Key Research Reagent Solutions for Introgression Studies
| Research Reagent / Material | Function and Role in Introgression Detection |
|---|---|
| Phased Haplotype Data | Essential for methods like RNDmin, Gmin, and dmin. Allows for the comparison of individual chromosomal segments to identify highly similar haplotypes between species [11]. |
| Outgroup Genome | A genomic sequence from a closely related species that diverged before the species pair of interest. Critical for methods like RNDmin and ABBA-BABA tests to polarize alleles and provide a scale for divergence [11]. |
| Whole-Genome Sequencing Data | Provides the comprehensive genomic coverage necessary to scan for introgressed regions across the entire genome, moving beyond candidate genes to unbiased discovery [11]. |
| Reference Genome Assembly | A high-quality, annotated genome for the studied species. Serves as a map for aligning sequencing reads, calling variants, and determining the genomic context of introgressed loci (e.g., coding vs. non-coding regions) [4]. |
| Coalescent Simulation Software | Used to generate null distributions of test statistics (e.g., for dmin) under a model of no migration, allowing researchers to determine the significance of observed values and avoid false positives [11]. |
| Population Genetic Data Analysis Tools | Software packages (e.g., for calculating D-statistics, FST, dXY) that implement the various summary statistics and probabilistic models for introgression detection [4]. |
Emerging Approaches and Future Directions
The field of introgression detection is rapidly evolving. Beyond summary statistics, two major categories of methods are expanding the toolkit for researchers:
- Probabilistic Modeling: This approach provides a powerful framework to explicitly incorporate evolutionary processes such as mutation, drift, and selection, and has yielded fine-scale insights across diverse species [4]. These models can be more complex but offer a principled way to compare different evolutionary scenarios.
- Supervised Learning: An emerging approach with great potential, particularly when the detection of introgressed loci is framed as a semantic segmentation task. Machine learning models can be trained on simulated or empirical data to identify complex patterns associated with introgression [4].
These advanced methods are being applied across various clades, revealing introgressed loci linked to critical traits like immunity, reproduction, and environmental adaptation, which are of particular interest in biomedical and evolutionary research [4].
This case study demonstrates that the choice of method profoundly impacts the detection of introgressed regions and the downstream evolutionary and biomedical conclusions. Summary statistics like RNDmin and Gmin offer a robust and powerful means to identify introgression between sister species, especially for recent gene flow, while methods like dmin are superior for detecting rare introgressed lineages. The limitations of each method—such as sensitivity to mutation rate variation or low power for low-frequency migrants—mean that an inappropriate choice can lead to both false negatives and false positives. As the field progresses, leveraging a combination of summary statistics, probabilistic models, and supervised learning, tailored to the specific research question and data available, will provide the most reliable insights into the genomic landscapes of introgression. This rigorous approach is essential for accurately understanding the role of hybridization in adaptation, disease vector competence, and genome evolution.
The detection of introgression—the incorporation of genetic material from one species into the gene pool of another through hybridization and backcrossing—has become a fundamental aspect of evolutionary genomics [41]. As research has revealed that hybridization is far more common than previously thought, affecting everything from rapidly diversifying clades to deeply divergent taxa, the development of robust detection methodologies has become increasingly important [41]. These methods must distinguish genuine introgression from other evolutionary phenomena that create similar genomic patterns, particularly incomplete lineage sorting (ILS), while accounting for complicating factors such as variation in evolutionary rates across lineages [41] [54]. The field has seen the emergence of three major methodological approaches: summary statistics, probabilistic models, and machine learning techniques, each with distinct strengths and limitations [4]. This comparative guide objectively evaluates the performance of these methodologies within the context of assessing statistical power for detecting introgression direction, providing researchers with evidence-based recommendations for method selection across diverse evolutionary scenarios.
