Beyond the D-Statistic: A Comparative Guide to Phylogenetic Networks for Detecting Reticulate Evolution
This article provides a comprehensive comparison for researchers and bioinformaticians between the widely used D-statistic (ABBA-BABA test) and modern phylogenetic network methods for detecting reticulate evolution.
Beyond the D-Statistic: A Comparative Guide to Phylogenetic Networks for Detecting Reticulate Evolution
Abstract
This article provides a comprehensive comparison for researchers and bioinformaticians between the widely used D-statistic (ABBA-BABA test) and modern phylogenetic network methods for detecting reticulate evolution. We explore the foundational principles of both approaches, detailing their methodological applications, strengths, and limitations. A troubleshooting guide addresses common challenges like computational scalability and interpreting complex results. Through a direct performance comparison and validation framework, we demonstrate that while the D-statistic offers a fast initial screening tool, phylogenetic networks provide a more robust and detailed inference of evolutionary history, especially in complex scenarios involving multiple reticulations or ghost lineages. This synthesis empowers more accurate detection of hybridization and introgression in genomic studies, with significant implications for understanding evolutionary trajectories in pathogen and drug target research.
Core Concepts: From Tree Discordance to Explicit Reticulate Models
Phylogenetics, the study of evolutionary relationships, has long relied on trees as its primary representational framework. However, the increasing recognition of reticulate evolutionary processes—such as hybridization, horizontal gene transfer, and introgression—has exposed the limitations of strictly tree-like models. This recognition has driven the development and adoption of phylogenetic networks, which can model these complex histories [1]. Phylogenetic networks are broadly categorized into two distinct paradigms: implicit networks and explicit networks [2] [1] [3]. Understanding the fundamental differences between these classes, their appropriate applications, and their performance characteristics is crucial for researchers aiming to accurately reconstruct evolutionary histories in the presence of gene flow.
This guide provides a objective comparison between implicit and explicit phylogenetic networks, situating them within the broader methodological landscape that includes popular heuristic approaches like the D-statistic. We synthesize current research to compare these frameworks based on their underlying assumptions, computational requirements, statistical foundations, and biological interpretability, supported by experimental data and detailed protocols.
Fundamental Definitions and Characteristics
Implicit Phylogenetic Networks
Implicit networks (also known as split networks or abstract networks) are primarily descriptive tools designed to visualize conflicting signals in phylogenetic data without attributing them to specific biological processes [1] [3]. They are typically unrooted graphs that summarize discordance based on genetic distances or conflicting tree topologies, regardless of the underlying biological cause [1].
Key Characteristics:
- Biological Interpretation: Internal nodes do not represent ancestral species or specific evolutionary events [3].
- Process Agnosticism: They visualize conflict but do not distinguish between different sources of discordance (e.g., ILS vs. hybridization) [1].
- Primary Use Case: Data exploration, visualization of conflicting signals, and analysis of datasets where the biological causes of discordance are unknown [1].
Explicit Phylogenetic Networks
In contrast, explicit networks are generative models of evolution that represent specific historical reticulate events [1] [3]. They are rooted, directed acyclic graphs whose internal nodes represent ancestral species, with reticulation nodes explicitly modeling events like hybridization or horizontal gene transfer [2] [1].
Key Characteristics:
- Biological Interpretation: Internal nodes represent explicit evolutionary events (speciation or reticulation) [3].
- Process Specificity: Reticulation vertices have two incoming edges, representing the fusion of genetic material from two ancestral populations [1].
- Model Parameters: Include inheritance probabilities (γ) that denote the proportion of genetic material contributed by each parent in a hybridization event [1] [3].
- Primary Use Case: Hypothesis testing about specific reticulate evolutionary histories [1].
Table 1: Core Conceptual Differences Between Implicit and Explicit Networks
| Feature | Implicit Networks | Explicit Networks |
|---|---|---|
| Rooting | Unrooted | Rooted, directed |
| Internal Nodes | No biological meaning | Represent ancestral species |
| Reticulations | Summarize conflict | Represent specific historical events |
| Inheritance Probabilities | Not applicable | Estimated (γ parameters) |
| Evolutionary Model | None (phenetic) | Multispecies Network Coalescent (MNSC) |
| Primary Strength | Fast data exploration & visualization | Biologically intuitive hypothesis testing |
The following diagram illustrates the fundamental structural and conceptual differences between these network types:
Figure 1: Conceptual classification of phylogenetic networks, highlighting core differences between implicit and explicit paradigms.
Methodological Comparison and Experimental Data
Performance and Scalability
A critical consideration for researchers is the computational performance and scalability of different inference methods. Experimental studies have quantified these metrics across various approaches.
Table 2: Experimental Performance Comparison of Phylogenetic Inference Methods
| Method (Category) | Representative Tools | Max Practical Taxa (Experimental) | Runtime for 25 Taxa | Key Limitation |
|---|---|---|---|---|
| Full-Likelihood Explicit | PhyloNet [4] | < 10 taxa [5] | > Weeks (CPU) [4] | Intractable likelihood calculations [4] [5] |
| Pseudolikelihood Explicit | SNaQ [3], MPL [4] | ~25 taxa [4] | Prohibitive beyond ~25 taxa [4] | Heuristic search, pseudo-likelihood approximation [4] [3] |
| Divide-and-Conquer Explicit | PhyloNet [5] | Infeasible otherwise [5] | Significantly reduced [5] | Relies on accurate subset inference [5] |
| Implicit Networks | SplitsTree [4] [6], Neighbor-Net [4] | 100s of taxa [1] | Fast (minutes/hours) [4] | No explicit biological interpretation [1] [3] |
| Hybrid Detection (D-statistic) | Various [1] | Subsets of 4 taxa [1] | Very Fast | Sensitive to assumptions; poor with multiple reticulations [1] |
Biological Interpretation and Identifiability
The biological interpretability of results and the theoretical identifiability of parameters are fundamental distinctions.
Explicit Networks provide a direct link between the graphical model and evolutionary processes. Reticulation nodes model hybridization, with inheritance probabilities (γ) quantifying the genomic contribution from each parent [1] [3]. A value of γ ≈ 0.5 suggests symmetrical hybridization, as in a diploid F1 hybrid, while values skewed toward 0 or 1 indicate asymmetrical introgression [1]. However, distinguishing between hybrid speciation and repeated backcrossing based on γ alone remains challenging [1].
Implicit Networks, being process-agnostic, do not offer this level of biological specificity. They are useful for initial data exploration but cannot delineate the exact nature of reticulate events [1] [3].
Under the Multispecies Network Coalescent (MNSC) model, which accounts for both ILS and hybridization, explicit network parameters have been proven to be theoretically identifiable given sufficient data [1] [3]. This means that, in theory, the true network can be distinguished from other networks based on the gene tree distribution it generates. Implicit networks lack such a formal identifiability guarantee.
Experimental Protocols for Method Evaluation
To ensure reproducible comparisons between methods, researchers must adhere to standardized experimental protocols. Below, we detail common workflows for evaluating phylogenetic network inference.
Protocol for Scalability Assessment
This protocol is derived from performance studies that quantify how methods handle increasing data size [4].
Dataset Simulation:
- Use a model phylogeny with a fixed number of reticulations (e.g., a single reticulation) to control for model complexity [4].
- Systematically vary the number of taxa (e.g., from 10 to 50) and the evolutionary divergence (sequence mutation rate) [4].
- Simulate multi-locus sequence alignments under the coalescent model with gene flow.
Method Execution:
Performance Metrics:
- Topological Accuracy: Compare the inferred network to the true simulated network using a distance metric (e.g., Robinson-Foulds distance for networks).
- Computational Requirements: Record CPU runtime and peak memory usage for each analysis.
- Success Rate: Note the proportion of analyses that complete within a reasonable time frame (e.g., 4 weeks).
The following diagram visualizes this experimental workflow:
Figure 2: Workflow for experimental assessment of phylogenetic network inference method scalability and performance.
Protocol for Inference from Multi-locus Data using SNaQ
SNaQ (Species Networks applying Quartets) is a representative pseudolikelihood method for explicit network inference. Its protocol highlights the steps common to many contemporary approaches [3].
Input Data Preparation:
- Obtain sequence alignments for multiple unlinked loci.
- For each locus, estimate a gene tree (topology and branch lengths). This step can be parallelized.
Summary of Gene Tree Discordance:
- For all possible combinations of four taxa (quartets), calculate the Concordance Factors (CFs). A CF is the proportion of genes whose true tree displays a specific quartet topology [3].
- The output is a set of observed CFs for the three possible quartets on every 4-taxon set.
Pseudolikelihood Optimization:
- The pseudolikelihood of a candidate network is computed based on the fit between its expected CFs (under the coalescent model with hybridization) and the observed CFs [3].
- A heuristic search is conducted over the space of phylogenetic networks (often level-1 networks) to find the topology, branch lengths (
t), and inheritance probabilities (γ) that maximize the pseudolikelihood.
This method bypasses the computationally intensive calculation of the full likelihood, enabling analysis of larger datasets than full-likelihood methods [3].
Selecting the appropriate software is essential for implementing the methodologies discussed. The table below catalogs major tools and their primary functions.
Table 3: Essential Software and Resources for Phylogenetic Network Analysis
| Tool / Resource | Category | Primary Function | Interpretation |
|---|---|---|---|
| PhyloNet [5] | Explicit Network | Maximum likelihood and parsimony inference from gene trees. | Infers explicit networks under the MNSC; includes full-likelihood and divide-and-conquer methods. |
| PhyloNetworks [3] | Explicit Network | Pseudolikelihood inference (SNaQ) from concordance factors or gene trees. | Infers explicit level-1 networks using a scalable quartet-based approach. |
| SplitsTree [4] [6] | Implicit Network | Computes split networks from distance matrices or tree collections. | Infers implicit networks for exploratory data analysis and conflict visualization. |
| D-statistic (ABBA-BABA) [1] | Hybrid Detection | Tests for gene flow among a set of four taxa. | A statistical test for introgression; does not infer a full network but can signal its necessity. |
| Sequence Aligner (e.g., MAFFT) | Data Preprocessing | Aligns raw nucleotide or amino acid sequences. | Creates the multiple sequence alignments used as input for gene tree estimation. |
| Gene Tree Estimator (e.g., RAxML) | Data Preprocessing | Estimates phylogenetic trees from sequence alignments. | Infers the gene trees that serve as input for many explicit network methods. |
Integrated Discussion: Placing D-Statistic and Network Methods in Context
The D-statistic and phylogenetic networks represent different points on a spectrum of methodological complexity and biological inference. The D-statistic is a targeted test for detecting gene flow between four taxa, serving as a useful and fast hypothesis-generation tool [1]. However, it operates on a limited taxonomic scale and can produce misleading results in the presence of multiple reticulations or ghost lineages [1].