Comparative Performance Analysis of Detection Methods
Table 1: Comparative performance of introgression detection methodologies
| Method Category | Specific Methods | Key Strengths | Key Limitations | Optimal Use Cases | Power for Direction Detection |
|---|---|---|---|---|---|
| Summary Statistics | D-statistic (ABBA-BABA) | Fast computation; Widely implemented; Effective for recent introgression [41] | High false positives with rate variation [41] [54]; Assumes no homoplasy [41] | Recently diverged taxa with constant evolutionary rates | Limited without additional tests |
| Q95 | Consistently high performance across scenarios [6] [21]; Simplicity and transparency [21] | May miss complex introgression patterns | Exploratory studies; Non-human systems [21] | Moderate to high with proper calibration | |
| Tree-Based Methods | Dsuite (Dtree) | More robust to homoplasies than site-based D [41] | Still vulnerable to rate variation effects [41] | Analyses with reliable local tree estimation | Moderate when phylogenetic signal is strong |
| Probabilistic Modeling | VolcanoFinder, MaLAdapt | Explicit modeling of evolutionary processes [4]; Fine-scale insights [4] | Computationally intensive; Model misspecification risk [6] | Well-defined demographic histories | High with correct model specification |
| Machine Learning | Genomatnn (CNN) | >95% accuracy on simulated data [12]; Handles phased/unphased data [12] | Requires extensive training data [4] [12]; Performance drops with scenario mismatch [6] | Large genomic datasets with known archetypes | High when trained on relevant scenarios |
Table 2: Performance across evolutionary scenarios based on benchmarking studies
| Method | Human-Like Scenarios | Old Divergence | Recent Gene Flow | Rate Variation | Adaptive Introgression |
|---|---|---|---|---|---|
| D-statistic | High [41] | High false positives [41] [54] | High [41] | Severe false positives [41] [54] | Limited |
| Q95 | Moderate to High [6] [21] | Maintains performance [21] | High [21] | Moderately robust [21] | High [6] [21] |
| Genomatnn | Very High [12] | Varies with training [6] | High [12] | Depends on training [6] | Very High (designed for AI) [12] |
| MaLAdapt | High with retraining [6] | Performance drops [6] | Moderate [6] | Moderate [6] | High in trained scenarios [6] |
| VolcanoFinder | Moderate [6] | Performance drops [6] | Moderate [6] | Moderate [6] | Moderate [6] |
Experimental Protocols and Benchmarking Approaches
Standardized Simulation Frameworks
Benchmarking studies employ sophisticated simulation protocols to evaluate method performance under controlled conditions. The most comprehensive approaches utilize the multispecies coalescent with introgression (MSci) model, which incorporates both incomplete lineage sorting and gene flow [54]. Simulations typically model a four-taxon system (((P1, P2), P3), O) where parameters can be systematically varied: divergence times (τ, measured in mutations per site), population sizes (θ), introgression proportion (γ), and substitution rate variation between lineages [54]. For assessing adaptive introgression methods, researchers implement forward-time simulations in SLiM (Simulation of Evolutionary Genetics) integrated with stdpopsim, adding selection coefficients to introgressed alleles [12]. These simulations generate genomic sequences under known evolutionary conditions, creating ground-truth datasets for quantifying classification accuracy, false positive rates, and statistical power.
Performance Metrics and Evaluation Criteria
Method evaluation employs standardized metrics calculated from confusion matrices: sensitivity (true positive rate), specificity (true negative rate), precision (positive predictive value), and overall accuracy ((TP+TN)/(TP+TN+FP+FN)) [6]. The area under the receiver operating characteristic curve (AUC-ROC) provides a comprehensive measure of classification performance across all threshold values [6]. Benchmarking studies particularly focus on the impact of evolutionary parameters on these metrics: divergence time (shallow to deep), population size, migration timing (recent vs. ancient), migration rate, selection strength, and rate variation between lineages [6] [54]. Performance is also assessed across different genomic contexts, including regions directly under selection, adjacent regions affected by hitchhiking, and unlinked neutral regions [6].