Explicit phylogenetic networks represent a more comprehensive inference framework. They aim to reconstruct the complete evolutionary history of all sampled taxa, simultaneously accounting for ILS and multiple reticulation events [1] [3]. The trade-off for this completeness is significantly higher computational cost and more complex model selection [4] [5]. Implicit networks occupy a middle ground, providing a rapid overview of data conflict that can help decide whether to pursue more rigorous explicit modeling [1].
A critical finding from recent research is that these methods can yield contradictory results. For example, a study of Xiphophorus fishes found that an explicit network inferred via SNaQ detected fewer reticulation events than a tree with added gene flow events suggested by D-statistic analyses [1]. This underscores the importance of method choice and suggests that explicit networks might provide a more conservative and coherent picture of evolutionary history by integrating signals across the entire phylogeny.
Implicit and explicit phylogenetic networks are complementary tools with distinct strengths and applications. Implicit networks are superior for rapid data exploration and visualization of conflicting phylogenetic signals. In contrast, explicit networks are indispensable for formulating and testing specific biological hypotheses about reticulate evolution, as they provide a statistically rigorous, model-based framework for inference, albeit at a higher computational cost.
The D-statistic remains a valuable initial test for gene flow, but its limitations in complex scenarios necessitate the use of more robust network inference methods for whole-genome data. Current research is focused on improving the scalability and statistical power of explicit network methods through techniques like divide-and-conquer and pseudolikelihood approximations [5] [3]. As these methods continue to mature, they are poised to become the standard for reconstructing the richly interconnected Tree of Life.
The detection of gene flow is crucial for constructing accurate evolutionary histories across diverse fields, from evolutionary biology to drug development. The D-statistic (ABBA-BABA test) is a widely used formal test for detecting gene flow, but it represents just one approach in a broader methodological landscape. This guide provides an objective comparison between the D-statistic and more complex phylogenetic network methods, evaluating their performance, scalability, and applicability based on current research. We summarize experimental data on computational demands, accuracy under simulation, and practical scope, providing researchers with a clear framework for selecting appropriate methods based on their specific study systems and data constraints.
The evolutionary history of species and populations is often not a simple branching tree. Processes like gene flow (hybridization, introgression) and incomplete lineage sorting (ILS) create conflicting signals in genomic data, necessitating methods that can explicitly model these reticulate events. The D-statistic is a powerful, widely-used population genetic test designed to detect signals of gene flow between closely related species or populations by measuring allele frequency patterns against a null hypothesis of a strictly bifurcating tree [4]. Its simplicity and computational efficiency have made it a staple in evolutionary studies.
In contrast, phylogenetic network inference methods aim to reconstruct explicit evolutionary graphs that represent the full history of speciation and gene flow events. These methods model the interaction of multiple evolutionary processes, such as sequence mutation, gene flow, and ILS, to infer a more complete phylogenetic hypothesis [4]. The choice between using a simple test like the D-statistic or investing in a full network inference is a critical decision that balances statistical power, computational cost, and biological interpretability. This guide objectively compares these approaches using published experimental data and simulation studies to inform researchers and drug development professionals.
Methodological Comparison: D-Statistic vs. Phylogenetic Networks
The D-statistic and phylogenetic network methods differ fundamentally in their goals, inputs, and underlying assumptions. The table below summarizes their core characteristics.
Table 1: Core Characteristics of the D-Statistic and Phylogenetic Network Methods
| Feature | D-Statistic | Phylogenetic Network Methods |
|---|---|---|
| Primary Goal | To test for the presence of a signal of gene flow. | To reconstruct an explicit evolutionary history that includes gene flow events. |
| Phylogenetic Scope | Typically operates on four taxa (a rooted triplet with an outgroup). | Can handle multiple taxa (dozens or more) to build a comprehensive network. |
| Output | A single statistic (D) and a p-value indicating deviation from a tree-like history. | A directed acyclic graph (network) showing species relationships and reticulations. |
| Key Assumption | Identifies gene flow that is inconsistent with a strictly bifurcating model; cannot easily distinguish gene flow from other processes like ancestral population structure. | Explicitly models gene flow and ILS; methods differ in their specific model assumptions (e.g., coalescent-based). |
| Data Input | Genome-wide counts of site patterns (ABBA, BABA). | Can use gene tree topologies, sequence alignments, or single-nucleotide polymorphisms. |
| Interpretation | Signal of gene flow between two specific lineages after their divergence from a third. | Visual representation of the evolutionary relationships, including the placement and number of hybridization events. |
Performance and Scalability: Experimental Data
The performance of these methods is critically evaluated based on their accuracy in recovering known evolutionary histories and their computational scalability. Simulation studies provide the primary evidence for these comparisons.
Accuracy and Computational Demand
A key scalability study tested state-of-the-art phylogenetic network methods on datasets of increasing size and complexity, including simulations with a single reticulation (gene flow event) [4]. The findings highlight a significant trade-off.
Table 2: Performance of Phylogenetic Network Methods on Empirical and Simulated Data [4]
| Method Type | Method Name (Example) | Accuracy (Topological) | Computational Limitations |
|---|---|---|---|
| Probabilistic (ML) | MLE, MLE-length | Most accurate | Prohibitive for >25 taxa (weeks of runtime, high memory) |
| Pseudo-Likelihood | MPL, SNaQ | High accuracy | More scalable than MLE, but still challenging for large datasets |
| Parsimony-Based | MP (Minimize Deep Coalescence) | Lower accuracy | More scalable than probabilistic methods |
| Distance-Based | Neighbor-Net, SplitsNet | Lower accuracy | Computationally fastest, produces a single network |
The study concluded that probabilistic methods, which maximize likelihood under coalescent-based models, are the most accurate [4]. However, this accuracy comes at a high computational cost. None of the probabilistic methods could complete analyses on datasets with 30 or more taxa within a practical timeframe, indicating that the field lags behind the needs of modern phylogenomic studies with dozens of genomes [4].
Impact of Data Scale on Performance
The same study found that the performance of all network inference methods is negatively impacted by two key dimensions of scale: the number of taxa and the evolutionary divergence (sequence mutation rate) [4]. As either factor increases, the topological accuracy of the inferred network degrades. This contrasts with the D-statistic, whose performance is less directly tied to the number of overall taxa but is constrained by its specific four-taxon requirement.
Experimental Protocols and Workflows
Understanding the typical workflows for these methods is essential for their application and for interpreting results from the literature.
Workflow for Phylogenetic Network Inference
The general process for inferring a phylogenetic network from molecular data involves multiple stages, from data collection to final tree evaluation [7].
A fundamental distinction in network inference lies in the choice of algorithm, which can be broadly categorized as either distance-based or character-based [7]. Character-based methods can be further divided into parsimony, maximum likelihood, and Bayesian approaches.
Table 3: Common Phylogenetic Tree/Network Construction Methods [7]
| Algorithm | Principle | Criteria for Final Tree | Scope of Application |
|---|---|---|---|
| Neighbor-Joining (NJ) | Minimal evolution; minimizes total branch length. | Produces a single tree. | Short sequences with small evolutionary distance. |
| Maximum Parsimony (MP) | Minimizes the number of evolutionary steps. | Tree with the smallest number of substitutions. | Sequences with high similarity. |
| Maximum Likelihood (ML) | Finds the tree that makes the data most probable under a model. | Tree with the maximum likelihood value. | Distantly related sequences. |
| Bayesian Inference (BI) | Uses Bayes' theorem to compute the probability of a tree. | The most sampled tree in MCMC analysis. | A small number of sequences. |
Protocol for the D-Statistic
The D-statistic workflow is more focused, as its goal is hypothesis testing rather than full phylogeny reconstruction. It does not require the iterative search for an optimal graph topology.
The core of the D-statistic protocol involves counting specific site patterns across the genome. For a four-taxon test (((P1, P2), P3), Outgroup), an "ABBA" site is one where P1 and the Outgroup share the ancestral allele (A), while P2 and P3 share the derived allele (B). A "BABA" site is the converse. The D-statistic is calculated as:
D = (Sum(ABBA) - Sum(BABA)) / (Sum(ABBA) + Sum(BABA))
A significant deviation from zero (assessed via a block jackknife or other resampling method) indicates an imbalance of site patterns inconsistent with a simple bifurcating tree, which is interpreted as evidence of gene flow between P2 and P3 [4].
The Scientist's Toolkit: Key Research Reagents and Solutions
Success in phylogenetic inference and detecting gene flow relies on a suite of software tools and data resources.
Table 4: Essential Research Reagents for Gene Flow Analysis
| Tool / Resource | Function | Application Context |
|---|---|---|
| PhyloNet | Software package for inferring and analyzing phylogenetic networks. | Implements methods like MLE, MLE-length, and MP for multi-locus data [4]. |
| SNaQ | Software for inferring species networks from quartets under coalescent models. | A pseudo-likelihood method that offers a balance of accuracy and scalability [4]. |
| ADMIXTOOLS | A software package suite for population genetics. | Contains tools for calculating D-statistics and other formal tests for admixture [4]. |
| High-Performance Computing (HPC) Cluster | Parallel computing environment. | Essential for running probabilistic network methods (ML, BI) on datasets of non-trivial size [4]. |
| Multi-Locus Sequence Data | Aligned DNA sequences from multiple independent loci. | The fundamental input for most phylogenetic network methods that account for ILS [7] [4]. |
| Reference Genomes | High-quality, assembled genomes. | Used as a baseline for mapping and calling variants for D-statistic analyses. |
The choice between the D-statistic and phylogenetic network methods is not a matter of which is universally better, but which is the right tool for the specific research question and data at hand.
Use the D-statistic when: Your goal is to test a specific hypothesis of gene flow between two lineages. It is ideal for initial screening, when working with very large genomic datasets (e.g., whole genomes), or when computational resources are limited. Its primary limitation is its inability to reconstruct the full network and its potential confusion of gene flow with other processes.
Use phylogenetic network methods when: Your goal is to reconstruct the complete evolutionary history of a group, including the number, placement, and direction of gene flow events. They are necessary when studying complex radiations with potential gene flow among multiple lineages. However, researchers must be aware of their severe computational constraints, which currently make them infeasible for large-scale phylogenomic studies with many taxa [4].
In conclusion, the D-statistic remains a powerful and efficient test for detecting gene flow, but it provides a limited, one-dimensional view. Phylogenetic network methods offer a powerful framework for reconstructing complex evolutionary histories but are currently constrained by scalability. The ongoing development of new algorithms, particularly those leveraging pseudo-likelihood and high-performance computing, is critical to bridging this methodological gap and fully realizing the potential of phylogenomic data.
Traditional phylogenetic analyses that assume strictly bifurcating trees often fail to capture the complexity of evolutionary histories involving processes such as hybridization, horizontal gene transfer, and introgression. In the presence of such gene flow, a phylogeny cannot be accurately described by a tree but instead requires the more general framework of a phylogenetic network—a directed acyclic graph that explicitly models reticulate evolutionary events [4] [8]. Phylogenetic networks are categorized as either explicit or implicit. Explicit networks directly represent specific evolutionary processes (e.g., gene flow through hybridization) at their reticulation nodes, whereas implicit networks merely summarize conflicting phylogenetic signals without specific biological interpretation [4]. This guide focuses on explicit phylogenetic networks, which provide a biologically meaningful framework for modeling reticulate history.
The advancement of high-throughput sequencing technologies has produced phylogenomic datasets that increasingly reveal the prevalence of gene flow across diverse taxa, including humans, ancient hominins, mice, and butterflies [4]. These developments have created two primary scalability challenges for phylogenetic inference: the number of taxa in a study and the evolutionary divergence among them [4]. While the impact of these scaling dimensions on phylogenetic tree inference has been well characterized, the scalability limits of phylogenetic network inference methods remain poorly understood until recently [4]. This guide provides a comprehensive comparison of approaches for detecting and quantifying gene flow, focusing specifically on the performance characteristics of the parsimony-based D-statistic (ABBA-BABA test) versus various phylogenetic network inference methods, with supporting experimental data from empirical studies.
Methodological Frameworks: D-Statistic vs. Phylogenetic Network Inference
The D-Statistic: A Parsimony-Based Test for Gene Flow
The D-statistic is a widely used parsimony-like method designed to detect gene flow between closely related species despite the presence of incomplete lineage sorting (ILS) [8]. This method operates on a four-taxon system (P1, P2, P3, O) with an established phylogeny of the form ((P1,P2),P3,O), where O is an outgroup. It compares the number of ABBA and BABA sites—parsimony-informative sites that support discordant phylogenies—to detect statistical evidence of gene flow [8].
- Mathematical Foundation: The D-statistic is calculated as D = (NABBA - NBABA) / (NABBA + NBABA), where NABBA and NBABA represent counts of these site patterns. Under pure ILS without gene flow, ABBA and BABA sites are equally likely, and D is expected to be zero. A significant deviation from zero indicates asymmetric gene flow, typically between P2 and P3 [8].
- Parameter Sensitivity: The effectiveness of the D-statistic is primarily determined by the relative population size (population size scaled by the number of generations since divergence). It is robust across a wide range of genetic distances but becomes less sensitive when population sizes are large relative to branch lengths in generations. The statistic is also affected by the direction, timing, and fraction of gene flow, as well as the number and size of loci analyzed [8].
- Associated f-statistics: To estimate the fraction of a genome affected by gene flow, several related statistics have been developed, including \( {\widehat{f}}G \), \( {\widehat{f}}{hom} \), and \( {\widehat{f}}_d \). However, these estimators often require precise knowledge of demographic parameters (divergence times, population sizes) and can exhibit high variance among loci, making them challenging to apply in practice without strong prior information [8].
Phylogenetic Network Inference: Modeling Reticulation Explicitly
Phylogenetic network methods aim to reconstruct explicit network structures that represent evolutionary histories involving reticulation. These methods can be broadly categorized into several classes based on their inference approach [4]:
- Concatenation Methods: Approaches such as Neighbor-Net and SplitsNet (SplitsTree) estimate a single phylogeny from combined sequence data across all loci. While computationally efficient, they typically account only for sequence mutation and may not adequately handle genealogical incongruence caused by ILS or gene flow [4] [9].
- Parsimony-based Methods: Methods like MP (Maximum Parsimony) utilize the minimize deep coalescence (MDC) criterion, seeking the species phylogeny that minimizes the number of deep coalescences needed to explain a given set of gene trees [4].
- Probabilistic Methods: These approaches perform inference under explicit evolutionary models that combine coalescent theory with biomolecular substitution models. They include:
- Full-Likelihood Methods: MLE and MLE-length (implemented in PhyloNet) calculate the full likelihood under the multispecies network coalescent (MSNC) model using gene tree topologies or topologies with branch lengths, respectively [4] [5].
- Pseudo-likelihood Methods: MPL and SNaQ (Species Networks applying Quartets) use approximations to the full model likelihood to improve computational efficiency while maintaining statistical consistency under appropriate conditions [4] [10].
Table 1: Comparison of Methodological Approaches to Detecting Gene Flow
| Method Category | Representative Methods | Theoretical Foundation | Data Input Requirements | Biological Processes Accounted For |
|---|---|---|---|---|
| D-Statistic | ABBA-BABA test [8] | Population genetics, parsimony | Genotype data for 4 taxa + outgroup | Gene flow, incomplete lineage sorting |
| Concatenation Networks | Neighbor-Net, SplitsNet [4] [9] | Distance-based, splits | Sequence alignments (concatenated) | Sequence mutation, some conflicting signal |
| Parsimony-based Networks | MP (Minimize Deep Coalescence) [4] | Parsimony, MDC criterion | Set of gene trees | Incomplete lineage sorting, gene flow |
| Probabilistic Networks | MLE, MLE-length (PhyloNet) [4] [5] | Coalescent theory, maximum likelihood | Gene trees or sequence alignments | ILS, gene flow, sequence mutation |
| Pseudo-likelihood Networks | SNaQ, MPL [4] [10] | Coalescent theory, quartets | Gene trees or concordance factors | ILS, gene flow |
Performance Comparison: Accuracy, Scalability, and Limitations
Detection Power and Accuracy
Empirical evaluations reveal significant differences in accuracy and detection power between methodological approaches:
D-Statistic Performance: The D-statistic effectively detects the presence of gene flow in a wide range of conditions, particularly for closely related species. However, its power is substantially diminished when population sizes are large relative to branch lengths in generations. The statistic serves primarily as a qualitative measure of gene flow, as estimating the actual fraction of introgressed genomic material (f) requires precise knowledge of divergence times and population sizes that is often unavailable in empirical studies [8].
Phylogenetic Network Accuracy: Probabilistic network inference methods generally demonstrate superior topological accuracy compared to parsimony-based or concatenation approaches. Methods maximizing likelihood under coalescent-based models or their pseudo-likelihood approximations consistently achieve the highest accuracy in recovering known phylogenetic networks in simulation studies [4]. The table below summarizes quantitative performance comparisons from empirical scalability studies.
Table 2: Performance and Scalability of Phylogenetic Network Methods on Large-Scale Datasets
| Method | Inference Type | Theoretical Guarantees | Maximum Practical Taxa | Key Limitations |
|---|---|---|---|---|
| D-Statistic | Gene flow detection | Statistical consistency under model assumptions [8] | Not applicable (fixed 4-taxon test) | Qualitative only; sensitive to population size; requires known species tree |
| Neighbor-Net | Concatenation network | None specifically for reticulation | Large datasets [9] | Does not explicitly model ILS or gene flow processes |
| MP (MDC) | Parsimony network | None known | >25 taxa [4] | Lower topological accuracy compared to probabilistic methods |
| MLE/MLE-length | Probabilistic network | Statistical consistency [5] | <25 taxa [4] | Prohibitive computational requirements beyond ~25 taxa |
| SNaQ/MPL | Pseudo-likelihood network | Statistical consistency for level-1 networks [10] | 30+ taxa with reduced accuracy [4] | Accuracy degrades with increasing taxa and mutation rate |
| ALTS | Tree-child network | Exact solution for displayed trees [11] | 50 taxa with 50 trees in ~15 minutes [11] | Limited to tree-child networks; performance depends on common clusters |
Scalability and Computational Requirements
Computational requirements represent a significant constraint for phylogenetic network inference, particularly for probabilistic approaches:
D-Statistic: Computationally efficient, allowing for genome-scale applications and bootstrap tests without significant computational burden [8].
Phylogenetic Network Methods: Scalability varies dramatically by approach. Concatenation methods like Neighbor-Net can handle large numbers of taxa but with biological interpretability limitations. Probabilistic methods face severe computational constraints, with full-likelihood methods often becoming prohibitive beyond 25 taxa. Even pseudo-likelihood methods show degraded accuracy with increasing taxon numbers and sequence mutation rates [4].
Innovative Scalable Approaches: Recent algorithmic developments aim to address these limitations:
- Divide-and-Conquer Inference: This approach infers networks on small, overlapping subsets of taxa (e.g., triplets) and merges them into a full network, enabling inference at scales previously infeasible for statistical methods [5].
- ALTS Algorithm: This method infers minimum tree-child networks by aligning lineage taxon strings from input trees, efficiently handling datasets with up to 50 taxa and 50 trees in approximately 15 minutes [11].
Experimental Protocols and Workflows
Standard Experimental Workflow for Phylogenetic Network Inference
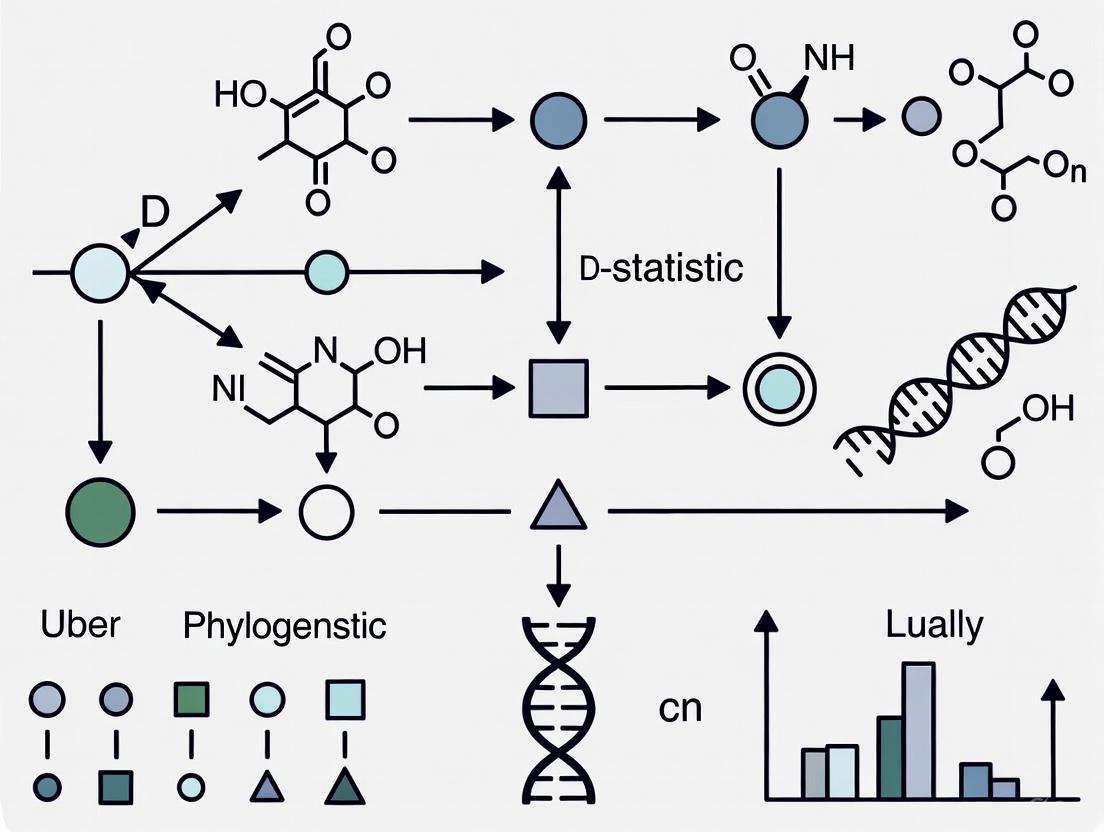
The following diagram illustrates a generalized workflow for inferring phylogenetic networks from genomic data, integrating both traditional and novel scalable approaches:
Diagram 1: Workflow for Phylogenetic Network Inference from Genomic Data. This workflow shows two pathways: traditional network inference methods and scalable approaches necessary for larger datasets. The process begins with multi-locus sequence data, proceeds through gene tree estimation, and culminates in biological interpretation of the inferred network. (Short Title: Phylogenetic Network Inference Workflow)
D-Statistic Analysis Protocol
The experimental protocol for implementing the D-statistic involves:
- Taxon Selection: Identify four taxa with a well-established species tree topology: ((P1, P2), P3, Outgroup).