Figure 1: Workflow for benchmarking introgression detection methods
Key Methodological Challenges and Limitations
Evolutionary Rate Variation and False Positives
A critical limitation affecting multiple methods, particularly summary statistics like the D-statistic, is sensitivity to variation in evolutionary rates across lineages [41] [54]. When sister lineages evolve at different rates, homoplasies (independent substitutions at the same site) occur more frequently in the faster-evolving lineage, creating ABBA-BABA asymmetry that mimics introgression signals [41]. Theoretical analyses and simulations demonstrate that even moderate rate variation (17-33% difference) in shallow phylogenies can inflate false positive rates to 35-100% with 500 Mb genomes [54]. This problem intensifies with increasing phylogenetic depth and when using distant outgroups [54]. While tree-based methods like Dsuite were developed to address this limitation, they remain vulnerable to rate variation effects, particularly when branch length information is unreliable [41].
Scenario-Specific Performance and Generalizability
Benchmarking studies reveal that method performance is highly context-dependent, with limited generalizability across evolutionary scenarios [6] [21]. Methods developed and trained on human genomic data (particularly Neanderthal and Denisovan introgression) often perform poorly when applied to other systems without retraining [6] [21]. For instance, machine learning approaches like Genomatnn and MaLAdapt show excellent performance in human-like scenarios but experience significant accuracy reductions when applied to older divergence times or different demographic histories [6]. This "training set bias" presents particular challenges for non-model organisms where appropriate training data may be limited. The Q95 statistic demonstrates more consistent performance across diverse scenarios, likely due to its simplicity and reduced reliance on specific demographic assumptions [21].
Figure 2: Comparison of summary statistics vs. machine learning approaches
Table 3: Key computational tools and resources for introgression research
| Tool/Resource | Category | Primary Function | Application Context |
|---|---|---|---|
| Dsuite | Software Package | Implements D-statistic and Dtree analyses [41] | Detection of introgression using site patterns and tree topologies |
| stdpopsim | Simulation Resource | Standardized population genetic simulations [12] | Method development and benchmarking with realistic parameters |
| SLiM | Simulation Software | Forward-time simulations with selection [12] | Modeling adaptive introgression and complex evolutionary scenarios |
| Genomatnn | Machine Learning Tool | CNN-based adaptive introgression detection [12] | High-accuracy identification of AI regions in genomic data |
| Q95 | Summary Statistic | Measures introgressed haplotype frequency [6] | Exploratory analysis and method performance comparison |
| VolcanoFinder | Probabilistic Model | Detects adaptive introgression using SFS [6] | Inference of selection on introgressed alleles |
| MaLAdapt | Machine Learning Tool | Random forest classification of AI [6] | Scenario-specific adaptive introgression detection |
| HyDe | Software Package | Hypothesis testing using site patterns [54] | Detection of hybrid speciation and ghost introgression |
Evidence-Based Guidelines for Method Selection
Recommendations for Different Research Contexts
Based on comprehensive benchmarking studies, method selection should be guided by specific research questions and biological systems. For exploratory analyses in non-human systems, the Q95 statistic provides a robust starting point due to its consistent performance across diverse scenarios and transparency of interpretation [21]. In systems with well-characterized demographic histories and constant evolutionary rates, the D-statistic remains effective for detecting recent introgression, though its vulnerability to rate variation must be considered [41] [54]. For targeted investigation of adaptive introgression in systems with sufficient training data, machine learning approaches like Genomatnn offer superior accuracy (≥95% on simulated data), particularly for distinguishing genuine adaptive introgression from background selection or neutral introgression [12]. When analyzing systems with known or suspected rate variation among lineages, tree-based methods like Dsuite provide greater robustness, though they require reliable phylogenetic estimation [41].
Future Method Development Priorities
Current methodological gaps highlight several priorities for future development. First, there is a critical need for methods specifically designed to handle rate variation among lineages, as this represents a major source of false positives across multiple approaches [41] [54]. Second, developing machine learning frameworks that require less scenario-specific training would significantly enhance applicability to non-model organisms [6]. Third, improved methods for detecting introgression directionality in complex phylogenetic networks would advance understanding of historical introgression patterns [4]. Finally, approaches that jointly model introgression and selection while accommodating heterogeneous genomic landscapes (e.g., recombination rate variation) would provide more biologically realistic inference [4] [12]. The continued benchmarking of new methods against standardized datasets, following the protocols established in recent comprehensive evaluations, will be essential for tracking progress in these areas [6] [21].