- Data Preparation: Obtain genome-wide SNP data or sequence alignments for the selected taxa.
- Site Pattern Counting: Scan the genome to count ABBA sites (where P1 and Outgroup share the ancestral allele, while P2 and P3 share the derived allele) and BABA sites (where P1 and P3 share the derived allele, while P2 and Outgroup share the ancestral allele).
- Statistical Testing: Calculate the D-value and perform a statistical test (e.g., jackknife or block bootstrap) to assess significance. A significant deviation from zero indicates gene flow between P2 and P3.
Protocol for Scalable Network Inference via Divide-and-Conquer
For large datasets where standard network inference fails, the divide-and-conquer protocol enables scalable analysis [5]:
- Subset Selection: Determine a collection of overlapping subsets of taxa (X₁, X₂, ..., Xₖ). When using three-taxon subsets (trinets), a hitting set algorithm can significantly reduce the number of subsets required without substantial accuracy loss.
- Subnetwork Inference: For each taxon subset, infer an accurate phylogenetic network (topology, divergence times, inheritance probabilities) using appropriate methods. With small subsets, even computationally intensive likelihood methods become feasible.
- Network Agglomeration: Combine the k subnetworks into a comprehensive phylogenetic network on the complete taxon set. This step involves reconciling topological features and parameters across overlapping subsets.
Table 3: Key Software Tools for Phylogenetic Network Analysis
| Tool Name | Methodology | Primary Function | Data Input | Applicable Scale |
|---|---|---|---|---|
| PhyloNet [5] | Probabilistic, Pseudo-likelihood | Network inference under MSNC | Gene trees, sequence alignments | Small to medium (up to ~30 taxa) |
| SNaQ [4] [10] | Pseudo-likelihood (quartets) | Network inference from quartets | Gene trees, concordance factors | Medium (dozens of taxa) |
| SplitsTree4 [9] | Splits, Neighbor-Net, Median-Joining | Network visualization and inference | Sequence alignments, distances | Large datasets |
| ALTS [11] | Lineage Taxon String alignment | Tree-child network inference | Set of phylogenetic trees | ~50 taxa, ~50 trees |
| HYBROSCALE [11] | Agreement forests | Network inference from trees | Set of phylogenetic trees | Limited by common clusters |
The choice between the D-statistic and phylogenetic network methods depends fundamentally on the biological question, dataset characteristics, and computational resources. The D-statistic provides a computationally efficient, robust method for detecting the presence of gene flow between specific taxon pairs in a four-taxon context, making it ideal for initial screening or focused hypothesis testing. However, it provides only qualitative evidence and requires a known species tree topology [8].
Phylogenetic network methods offer a more comprehensive approach for reconstructing explicit reticulate histories across multiple taxa. Probabilistic methods provide the highest accuracy but face severe computational constraints, while concatenation approaches sacrifice biological interpretability for scalability. Recent innovations in divide-and-conquer strategies [5] and novel algorithms like ALTS [11] are significantly expanding the feasible scale of network inference, enabling analyses previously limited to tree-based methods.
For researchers investigating gene flow, a strategic approach might combine both methodologies: using the D-statistic for initial detection and validation of gene flow, followed by phylogenetic network inference to reconstruct complete reticulate histories when applicable. As theoretical developments continue to expand the identifiability of more complex network structures [10] and computational methods overcome current scalability limitations, explicit phylogenetic networks are poised to become the standard framework for modeling reticulate evolution across the tree of life.
A fundamental challenge in modern evolutionary biology is deciphering the true history of species from genomic data. When gene trees constructed from different DNA sequences conflict with each other or with the hypothesized species tree, this phylogenetic incongruence signals complex evolutionary histories. The two primary processes responsible for such patterns are incomplete lineage sorting (ILS) and introgression/hybridization (IH) [12]. Both processes can produce remarkably similar patterns of gene tree discordance, making them difficult to distinguish without sophisticated analytical approaches [13]. Understanding which process explains observed genetic patterns is crucial for reconstructing accurate evolutionary histories and has implications for species delimitation, conservation biology, and understanding adaptive evolution.
ILS occurs when ancestral genetic polymorphisms persist through multiple speciation events and are randomly sorted into descendant lineages [12]. In contrast, introgression involves the transfer of genetic material from one species to another through hybridization and backcrossing [14]. This guide provides a comprehensive comparison of the leading methods used to distinguish between these processes, focusing on their theoretical foundations, application protocols, and performance characteristics.
Theoretical Foundations and Methodological Principles
The D-Statistic (ABBA-BABA Test)
The D-statistic is a parsimony-based method that detects gene flow by comparing frequencies of discordant site patterns in a four-taxon system [8]. The method operates on a rooted quartet ((P1, P2), P3, O), where P1 and P2 are sister species, P3 is a more distantly related ingroup, and O is an outgroup. The core principle involves counting sites with specific patterns:
- ABBA sites: Sites where P1 and O share the ancestral allele, while P2 and P3 share the derived allele.
- BABA sites: Sites where P1 and O share the derived allele, while P2 and P3 share the ancestral allele [8].
Under pure ILS without gene flow, ABBA and BABA sites are equally likely, resulting in a D-statistic value not significantly different from zero. A significant excess of either pattern indicates introgression [8]. The D-statistic is calculated as: D = (NABBA - NBABA) / (NABBA + NBABA)
where NABBA and NBABA represent the counts of ABBA and BABA sites, respectively.
Phylogenetic Network Methods
Phylogenetic network methods provide a framework for representing evolutionary histories that include reticulate events such as hybridization. Unlike traditional phylogenetic trees, networks can incorporate nodes with multiple ancestors, explicitly modeling gene flow [4]. These methods can be broadly categorized into:
- Distance-based methods: Such as Neighbor-Net, which construct split networks summarizing conflicting signals [4].
- Parsimony-based methods: Like the MP method implemented in PhyloNet, which minimizes deep coalescences [4].
- Probabilistic methods: Including MLE, MLE-length, MPL, and SNaQ, which operate under coalescent models with explicit parameters for population sizes, divergence times, and gene flow [4].
Probabilistic methods fit the network to gene tree distributions or sequence data using maximum likelihood or Bayesian frameworks, providing statistical support for inferred reticulations [4].
Method Comparison and Performance Analysis
Table 1: Key Characteristics of D-Statistic and Phylogenetic Network Methods
| Feature | D-Statistic | Phylogenetic Network Methods |
|---|---|---|
| Theoretical basis | Parsimony-based site pattern counting | Coalescent-based model fitting |
| Data requirements | SNP data or sequence alignments; minimal sampling (single individual per species often sufficient) | Multi-locus sequence data or gene trees; better performance with multiple individuals |
| Computational requirements | Low; efficient even for genome-scale data | High; especially for probabilistic methods (MLE, MPL) which become prohibitive beyond 25-30 taxa [4] |
| Primary output | Test statistic (D) with significance assessment | Reticulate phylogeny with estimated hybridization events |
| Key assumptions | Correct rooting; no ancestral population structure; constant substitution rates | Correct gene tree estimation; neutral evolution; no recombination within loci |
| Detection power | High sensitivity to recent gene flow; powerful for testing specific hypotheses | Better for characterizing complex reticulation histories; infers direction and timing of gene flow |
Table 2: Performance Comparison Across Evolutionary Scenarios
| Scenario | D-Statistic Performance | Phylogenetic Network Performance |
|---|---|---|
| Recent divergence | High power if population sizes small relative to divergence time [8] | High accuracy, but computational constraints with many taxa [4] |
| Deep divergence | Robust across wide genetic distances, but sensitive to large population sizes [8] | Degrades with increased sequence divergence [4] |
| Recent gene flow | Excellent detection power [8] | Good detection and characterization ability |
| Ancient gene flow | Limited power for very ancient events | Can detect older hybridization events |
| Multiple reticulations | Limited to four-taxon tests; complex extensions needed for multiple events | Theoretically can handle multiple reticulations, but practice limited to few events [4] |
Sensitivity Analysis and Limitations
The D-statistic's effectiveness is primarily determined by the relative population size (population size scaled by generations since divergence) [8]. It performs best when population sizes are small relative to branch lengths in generations. The method is robust across a wide range of divergence times but becomes less reliable when population sizes are large [8].
Phylogenetic network methods face scalability challenges. Probabilistic methods like MLE and MPL are most accurate but become computationally prohibitive with more than 25 taxa, with runtimes extending to weeks for datasets with 30+ taxa [4]. Accuracy degrades with both increasing taxon numbers and higher sequence divergence [4].
Experimental Protocols and Workflows
Implementing the D-Statistic
dot 4.1: D-Statistic Analysis Workflow
Step-by-Step Protocol:
- Taxon selection: Identify four taxa with established phylogenetic relationships: two sister species (P1, P2), a closely related species (P3), and an outgroup (O).
- Data preparation: Obtain whole-genome sequences or SNP data. Align sequences using tools like MUSCLE or MAFFT.
- Variant calling: Identify homologous sites across all four taxa.
- Site pattern classification: For each informative site, determine whether it fits ABBA, BABA, or other patterns.
- D-statistic calculation: Compute D = (NABBA - NBABA) / (NABBA + NBABA).
- Significance testing: Assess statistical significance using block jackknife or bootstrap resampling to account for linked sites.
- Interpretation: A significantly positive D suggests gene flow between P3 and P2; negative D suggests gene flow between P3 and P1.
Phylogenetic Network Inference
dot 4.2: Phylogenetic Network Inference Pipeline
Step-by-Step Protocol:
- Locus selection: Select dozens to hundreds of independent loci distributed across the genome.
- Gene tree estimation: Infer gene trees for each locus using maximum likelihood or Bayesian methods.
- Network inference: Input gene trees into network inference software (e.g., PhyloNet for MLE/MPL, PhyloNetworks for SNaQ).
- Model selection: Compare networks with different numbers of reticulations using information criteria (AIC, BIC) or likelihood-ratio tests.
- Validation: Assess support using bootstrap resampling of loci or posterior probabilities.
- Interpretation: Identify hybridization events, direction of gene flow, and evolutionary relationships.
Case Studies in Empirical Research
Distinguishing ILS and Introgression in European Bison
The evolutionary history of the wisent (European bison) presents a classic case study. Mitochondrial DNA analysis placed wisent closer to cattle than to American bison, suggesting hybridization [15]. However, whole-genome analysis revealed that only a small portion (1.0-4.0%) of wisent nuclear genome showed cattle ancestry, with ABBA-BABA tests indicating recent rather than ancient introgression [15].
Nuclear gene trees displayed heterogeneous topologies, with the relative frequencies of different tree topologies consistent with expectations from ILS rather than widespread hybridization. Coalescent simulations confirmed that ILS alone could explain the anomalous mtDNA phylogeny as a rare event [15]. This case demonstrates the importance of genome-wide data and coalescent modeling for distinguishing these processes.
Complex Reticulation in Cobitis Fish Complex
The Cobitis fish complex exemplifies deep reticulate evolution. Phylogenomic analysis revealed mito-nuclear discordance, with C. tanaitica exhibiting mtDNA clustering with C. elongatoides but nuclear similarity to C. taenia [14]. Application of multiple methods (D-statistic, coalescent simulations, phylogenetic networks) indicated this pattern resulted from ancient hybridization and mitochondrial capture rather than ILS [14].
Interestingly, contemporary hybrids in this complex reproduce clonally (gynogenesis), preventing ongoing introgression. This suggests the detected hybridization events were ancient episodes mediated by previously existing hybrids with non-clonal inheritance [14]. This case highlights how method integration can unravel complex evolutionary histories.
Pine Species Divergence with Secondary Contact
Population genomic analysis of Pinus massoniana and P. hwangshanensis demonstrated the power of comparing allopatric versus parapatric populations. The finding of significantly more admixture in parapatric populations than allopatric ones provided evidence for secondary contact and introgression rather than ILS [13]. Approximate Bayesian Computation (ABC) modeling supported a scenario of long isolation followed by secondary contact during Pleistocene range expansions [13].
Research Toolkit: Essential Materials and Reagents
Table 3: Key Research Reagents and Computational Tools
| Tool/Reagent | Function/Purpose | Application Context |
|---|---|---|
| Whole-genome sequencing data | Provides comprehensive genomic coverage for site pattern analysis and gene tree estimation | Both D-statistic and network methods |
| Targeted sequence capture | Enriches specific loci across multiple individuals for multi-locus analyses | Phylogenetic network methods |
| BEAST/BEAST2 | Bayesian evolutionary analysis; divergence time estimation under coalescent models | Demographic inference for contextualizing ILS/IH |
| PhyloNet | Infers phylogenetic networks from gene trees under coalescent models | Network inference (MLE, MPL methods) |
| ADMIXTOOLS | Implements D-statistic and related f-statistics for detecting gene flow | D-statistic analysis |
| SplitsTree4 | Constructs phylogenetic networks from distance matrices | Distance-based network inference |
| SNaQ | Infers phylogenetic networks using quartet-based pseudo-likelihood | Network inference for larger datasets |
| HyDe | Detects hybridization using site pattern probabilities | Hybridization detection in multi-species systems |
Integrated Analysis Framework
dot 7.1: Decision Framework for Method Selection
For comprehensive analysis, researchers should consider an integrated approach:
- Initial screening: Use D-statistic to test for significant gene flow in specific taxon quartets.
- Detailed characterization: Apply phylogenetic network methods to infer detailed reticulate histories.
- Validation: Use coalescent simulations to assess whether inferred parameters could generate observed patterns through ILS alone.
- Contextualization: Incorporate demographic history (divergence times, population sizes) to interpret results biologically.
The most robust conclusions emerge from consistency across multiple methods and careful consideration of biological context, sampling design, and methodological assumptions.
The study of evolutionary history has been revolutionized by methods designed to detect past gene flow. Two prominent approaches have emerged: the D-statistic (ABBA-BABA test) and Phylogenetic Network inference methods. The D-statistic operates as a targeted hypothesis test for specific admixture events, providing a statistical signal of gene flow between four predefined populations or species [16]. In contrast, phylogenetic network methods aim to reconstruct comprehensive evolutionary histories that explicitly represent both divergence and hybridization events as reticulate networks [4]. This guide provides an objective comparison of these methodologies, examining their performance, underlying assumptions, and suitability for different research scenarios in evolutionary biology and genomics.
Methodological Foundations and Comparison
Core Principles of Each Approach
The D-statistic Framework: The D-statistic tests the correctness of a hypothetical phylogenetic relationship between four populations (P1, P2, P3, and an outgroup P4) by evaluating specific allelic patterns [16]. It operates by comparing the frequencies of two discordant site patterns: "ABBA" sites, where P1 and P4 share allele A while P2 and P3 share allele B, and "BABA" sites, where P1 and P3 share allele A while P2 and P4 share allele B [16]. Under the null hypothesis of no gene flow, these patterns should occur with equal probability. Significant deviation from this expectation, measured by the D-statistic, provides evidence of gene flow, typically between P3 and P2 or P1 [16].
Phylogenetic Network Framework: Phylogenetic network methods represent evolutionary histories as directed acyclic graphs that can incorporate both vertical descent and horizontal gene flow through reticulation nodes [4]. Unlike the D-statistic which tests a specific hypothesis, network methods perform full inference by searching among all possible phylogenies defined on a set of taxa. Explicit networks attribute reticulations to specific evolutionary processes like gene flow, while implicit networks merely summarize conflicting phylogenetic signal without specific biological interpretation [4].
Performance Comparison: Accuracy and Scalability
Experimental comparisons on both simulated and empirical datasets reveal significant performance differences between these approaches and among specific implementation methods.
Table 1: Performance Comparison of Phylogenetic Network Inference Methods [4]
| Method Category | Representative Methods | Topological Accuracy | Computational Efficiency | Maximum Practical Taxa |
|---|---|---|---|---|
| Probabilistic (Full-likelihood) | MLE, MLE-length | Highest | Lowest (weeks for >25 taxa) | ~25 taxa |
| Probabilistic (Pseudo-likelihood) | MPL, SNaQ | High | Medium | Larger than full-likelihood |
| Parsimony-based | MP (Minimize Deep Coalescence) | Moderate | Medium | Larger than probabilistic |
| Concatenation | Neighbor-Net, SplitsNet | Lower | Highest | 50+ taxa |
Table 2: D-statistic Performance Characteristics [16]
| Performance Aspect | Traditional D-statistic | Improved D-statistic |
|---|---|---|
| Data Requirements | Single read per population | All reads, multiple individuals |
| Sequencing Depth | Inefficient for 1-10x depth | Optimal power at 2x depth |
| Error Correction | Limited | Type-specific error correction |
| Distribution | Standard normal approximation | Standard normal approximation |
Key findings from empirical evaluations indicate that probabilistic phylogenetic network methods (MLE, MLE-length) achieve the highest accuracy but become computationally prohibitive beyond approximately 25 taxa, requiring weeks of runtime without completion [4]. The improved D-statistic significantly outperforms the traditional approach for low and medium sequencing depths (1-10×), with performance comparable to perfectly called genotypes at just 2× sequencing depth [16].
Experimental Protocols and Methodologies
Standard D-Statistic Implementation Protocol
Workflow Overview:
- Population Selection: Identify four populations with a clear phylogenetic hypothesis to test, typically involving P1, P2, P3, and an outgroup P4 [16].
- Sequence Data Processing: Process high-throughput sequencing data, often requiring special handling for ancient DNA with deamination patterns [16].
- Site Pattern Counting: For each informative site, count ABBA and BABA patterns across all reads or a sampled base [16].
- Statistical Testing: Calculate the D-statistic as D = (ΣABBA - ΣBABA) / (ΣABBA + ΣBABA) and assess significance against a standard normal distribution [16].
Advanced Considerations: The improved D-statistic protocol incorporates multiple individuals per population without genotype calling, uses all available reads rather than sampling a single base, applies type-specific error correction for sequencing errors, and can correct for introgression from external populations not part of the supposed genetic relationship [16].
Phylogenetic Network Inference Protocol
Workflow Overview:
- Sequence Collection and Alignment: Collect homologous DNA sequences through experiments or public databases and perform multiple sequence alignment [7].
- Data Trimming: Precisely trim aligned sequences to remove unreliable regions while preserving genuine phylogenetic signals [7].
- Evolutionary Model Selection: Select appropriate substitution models (e.g., JC69, K80, TN93, HKY85) based on sequence characteristics [7].
- Network Inference: Apply specific network inference algorithms (e.g., MLE, MPL, SNaQ) to estimate phylogenetic networks [4].
- Topology Evaluation: Assess inferred networks through statistical support measures such as bootstrap values [17].
Method-Specific Variations:
- MLE Approach: Uses maximum likelihood estimation under coalescent-based models with branch length information [4].
- MPL Approach: Applies pseudo-likelihood approximations to full model likelihood calculations [4].
- SNaQ Approach: Combines pseudo-likelihoods under a coalescent model with quartet-based concordance analysis [4].
Figure 1: Comparative Workflows of D-Statistic and Phylogenetic Network Methods
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents and Computational Tools
| Category | Specific Tools/Reagents | Function/Purpose |
|---|---|---|
| D-Statistic Implementation | ANGSD (doAbbababa2) [16], ADMIXTOOLS (qpDstat) [16] | Implements D-statistic on low-depth NGS data with error correction |
| Phylogenetic Network Software | PhyloNet [4], SNaQ [4] | Infers explicit phylogenetic networks under coalescent models |
| Sequence Alignment | MEGA10 [17], Bio Edit [17] | Multiple sequence alignment and editing |
| Tree/Network Visualization | MicrobeTrace [17] | Visualizes molecular transmission networks |
| Statistical Framework | R Language [7] | Provides statistical analysis and custom algorithm implementation |
Discussion: Strategic Selection for Evolutionary Analysis
The choice between D-statistic and phylogenetic network methods depends critically on research goals, dataset characteristics, and computational resources.