The rapid expansion of genomic datasets across diverse taxa has created unprecedented opportunities to investigate the impact of introgression—the transfer of genetic material between species or populations—on evolution and adaptation. However, this growth has outpaced the development of standardized frameworks for validating introgression signals, creating a critical bottleneck in comparative evolutionary genomics. As noted in a recent assessment, the most frequently used metrics to detect introgression are "difficult to compare across studies and even more so across biological systems due to differences in study effort, reporting standards, and methodology" [53]. This lack of standardization persists despite the recognition that introgression can have myriad effects, from providing raw genetic material for adaptation [53] to potentially blurring species boundaries in various lineages [2].
The methodological landscape for detecting introgression has diversified considerably, spanning summary statistics, probabilistic modeling, and emerging machine learning approaches [4]. Each category offers distinct advantages and limitations, yet researchers lack consensus on appropriate application scenarios, performance benchmarks, or reporting standards. This fragmentation is particularly problematic for studies aiming to detect the direction of introgression, where methodological inconsistencies can directly impact biological interpretations. Recent research emphasizes that "differences in sequencing technologies may bias values of Patterson's D" and that "introgression may differ throughout the course of the speciation process" [53], further complicating cross-study comparisons.
This guide provides a systematic comparison of current methods for validating introgression signals, with particular emphasis on assessing their power to detect introgression directionality. By synthesizing experimental data, detailing methodological protocols, and identifying critical research reagents, we aim to advance toward field-wide best practices that will enhance the reliability, reproducibility, and interpretability of introgression research.
Methodological Landscape: Approaches for Detecting Introgression
The detection of introgressed genomic regions relies on identifying patterns of shared genetic variation that deviate from expectations under strict divergence without gene flow. Methodologies have evolved from single-statistic approaches to integrated frameworks that combine multiple lines of evidence [4]. These can be broadly categorized into three paradigms: summary statistics, probabilistic modeling, and supervised learning approaches, each with distinct strengths for specific evolutionary scenarios.
Summary statistics represent some of the earliest and most widely used approaches for introgression detection. Methods such as Patterson's D (the ABBA-BABA test) and related f-statistics identify introgression by measuring asymmetries in allele sharing patterns between populations or species [53]. These methods are computationally efficient and can be applied to genome-wide data, but they have inherent limitations, including sensitivity to population structure and an inability to identify specific introgressed loci in early implementations [53] [11]. The RNDmin statistic offers a modest increase in power for detecting recent introgression while remaining robust to variation in mutation rates [11]. These statistics are particularly useful for initial genome scans but may require complementary approaches for fine-scale validation.
Probabilistic modeling approaches provide a more powerful framework for explicit incorporation of evolutionary processes. Methods in this category use coalescent theory or hidden Markov models to infer the posterior probability of introgression along genomic regions while accounting for confounding factors like incomplete lineage sorting [4]. These models can incorporate information about population size changes, divergence times, and migration rates, offering nuanced insights across diverse species [4]. The trade-off for this increased statistical power is greater computational demand and more complex implementation requirements.
Supervised learning represents an emerging frontier in introgression detection, where models are trained on simulated genomic data to classify regions as introgressed or not introgressed [4]. When framed as a semantic segmentation task, these methods show particular promise for identifying precise boundaries of introgressed segments [4]. The performance of these approaches depends heavily on the biological realism of training simulations and appropriate feature selection, but they offer scalability to large genomic datasets once trained.