D-statistic is optimal when:
- Testing specific gene flow hypotheses between defined populations
- Working with low-coverage sequencing data (1-10×)
- Analyzing datasets with more than 30 taxa where network methods become computationally prohibitive [16] [4]
- Requiring rapid assessment of admixture signals
Phylogenetic network methods are preferable when:
- Reconstructing comprehensive evolutionary histories without predefined hypotheses
- Analyzing smaller datasets (≤25 taxa) where probabilistic methods remain feasible [4]
- Working with closely related species where incomplete lineage sorting is prevalent
- Visualizing complex evolutionary relationships with multiple reticulations
Emerging trends point toward normal networks as a promising class that balances biological relevance with mathematical tractability [18]. Furthermore, methodological improvements continue to enhance both approaches, such as the development of D-statistic implementations that utilize all available reads rather than sampling single bases [16].
For researchers in drug development and infectious disease studies, phylogenetic networks have proven particularly valuable for tracing transmission dynamics of pathogens like HIV and understanding the spread of drug resistance mutations [17]. The D-statistic remains widely applied in evolutionary studies of ancient DNA and population genomics where specific admixture hypotheses need testing [16].
Both D-statistic and phylogenetic network methods provide powerful approaches for detecting gene flow, yet they occupy distinct niches in evolutionary biology research. The D-statistic offers a targeted, statistically robust framework for testing specific admixture hypotheses, with particular strengths for low-coverage sequencing data. Phylogenetic network methods provide comprehensive evolutionary reconstruction but face computational constraints that limit their application to smaller datasets. Researchers should select methods based on their specific experimental questions, dataset scale, and computational resources, with the understanding that ongoing methodological developments continue to enhance the biological interpretability of both approaches.
A Practical Guide to Implementation and Workflow Integration
The D-statistic, or ABBA-BABA test, provides a powerful parsimony-based approach for detecting deviations from strict bifurcating evolutionary histories, most commonly used to identify gene flow between closely related species or populations. This methodology compares the frequencies of two discordant allele patterns ("ABBA" and "BABA") across genomes to test for significant deviations from the null expectation of equal frequencies, which would indicate introgression. This guide details the experimental workflow for implementing D-statistic analyses, provides a direct comparison with phylogenetic network methods, and presents empirical data evaluating their relative performance across different biological scenarios. The comparative analysis reveals that while D-statistic offers computational efficiency and simplicity for specific introgression tests, phylogenetic network methods provide more comprehensive evolutionary models at the cost of significantly greater computational resources.
In the era of genomics, detecting gene flow between species—introgression—has become fundamental to understanding evolutionary processes. The D-statistic and phylogenetic network methods represent two complementary approaches for identifying these complex evolutionary signals. The D-statistic operates as a targeted test for a specific four-taxon introgression scenario, quantifying deviations in allele pattern frequencies that suggest gene flow between non-sister taxa [19]. In contrast, phylogenetic network methods aim to reconstruct complete evolutionary histories that may include multiple reticulation events, explicitly modeling processes like hybridization and horizontal gene transfer that violate tree-like ancestry [4].
These methodologies differ fundamentally in their scope and underlying assumptions. The D-statistic tests a specific hypothesis about relationships between four predefined populations, requiring researchers to specify the exact phylogenetic context upfront [20] [19]. Phylogenetic network methods attempt to infer the overall phylogenetic structure from sequence data, potentially identifying unexpected relationships without requiring pre-specified hypotheses about which taxa might be involved in gene flow events [4]. This distinction makes D-statistic ideal for focused hypothesis testing, while network methods serve better for exploratory analysis of complex evolutionary scenarios.
The D-Statistic Workflow: From Sequences to Interpretation
Conceptual Foundation of the ABBA-BABA Test
The D-statistic operates on a simple but powerful principle: under a strictly bifurcating evolutionary tree with no gene flow, two specific discordant allele patterns should occur at approximately equal frequencies. The test requires four taxa with established phylogenetic relationships: (((P1, P2), P3), Outgroup) [20] [19]. The outgroup provides the ancestral state reference.
- ABBA sites: Sites where P2 and P3 share a derived allele ("B") while P1 retains the ancestral state ("A")
- BABA sites: Sites where P1 and P3 share a derived allele ("B") while P2 retains the ancestral state ("A")
Under the null hypothesis of no introgression, both patterns arise equally through incomplete lineage sorting (ILS). A significant excess of either pattern indicates gene flow, specifically between the taxa sharing derived alleles in the overrepresented pattern [19]. The D-statistic quantifies this deviation:
D = (ΣABBA - ΣBABA) / (ΣABBA + ΣBABA) [20]
A significant positive D value suggests gene flow between P2 and P3, while a significant negative value suggests gene flow between P1 and P3 [19].
Experimental Protocol and Computational Implementation
Data Preparation and Allele Frequency Estimation
The initial phase involves processing raw sequence data into analyzable allele frequencies:
Input Data: The workflow begins with genotype data from multiple individuals across populations, typically in variant call format (VCF) or similar. The data should be filtered for bi-allelic sites with sufficient quality [20].
Population Definition: Individuals are grouped into populations based on the biological hypothesis. For example, in a study of Heliconius butterflies, populations might represent different geographical races or species [20].
Derived Allele Frequency Calculation: Using the outgroup to determine ancestral states, compute the frequency of derived alleles in each population at each site. This can be accomplished using tools like the
freq.pyscript from the genomics_general package [20]:
This process generates a table of derived allele frequencies across all polymorphic sites for downstream analysis.
ABBA-BABA Calculation and Statistical Testing
With allele frequencies prepared, the analysis proceeds to pattern counting and significance testing:
ABBA/BABA Proportion Calculation: For each SNP site, compute the proportion that follows ABBA and BABA patterns using population allele frequencies rather than individual genotypes [20]:
ABBA = (1 - p1) * p2 * p3BABA = p1 * (1 - p2) * p3where p1, p2, p3 represent derived allele frequencies in populations P1, P2, and P3, respectively.
D-Statistic Computation: Sum ABBA and BABA proportions across all sites and calculate the D-statistic using the formula above [20].
Block Jackknife Significance Testing: To account for linkage disequilibrium and non-independence among sites, perform a block jackknife procedure with typically 1 Mb blocks [20]:
- Calculate pseudovalues of D by successively excluding each block
- Compute standard error from the pseudovalue variance
- Derive a Z-score to assess significance (|Z| > 3 generally considered significant) [21]
This workflow can be implemented in R or using specialized tools like the ipyrad-analysis toolkit [21].
Visualization of the D-Statistic Workflow
The following diagram illustrates the complete analytical pipeline from raw data to interpretation:
Figure 1: Complete D-statistic workflow from raw data processing through statistical testing and biological interpretation.
Comparative Performance Analysis
Methodological Comparison Framework
To objectively evaluate the performance characteristics of D-statistic versus phylogenetic network methods, we analyzed their behavior across multiple dimensions including computational efficiency, detection power, scalability, and implementation requirements. The comparison draws from both empirical studies and theoretical considerations.
Table 1: Methodological Comparison Between D-Statistic and Phylogenetic Network Approaches
| Characteristic | D-Statistic | Phylogenetic Network Methods |
|---|---|---|
| Computational Demand | Low to moderate; suitable for genome-scale data [8] | High; often prohibitive beyond 25-30 taxa [4] |
| Primary Function | Hypothesis testing for specific introgression scenarios [19] | Full phylogenetic inference including reticulations [4] |
| Data Requirements | Four predefined populations with known relationships [20] | Multiple loci across any number of taxa [4] |
| Detection Power | Robust for recent gene flow; sensitive to population size [8] | Varies by method; probabilistic approaches most accurate [4] |
| Key Limitations | Cannot distinguish ancestral structure from gene flow [22] | Computational limitations with increasing taxa [4] |
| Optimal Use Case | Testing specific gene flow hypotheses between known taxa | Inferring complex evolutionary histories with multiple reticulations |
| Implementation Tools | genomics_general, ipyrad-analysis [20] [21] | PhyloNet, SNaQ, Neighbor-Net [4] |
Empirical Performance Data
Several studies have quantitatively evaluated the performance of these methods under controlled conditions. The D-statistic demonstrates particular robustness to certain parameters while showing sensitivity to others:
Table 2: Empirical Performance Characteristics of D-Statistic Based on Simulation Studies
| Parameter | Effect on D-Statistic | Performance Implications |
|---|---|---|
| Population Size | Primary determinant of sensitivity; large populations reduce power [8] | Most effective when population sizes are small relative to branch lengths [8] |
| Genetic Distance | Robust across wide range of divergence times (0.3%-5% sequence divergence) [8] | Applicable to both recently diverged and moderately distant taxa |
| Gene Flow Direction | Asymmetric detection power; more powerful for certain directions [8] | Important to test multiple taxon arrangements |
| Locus Count | Power increases with more loci; minimal window size requirements [22] | Requires substantial genomic data for reliable inference |
| Outgroup Distance | Moderate effect; very distant outgroups can reduce power [8] | Optimal with appropriately chosen outgroup |
A critical finding from simulation studies is that the D-statistic is not an unbiased quantitative estimator of gene flow proportion. Its expected value increases non-linearly with the actual proportion of introgression (f) and is influenced by population size and divergence times [22]. This makes it most appropriate for detecting the presence rather than quantity of gene flow.
For phylogenetic network methods, performance varies considerably by implementation. Probabilistic methods (MLE, MLE-length) generally provide the highest accuracy but become computationally prohibitive with more than 25 taxa, often failing to complete analyses with 30+ taxa even after weeks of computation [4]. Pseudo-likelihood approximations (MPL, SNaQ) offer better scalability with moderate accuracy trade-offs [4].
Advanced D-Statistic Derivatives and Extensions
To address limitations of the standard D-statistic, several modified statistics have been developed:
- fd statistic: Better identifies introgressed loci while avoiding biases that affect D in regions of reduced diversity [22]
- fG and fhom: Estimate the fraction of genome affected by gene flow, though with high variance across loci [8]
- D3 statistic: Three-sample test using genetic distances rather than outgroup, useful when no appropriate outgroup is available [19]
These derivatives maintain the computational efficiency of the original D-statistic while addressing specific limitations, though they may introduce new assumptions or requirements.