Table 1: Comparative Analysis of Major Introgression Detection Method Categories
| Method Category | Example Methods | Key Advantages | Key Limitations | Power to Detect Direction |
|---|---|---|---|---|
| Summary Statistics | Patterson's D, f-statistics, RNDmin, Gmin [53] [11] | Computationally efficient; simple implementation; good for initial screening | Difficult to compare across studies; sensitive to population structure; limited resolution [53] | Moderate (requires specific phylogenetic sampling) |
| Probabilistic Modeling | IBD-based methods, CoalHMM, Approximate Bayesian Computation [4] [60] | Accounts for evolutionary processes; provides confidence estimates; handles uncertainty | Computationally intensive; complex implementation; model misspecification risk [4] | High (explicitly models directionality) |
| Supervised Learning | Semantic segmentation networks, classifier-based approaches [4] | High scalability; pattern recognition capability; rapid application once trained | Dependent on training data quality; limited interpretability; data requirements [4] | Variable (depends on feature selection) |
Quantitative Comparison of Method Performance
Empirical assessments of introgression detection methods reveal significant variation in performance across different evolutionary scenarios. A comprehensive analysis examining patterns of introgression across eukaryotes collated Patterson's D values from 123 studies, highlighting that this statistic "is not a precise estimator of the fraction of the genome that has introgressed, it is at least proportional to this quantity" [53]. This meta-analysis found that introgression has been most frequently measured in plants and vertebrates, with less attention given to other eukaryotic groups, creating significant taxonomic biases in our understanding of introgression frequency [53].
Performance benchmarks indicate that summary statistics generally have high power to detect introgressed loci "when migration is recent and strong" [11]. The RNDmin statistic, which calculates the minimum pairwise sequence distance between two population samples relative to divergence to an outgroup, offers "a modest increase in power over other, related tests" while remaining "robust to variation in the mutation rate" [11]. This robustness to rate variation is particularly valuable for comparative analyses across genomic regions with different evolutionary constraints.
For bacterial systems, where introgression occurs through homologous recombination rather than meiotic processes, a recent large-scale analysis of 50 major bacterial lineages revealed that "bacteria present various levels of introgression, with an average of 2% of introgressed core genes and up to 14% in Escherichia–Shigella" [2]. This study utilized a phylogeny-based approach that detected introgression based on "phylogenetic incongruency between gene trees and the core genome tree" [2], demonstrating how methodological adaptation is necessary for different biological systems.
The impact of reference genome quality on introgression detection has been quantitatively demonstrated in studies comparing different human genome assemblies. Research leveraging the complete T2T-CHM13 reference genome identified "approximately 51 Mb of Neanderthal sequences unique to T2T-CHM13, predominantly in genomic regions where GRCh38 and T2T-CHM13 assemblies diverge" [60]. This represents a substantial improvement over previous references, with T2T-CHM13 significantly improving "read mapping quality in archaic samples" and showing "a significant reduction in the standard deviation of read depth" [60], a key metric for mapping quality in complex genomic regions.
Table 2: Quantitative Performance Metrics for Introgression Detection Methods
| Method | Statistical Power | False Positive Rate | Direction Detection Accuracy | Optimal Application Scenario |
|---|---|---|---|---|
| Patterson's D | Variable; depends on timing and strength of introgression [53] | High if population structure not accounted for [53] | Limited; requires specific sister species relationships [53] | Initial screening for asymmetric introgression |
| RNDmin | High for recent and strong migration [11] | Robust to mutation rate variation [11] | Moderate with appropriate outgroup [11] | Detection of recent introgression between sister taxa |
| IBD-based Methods | High for detecting segments >0.5 cM [60] | Low when reference panels are available [60] | High through haplotype matching [60] | Analysis of archaic introgression in modern populations |
| IntroMap | High for structural variants and large introgressions [14] | Low due to signal processing approach [14] | Limited without additional directional tests [14] | Plant breeding applications with reference genome |
Experimental Protocols for Key Methodologies
The ABBA-BABA test, also known as Patterson's D, detects introgression by measuring an excess of shared derived alleles between non-sister populations. The protocol begins with variant calling from whole-genome sequencing data, followed by phylogenetic inference to establish population relationships. The test statistic D = (ABBA - BABA) / (ABBA + BABA) is calculated, where ABBA represents sites where populations P1 and P3 share a derived allele, and BABA represents sites where P2 and P3 share a derived allele, with P1 and P2 being sister populations [53]. Significance is typically assessed using block jackknife resampling to generate confidence intervals, with |D| > 0 and Z-scores > 3 often considered significant evidence of introgression [53]. Critical considerations for this protocol include appropriate sampling design (avoiding sister species introgression which D cannot detect) and accounting for ancestral population structure that can generate false positives [53].
Protocol 2: Phylogeny-Based Introgression Detection in Bacteria
For detecting introgression in bacterial core genomes, a robust protocol involves first defining species boundaries using Average Nucleotide Identity (ANI) with cutoffs of 94-96% [2]. Researchers then construct a maximum-likelihood phylogenomic tree using concatenated core genome alignments. Introgression events are inferred based on "phylogenetic incongruency between gene trees and the core genome tree" [2]. A gene sequence is classified as introgressed when it forms a monophyletic clade inconsistent with the core genome phylogeny and is "statistically more similar to the sequence of a different ANI-species than at least one sequence of the genomes of its own species" [2]. Levels of introgression are expressed as the fraction of core genes satisfying these criteria, with validation through comparison with gene-flow based species definitions [2].
Protocol 3: The IntroMap Pipeline for Plant Breeding Applications
IntroMap provides a specialized protocol for detecting introgressed regions in plant breeding contexts without requiring variant calling [14]. The protocol begins with aligning NGS reads to a reference genome using standard aligners like Bowtie2. The pipeline then "determines a score for each nucleotide position in the reference genome by parsing the MD tags present in each alignment record of the BAM file" [14]. This information is converted to a binary match/mismatch vector for each read. A sparse matrix is constructed, and mean values for all columns are computed to generate "per-base calling scores for the overall alignment of that chromosome at each nucleotide position" [14]. A low-pass filter is applied via convolution with a window function to remove high-frequency noise, followed by locally weighted linear regression to fit a signal representing homology between the sequenced cultivar and reference. Regions where scores drop below a set threshold are identified as putative introgressions, with optimal parameters depending on sequencing depth and divergence [14].
Protocol 4: Reference-Free Archaic Introgression Detection with IBDmix
IBDmix enables detection of Neanderthal-introgressed sequences in modern humans without requiring an unadmixed reference population [60]. The protocol begins with high-coverage sequencing data (≥30×) from modern individuals, with careful attention to pre-phasing filtering strategies that can substantially influence ancestry estimates [60]. The method identifies segments identical-by-descent (IBD) between modern individuals and high-quality archaic genomes (e.g., Altai Neanderthal). Key steps include: (1) remapping modern and archaic sequencing reads to a consistent reference genome (T2T-CHM13 recommended); (2) joint variant calling; (3) phasing using Shapeit; and (4) applying the IBDmix algorithm to identify IBD segments [60]. Critical parameters include Minor Allele Count (MAC) cutoffs and Variant Quality Score Log Odds (VQSLOD) thresholds, with stringent thresholds potentially introducing systematic biases by excluding genuine variants [60].
Visualization of Method Workflows
IntroMap Bioinformatics Pipeline
Table 3: Essential Research Reagents and Computational Tools for Introgression Studies
| Resource Category | Specific Tools/Reagents | Function/Purpose | Application Context |
|---|---|---|---|
| Reference Genomes | T2T-CHM13, GRCh38, lineage-specific assemblies [60] | Provides mapping foundation; improved references enhance detection sensitivity | All introgression studies; T2T-CHM13 shows superior mapping for archaic DNA [60] |
| Variant Callers | GATK, BCFtools, specialized ancient DNA pipelines [60] | Identifies genetic variants from sequencing data; quality filtering critical for downstream analysis | Initial data processing; variant quality filters significantly impact introgression detection [60] |
| Introgression Detection Software | PLINK, ADMIXTOOLS, IntroMap, IBDmix [14] [60] | Implements specific detection algorithms; varies in statistical approach and assumptions | Method-specific applications; IntroMap for plant breeding without variant calling [14] |
| Visualization Platforms | ASH (Arc-Seq Hub), custom genome browsers [60] | Enables exploration of introgressed segments and their functional implications | Data interpretation; ASH provides interactive resource for archaic sequences [60] |
| Simulation Tools | ms, SLiM, stdpopsim [4] | Generates synthetic genomic data under evolutionary scenarios for method validation | Power analysis; benchmarking false positive rates; training machine learning models [4] |
Our systematic comparison reveals significant progress in methodological development for introgression detection, yet also highlights persistent challenges in standardization and validation. The field would benefit from community-established benchmarks, standardized reporting metrics, and explicit validation frameworks that account for taxonomic diversity and varying evolutionary scenarios. Future methodological development should prioritize approaches that not only detect introgression but also accurately determine its direction, timing, and functional consequences across diverse biological systems.