Essential Research Reagents and Computational Tools
Successful implementation of introgression analyses requires both biological materials and computational resources. The following toolkit represents essential components for designing and executing these studies.
Table 3: Essential Research Toolkit for Introgression Analysis
| Tool/Resource | Function | Implementation Notes |
|---|---|---|
| Population Genomic Data | Source of genetic variation for analysis | Multiple individuals per population recommended; whole-genome sequencing preferred |
| genomics_general Package | Python utilities for frequency calculation and D-statistic | Provides freq.py for allele frequency calculation [20] |
| ipyrad-analysis Toolkit | Implementation of ABBA-BABA tests with visualization | Supports automated test generation from tree topology [21] |
| R Statistical Environment | Data manipulation, visualization, and custom analysis | Essential for block jackknife implementation and result visualization [20] |
| PhyloNet Software | Phylogenetic network inference under coalescent model | Provides multiple inference algorithms including MLE and MP [4] |
| Outgroup Sequence | Polarizes alleles as ancestral or derived | Should be appropriately diverged; critical for accurate pattern identification [20] |
Integrated Analysis Strategy
For comprehensive introgression analysis, we recommend a hierarchical approach that leverages the complementary strengths of both methodologies:
- Initial Screening: Apply D-statistic tests to multiple taxon quartets to identify potential gene flow events
- Validation: Use fd statistics to confirm introgression in identified regions and avoid biases [22]
- Contextualization: Implement phylogenetic network methods on subsets of taxa with strong signals to model complex relationships
- Interpretation: Integrate results with additional evidence (e.g., dXY, linkage disequilibrium) to distinguish introgression from ancestral structure
This integrated strategy balances computational efficiency with comprehensive inference, leveraging the hypothesis-testing strength of D-statistic while contextualizing results within broader evolutionary patterns.
The D-statistic provides a computationally efficient, targeted approach for detecting specific introgression scenarios, with particular strength in analyzing genome-scale data across moderate evolutionary distances. Its implementation through a standardized workflow of allele frequency calculation, pattern counting, and block jackknife validation offers robust detection of gene flow signals. However, its limitations in distinguishing gene flow from ancestral structure and its sensitivity to population size parameters necessitate complementary approaches. Phylogenetic network methods offer more comprehensive evolutionary modeling but face severe computational constraints with increasing taxonomic sampling. The choice between these approaches should be guided by specific research questions, dataset characteristics, and computational resources, with integrated strategies often providing the most insightful resolution of complex evolutionary histories.
This guide provides an objective comparison between maximum pseudolikelihood (MPL) and maximum likelihood estimation (MLE) for inferring complex networks in social, phylogenetic, and psychometric research. MLE is generally superior in statistical accuracy (lower bias, better coverage) for models with strong dependence structures but is often computationally intractable for large or complex problems [23] [24]. MPL offers a computationally efficient and viable approximation, enabling the analysis of large-scale datasets (e.g., many taxa or variables) that are prohibitive for full-likelihood methods, though it can exhibit higher bias and underestimate standard errors [25] [26] [27]. The choice between them involves a fundamental trade-off between statistical precision and computational feasibility, heavily influenced by the specific data structure and research goals.
The following table summarizes the key performance characteristics of MLE and MPL established across various fields.
Table 1: Comparative Performance of MLE and MPL Across Domains
| Domain | Criterion | Maximum Likelihood (MLE) | Maximum Pseudolikelihood (MPL) |
|---|---|---|---|
| Exponential Random Graph Models (ERGMs) | Bias & Coverage | Lower bias, better coverage rates [23] [24] | Higher bias, especially with complex dependence [23] [24] |
| Standard Errors | Accurate estimation [23] | Tends to underestimate standard errors [23] | |
| Phylogenetic Network Inference | Topological Accuracy | High accuracy where computationally feasible [27] [4] | High accuracy, often comparable to MLE [25] [27] |
| Computational Scalability | Limited to small networks (e.g., ~10-25 taxa) [27] [4] | Scales to large networks (dozens of taxa) [25] [27] | |
| Runtime | Prohibitive for large datasets (weeks of CPU) [27] | Fast and practical for large analyses [25] | |
| Ising Model (Psychometrics) | Parameter Estimation | Most accurate, but only feasible for small graphs [26] | A stable, consistent approximation for large graphs [26] |
| Method Preference | Gold standard when possible [26] | JPL for dense networks, DPL for sparse networks [26] |
Detailed Experimental Comparisons
Performance in Exponential Random Graph Models (ERGMs)
Experimental Protocol: A simulation study compared MLE and MPL using social network data based on Lazega's law firm network. Simulations involved two model versions: one representing the original law firm data and another with increased transitivity to amplify dependency. The study evaluated estimators for bias, standard errors, coverage rates, and efficiency for both natural and mean-value parameterizations [23] [24].
Key Findings:
- Bias and Coverage: MLE estimators consistently showed lower bias and better coverage rates for the true parameter values compared to MPL estimators [23] [24].
- Impact of Dependence: The performance gap between MLE and MPL widened in the model with increased transitivity, indicating that MPL struggles more with complex dependency structures [23].
- Bias-Reduced MPL: A modified MPL estimator, using an approach aimed at reducing bias (e.g., Firth's method), was proposed and shown to outperform standard MPL, though it still did not match MLE performance [23].
Performance in Phylogenetic Network Inference
Experimental Protocol: Studies evaluated phylogenetic network inference methods on both simulated and empirical datasets (e.g., from natural mouse populations). Simulations used model phylogenies with a single reticulation event. Methods were compared on their ability to recover the correct network topology from multi-locus sequence data. Key metrics included topological accuracy and computational requirements (runtime and memory usage) [27] [4].
Key Findings:
- Accuracy: Probabilistic methods (both MLE and MPL-based like SNaQ) were the most accurate for topological inference [27] [4].
- Scalability: MLE methods (e.g., in PhyloNet) became computationally prohibitive, failing to complete analyses beyond 25-30 taxa. In contrast, MPL methods (e.g., SNaQ) successfully scaled to analyses involving dozens of taxa [25] [27] [4].
- Robustness: The quartet-based pseudolikelihood method SNaQ was found to be more robust to gene tree estimation errors compared to some full-likelihood approaches [25].
Table 2: Scalability of Phylogenetic Network Methods (Based on [27] [4])
| Method | Inference Criterion | Max Practical Taxa | Runtime | Key Limitation |
|---|---|---|---|---|
| MLE (PhyloNet) | Full Likelihood | ~25 taxa | Many weeks (CPU) | Computationally prohibitive beyond small datasets |
| MLE-length (PhyloNet) | Full Likelihood (with branch lengths) | ~25 taxa | Many weeks (CPU) | Computationally prohibitive beyond small datasets |
| SNaQ (PhyloNetworks) | Pseudolikelihood (Quartets) | Dozens of taxa | Fast | Enabled large-scale network inference |
| MPL (PhyloNet) | Pseudolikelihood | ~25 taxa | More scalable than MLE | Less robust to gene tree error than SNaQ [25] |
Performance in Ising Models (Network Psychometrics)
Experimental Protocol: An extensive simulation study compared estimators for the Ising model, which is used for binary data (e.g., symptom networks). The study varied the number of variables, sample size, and network type. Estimators based on the exact likelihood (MLE) were compared against two pseudolikelihood approximations: the joint pseudolikelihood (JPL) and the disjoint pseudolikelihood (DPL), also known as nodewise regression [26].
Key Findings:
- Feasibility: Exact MLE is only computationally feasible for small graphs, while pseudolikelihood methods are necessary for larger networks [26].
- JPL vs. DPL: JPL is a stable estimation method that accurately approximates MLE estimates. DPL, while popular and simple, only performs well with large sample sizes [26].
- Network Structure: The choice between JPL and DPL depends on network structure. DPL is more efficient for sparse networks, while JPL performs better for dense networks [26].
The Scientist's Toolkit
Table 3: Essential Software and Reagents for Network Inference
| Tool Name | Primary Function | Application Context | Key Features |
|---|---|---|---|
| statnet (R suite) | ERGM estimation and analysis | Social Network Analysis | Implements both MLE (via MCMC) and MPLE for ERGMs [23] |
| PhyloNet | Phylogenetic network inference | Evolutionary Biology | Implements full-likelihood MLE methods (and MPL) [27] [4] |
| PhyloNetworks | Phylogenetic network inference | Evolutionary Biology | Implements SNaQ, a quartet-based pseudolikelihood method [25] |
| bgms (R package) | Bayesian Ising model estimation | Network Psychometrics | Uses the Joint Pseudolikelihood (JPL) approach [26] |
| IsingFit / MGM (R packages) | Ising model estimation | Network Psychometrics | Use the Disjoint Pseudolikelihood (DPL) approach [26] |
| BPP Software | Multi-species coalescent simulation | Evolutionary Biology | Used to simulate gene trees under the coalescent model for phylogenetic studies [28] |
In the field of phylogenetics, scalability—the ability to handle increasing numbers of taxa—is a critical challenge that separates theoretical methodology from practical application. As phylogenomic studies regularly encompass hundreds of genomes, the performance characteristics of analytical methods become as important as their statistical properties. This guide objectively compares the scalability of two predominant approaches for detecting gene flow: the D-statistic (and its extensions) and phylogenetic network inference methods.
The D-statistic operates as a test for gene flow between specific taxa within a hypothesized phylogenetic tree, requiring researchers to formulate explicit evolutionary hypotheses in advance. In contrast, phylogenetic network methods perform full inference by searching among all possible phylogenetic relationships to reconstruct explicit networks that represent evolutionary history with reticulations. While network methods offer more comprehensive insights, they face significant computational constraints that the D-statistic approach largely avoids [4].
This comparison examines how both approaches perform as taxon numbers scale from dozens to hundreds, providing researchers with actionable data to select appropriate methods for their study systems and computational resources.