Emerging opportunities include the integration of multiple methodological approaches in consensus frameworks, leveraging the complementary strengths of different methods. Additionally, the development of taxon-specific best practices—recognizing the distinct mechanisms of introgression in sexual eukaryotes, bacteria, and plants—will enhance biological insights. As genomic datasets continue to expand in size and taxonomic coverage, standardized practices for validating introgression signals will be essential for advancing our understanding of this fundamental evolutionary process.
Conclusion
The power to detect introgression direction is not inherent to a single method but emerges from a careful, multi-faceted approach. This analysis demonstrates that while a core set of introgressed regions can be reliably identified by nearly all algorithms, substantial heterogeneity exists between maps produced by different methods, which can directly impact downstream biological interpretations. Researchers must therefore move beyond reliance on a single algorithm and instead adopt a consensus strategy that utilizes multiple, complementary detection maps to ensure robustness. Future directions should focus on developing integrated prediction sets, creating standardized benchmarks with simulated data, and exploring the implications of introgressive haplotypes for complex trait mapping and drug target identification in clinical genomics. Embracing this rigorous, multi-method framework is essential for unlocking the full potential of introgression studies in evolutionary and biomedical research.
References
- 1. Adaptive Introgression as an Evolutionary Force: A Meta ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12181397/]
- 2. Introgression impacts the evolution of bacteria, but species ... [https://www.nature.com/articles/s41467-025-64947-1]
- 3. Gene flow and introgression are pervasive forces shaping ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9650840/]
- 4. Review Decoding genomic landscapes of introgression [https://www.sciencedirect.com/science/article/pii/S0168952525001660]
- 5. Model-based detection and analysis of introgressed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6165692/]
- 6. Performance evaluation of adaptive introgression ... [https://peercommunityjournal.org/articles/10.24072/pcjournal.617/]
- 7. The contribution of Neanderthal introgression to modern ... [https://www.sciencedirect.com/science/article/pii/S0960982222013045]
- 8. Analysis of bacterial genomes from an evolution experiment ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1007199]
- 9. Low statistical power in biomedical science: a review of three ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5367316/]
- 10. Phylogenomic approaches to detecting and characterizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9208645/]
- 11. Powerful methods for detecting introgressed regions from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4899106/]
- 12. Detecting adaptive introgression in human evolution using ... [https://elifesciences.org/articles/64669]
- 13. Inference of Gene Flow between Species from Genomic Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12203007/]
- 14. IntroMap: a signal analysis based method for the detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5716257/]
- 15. A statistical model for reference-free inference of archaic local ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6555542/]
- 16. Detecting adaptive introgression in human evolution using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8192126/]
- 17. Comparative Performance of Popular Methods for Hybrid ... [https://pubmed.ncbi.nlm.nih.gov/33404632/]
- 18. Assessing simulation-based supervised machine learning ... [https://www.nature.com/articles/s41437-025-00773-x]
- 19. Inferring the Direction of Introgression Using Genomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10439365/]
- 20. Gene flow analysis method, the D-statistic, is robust in a wide ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-2002-4]
- 21. Benchmarking the detection of adaptive introgression from ... [https://evolbiol.peercommunityin.org/articles/rec?articleId=835]
- 22. De novo annotation reveals transcriptomic complexity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12501010/]
- 23. Mapping gene flow between ancient hominins through ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1008895]
- 24. Identifying and Interpreting Apparent Neanderthal Ancestry ... [https://www.sciencedirect.com/science/article/pii/S0092867420300593]
- 25. New Genomic Methods Offer Insight into the Legacy of Archaic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8480178/]
- 26. Spatial and temporal analyses identify two introgression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12137868/]
- 27. A practical comparison of the next-generation sequencing ... [https://www.life-science-alliance.org/content/6/4/e202201744]
- 28. Performance evaluation of pipelines for mapping, variant ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04144-1]
- 29. Comparing Neanderthal introgression maps reveals core ... [https://pubmed.ncbi.nlm.nih.gov/?linkname=pubmed_pubmed_citedin&from_uid=41040238]
- 30. Supervised machine learning reveals introgressed loci in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5933812/]
- 31. Refining models of archaic admixture in Eurasia with ... [https://www.nature.com/articles/s41467-021-26503-5]
- 32. Download flat bibtex file of all 1625 entries [http://www.esp.org/recommended/literature/paleo-neander/inc.files/bt-flat.txt]
- 33. Detecting archaic introgression and modeling multiple- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9038769/]
- 34. Whole-Genome Alignment: Methods, Challenges, and ... [https://www.mdpi.com/2076-3417/14/11/4837]
- 35. IQ-TREE: A Fast and Effective Stochastic Algorithm for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4271533/]
- 36. smirarab/ASTRAL: Accurate Species TRee ALgorithm [https://github.com/smirarab/ASTRAL]
- 37. ASTRAL-III: polynomial time species tree reconstruction from ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2129-y]
- 38. Using all Gene Families Vastly Expands Data Available for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9178227/]
- 39. Estimates of introgression as a function of pairwise distances [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2747-z]
- 40. Comparing Neanderthal introgression maps reveals core ... [https://pubmed.ncbi.nlm.nih.gov/41040238/]
- 41. Towards Reliable Detection of Introgression in the Presence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11639170/]
- 42. A genomic assessment of directional biases in gene flow ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5468134/]
- 43. Genomic introgression underlies environmental adaptation ... [https://www.sciencedirect.com/science/article/pii/S2468265925000563]
- 44. A New Method to Scan Genomes for Introgression in a ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0118621]
- 45. Extensive Homoplasy , Nonstepwise Mutations, and Shared Ancestral... [https://www.academia.edu/75005650/Extensive_Homoplasy_Nonstepwise_Mutations_and_Shared_Ancestral_Polymorphism_at_a_Complex_Microsatellite_Locus_in_Lake_Malawi_Cichlids]
- 46. Does Asymmetric Reproductive Isolation Predict the ... [https://www.mdpi.com/2073-4425/16/2/124]
- 47. Hyperparameter Optimization and Evaluation Metrics for ... [https://link.springer.com/article/10.1007/s10922-025-09973-6]
- 48. Comparison of methods for tuning machine learning model ... [https://bmcmedresmethodol.biomedcentral.com/articles/10.1186/s12874-025-02561-x]
- 49. Hybrid Machine Learning Model for the Prediction of ... [https://www.sciencedirect.com/science/article/pii/S2666827025001240]
- 50. What Is Data Completeness? Definition, Examples, And KPIs [https://www.montecarlodata.com/blog-what-is-data-completeness/]
- 51. The 6 Data Quality Dimensions with Examples [https://www.collibra.com/blog/the-6-dimensions-of-data-quality]
- 52. The effects of introgression across thousands ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8601620/]
- 53. A need for standardized reporting of introgression [https://pmc.ncbi.nlm.nih.gov/articles/PMC9554761/]
- 54. Detecting Introgression in Shallow Phylogenies [https://pmc.ncbi.nlm.nih.gov/articles/PMC12485364/]
- 55. Genomic data reveal a north-south split and introgression ... [https://www.nature.com/articles/s41467-025-58543-6]
- 56. Modelling the Consequences of Domestication‐Introgression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12446728/]
- 57. Genetic introgression between different groups reveals the ... [https://www.nature.com/articles/s41598-022-22674-3]
- 58. IntroUNET: Identifying introgressed alleles via semantic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10906877/]
- 59. VEGA: visual of phylogenetic trees for comparison ... evolutionary [https://link.springer.com/article/10.1007/s12650-020-00635-0]
- 60. A refined analysis of Neanderthal-introgressed sequences in ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03502-z]