Performance Comparison Across Taxa Scales
Quantitative Performance Metrics
Table 1: Phylogenetic Network Method Performance Across Taxon Scales
| Method Category | Representative Tools | Practical Taxon Limit | Runtime Performance | Memory Requirements | Accuracy Trends |
|---|---|---|---|---|---|
| Probabilistic (Full-likelihood) | MLE, MLE-length (PhyloNet) | <25 taxa [4] | Prohibitive (>weeks) beyond 25 taxa [4] | Very high | Highest accuracy below scalability limits [4] |
| Pseudo-likelihood | MPL, SNaQ [4] | ~30 taxa [4] | Days to weeks at 30 taxa [4] | High | High accuracy, degrades with increased sequence mutation [4] |
| Parsimony-based | MP (Minimize Deep Coalescence) [4] | 25-30 taxa [4] | More efficient than probabilistic methods [4] | Moderate | Lower than probabilistic methods [4] |
| Tree-child Networks | ALTS [11] | 50 taxa [11] | ~15 minutes for 50 taxa/50 trees [11] | Moderate | Accurate for tree-child networks with trivial common clusters [11] |
| Concatenation | Neighbor-Net, SplitsNet [4] | 50+ taxa [4] | Most efficient [4] | Low | Lower accuracy in presence of gene flow [4] |
Table 2: D-Statistic vs. Network Methods Scalability Profile
| Performance Dimension | D-Statistic | Phylogenetic Network Methods |
|---|---|---|
| Theoretical scaling | Linear with number of test quadruples [4] | NP-hard problem [4] |
| Practical taxon limits | Hundreds to thousands [4] | 25-50 taxa for most methods [4] |
| Runtime performance | Minutes to hours regardless of taxon number | Prohibitive beyond method-specific limits [4] |
| Hypothesis testing | Requires a priori hypotheses [4] | Infers networks without pre-specified hypotheses [4] |
| Output complexity | Single statistic with significance test [4] | Complex phylogenetic network [4] |
| Best use case | Testing specific gene flow hypotheses | Comprehensive exploration when gene flow patterns are unknown |
Experimental Performance Data
Topological Accuracy Degradation: Experimental studies demonstrate that topological accuracy of phylogenetic network methods systematically degrades as taxon numbers increase. Similarly, increased sequence mutation rates negatively impact performance, particularly for pseudo-likelihood methods [4].
Computational Bottlenecks: For probabilistic phylogenetic network methods, model likelihood calculations represent the primary performance bottleneck. One study found that none of the probabilistic methods completed analyses of datasets with 30 taxa or more, even after many weeks of CPU runtime [4].
Algorithmic Innovations: New approaches like the ALTS method, which aligns lineage taxon strings from phylogenetic trees, demonstrate that algorithmic improvements can extend practical limits. This method can infer tree-child networks for up to 50 taxa with 50 input trees in approximately 15 minutes [11].
Experimental Protocols for Scalability Assessment
Standardized Scalability Benchmarking
Table 3: Essential Research Reagents and Computational Tools
| Research Reagent / Software | Function in Scalability Research | Implementation Considerations |
|---|---|---|
| PhyloNet Software Package [4] | Implements MLE, MLE-length, and MPL methods | Requires Java; memory-intensive for large analyses |
| ALTS [11] | Infers tree-child networks by aligning lineage taxon strings | Efficient for trees with trivial common clusters |
| SNaQ [4] | Implements pseudo-likelihood with quartet concordance | Better scaling than full-likelihood methods |
| Empirical Mouse Population Datasets [4] | Provide biological validation for scalability studies | Natural population data with known evolutionary history |
| Simulated Phylogenies with Single Reticulations [4] | Enable controlled scalability testing | Model networks with known properties for accuracy assessment |
Detailed Methodological Framework
Dataset Selection and Preparation:
- Empirical datasets: Studies should include natural population data with known evolutionary relationships, such as mouse populations that have been well-characterized for gene flow patterns [4]
- Simulated phylogenies: Controlled simulations should model phylogenies with single reticulation events across varying taxon numbers (dozens to hundreds) and evolutionary divergence levels [4]
- Sequence data parameters: Vary mutation rates systematically to evaluate interaction between sequence divergence and taxon number [4]
Performance Metric Collection:
- Runtime tracking: Measure computational time until completion or until predetermined timeout (e.g., 4 weeks) [4]
- Memory usage: Monitor peak memory consumption during analysis [4]
- Topological accuracy: Compare inferred networks to true simulated networks using topological distance measures [4]
- Scalability curves: Plot performance metrics against increasing taxon numbers to identify breaking points [4]
Method Implementation Protocols:
- Probabilistic methods: Configure MLE and MLE-length implementations in PhyloNet with identical convergence criteria for fair comparison [4]
- Pseudo-likelihood methods: Implement SNaQ with standard quartet-based concordance analysis [4]
- Tree-child networks: Apply ALTS method using lineage taxon string alignment with all possible taxon orderings [11]
Methodological Workflows and Logical Relationships
Discussion and Research Implications
Interpretation of Performance Data
The experimental data reveal a fundamental trade-off between methodological completeness and scalability. Phylogenetic network methods provide comprehensive inference but face severe computational constraints that limit their application to studies with approximately 50 taxa or fewer [4] [11]. In contrast, the D-statistic approach maintains practically linear scaling with taxon numbers, enabling analyses with hundreds of taxa, but requires researchers to pre-specify testable hypotheses about gene flow [4].
The accuracy degradation observed in network methods with increasing taxon numbers stems from two primary factors: (1) the exponential growth of network space that must be searched, and (2) increased phylogenetic complexity that challenges heuristic search strategies. This degradation manifests particularly strongly in scenarios with elevated sequence mutation rates, where signal becomes increasingly difficult to distinguish from noise [4].
Practical Recommendations for Researchers
For studies with ≤50 taxa: Network inference methods (particularly tree-child approaches like ALTS and pseudo-likelihood methods like SNaQ) provide the most comprehensive insights, assuming adequate computational resources [11].
For studies with 50-200 taxa: A hybrid approach works best—use fast screening with D-statistics to identify potential gene flow patterns, then apply network methods to specific clades of interest where reticulation is suspected [4].
For studies with >200 taxa: D-statistic and related hypothesis-testing approaches represent the only currently feasible method for genome-wide gene flow detection, though this requires careful formulation of evolutionary hypotheses [4].
Future Methodological Directions
The scalability gap between hypothesis-testing approaches like the D-statistic and comprehensive network inference represents a critical methodological challenge. Promising research directions include:
- Improved heuristics that better navigate the vast network space while maintaining topological accuracy [4]
- Algorithmic innovations like the ALTS approach that reframe the computational problem to avoid explicit likelihood calculations [11]
- Hybrid methods that combine the scalability of D-statistic screening with the inferential power of network methods
- High-performance computing implementations that leverage parallelization and GPU acceleration for likelihood calculations
Until these innovations mature, researchers must carefully match their methodological choices to their specific research questions, taxon sampling, and computational resources, while clearly acknowledging the limitations imposed by scalability constraints in their inferences about evolutionary history.
The genomic revolution has provided an abundance of data for phylogenetic studies, shifting the primary challenge from data acquisition to methodological analysis [29]. In this new era, a major hurdle is resolving incongruence—where different datasets or analytical methods yield conflicting evolutionary trees. This incongruence can stem from two primary sources: biological processes such as horizontal gene transfer, hybridization, and incomplete lineage sorting, or methodological issues including model violations and misassigned data [29]. This guide focuses on comparing two principal methodological approaches for detecting gene flow: the D-statistic (ABBA-BABA test) and phylogenetic network methods, providing researchers with a clear framework for selecting and applying these tools effectively.
The D-statistic and phylogenetic network methods represent distinct philosophical and technical approaches to identifying evolutionary histories that deviate from a strictly branching tree.
The D-statistic (ABBA-BABA Test)
The D-statistic is a population genomics tool designed to test a specific phylogenetic hypothesis regarding gene flow or ancient introgression. It operates on the principle of analyzing patterns of allele sharing among four taxa (P1, P2, P3, and an outgroup) to detect statistical deviations from a strictly bifurcating tree [27]. Its primary strength lies in its simplicity and computational efficiency, allowing for genome-scale scans to identify specific loci involved in gene flow events [27].
Phylogenetic Network Inference Methods
Phylogenetic network methods aim to reconstruct explicit evolutionary histories that can include both divergence and hybridization events. These methods represent the phylogeny as a directed acyclic graph, providing a more comprehensive model of evolution when gene flow occurs [27]. They can be broadly categorized as follows:
- Concatenation Methods: Approaches like Neighbor-Net analyze combined sequence data from all loci to infer a network that summarizes conflicting signals [27].
- Multi-locus Methods: These methods, which include parsimony-based (MP), maximum likelihood (MLE), and pseudo-likelihood (MPL, SNaQ) approaches, use individual gene trees as input to infer a species network under models that account for both incomplete lineage sorting and gene flow [27].
Diagram 1: A workflow comparing the general analytical paths for the D-statistic and phylogenetic network inference methods.
Quantitative Comparison of Methods
The table below summarizes the core characteristics, requirements, and outputs of the D-statistic versus general phylogenetic network methods.
Table 1: Method Comparison: D-statistic vs. Phylogenetic Network Inference
| Feature | D-statistic | Phylogenetic Network Inference |
|---|---|---|
| Core Function | Statistical test for a specific gene flow hypothesis [27] | Reconstruction of explicit phylogenetic history with reticulations [27] |
| Input Data | Genomic variant data from 4 taxa (P1, P2, P3, Outgroup) [27] | Multi-locus sequence alignments or pre-estimated gene trees [27] |
| Computational Demand | Low to Moderate | High to Very High [27] |
| Scalability | Highly scalable to genome-wide data [27] | Scalability is a major challenge; probabilistic methods often prohibitive beyond ~25 taxa [27] |
| Primary Output | Test statistic (D) and p-value [27] | A directed phylogenetic network with inferred reticulation nodes [27] |
| Key Advantage | Efficient for targeted testing and scanning for introgression signals [27] | Provides a comprehensive, visual evolutionary model incorporating gene flow [27] |
| Main Limitation | Requires an a priori hypothesis; does not provide a full phylogeny [27] | Computationally intensive, limiting application to large datasets [27] |
A critical scalability study has highlighted the performance limitations of phylogenetic network methods. The table below synthesizes key findings on the topological accuracy and computational runtimes of different network inference classes.
Table 2: Scalability and Performance of Network Inference Methods (Adapted from [27])
| Method Class | Representative Examples | Reported Topological Accuracy | Computational Limitations |
|---|---|---|---|
| Concatenation | Neighbor-Net, SplitsNet | Lower than probabilistic methods [27] | Less computationally intense, but may not adequately model process causing gene tree discordance [27] |
| Parsimony-based Multi-locus | MP (Minimize Deep Coalescence) | Less accurate than probabilistic methods [27] | More efficient than likelihood methods, but less accurate [27] |
| Probabilistic Multi-locus (Full Likelihood) | MLE, MLE-length | Most accurate in studies [27] | Prohibitive for datasets with ≥30 taxa; runtime can extend to weeks [27] |
| Probabilistic Multi-locus (Pseudo-likelihood) | MPL, SNaQ | High accuracy, though potentially lower than full-likelihood methods [27] | More efficient than full-likelihood, but still computationally demanding for larger datasets [27] |
Experimental Protocols for Phylogenomic Analysis
Basic Protocol: Detecting Gene Flow with the D-statistic
The D-statistic provides a statistical framework for testing gene flow between two species (P2 and P3) that are sister lineages relative to P1.
- Taxon Selection: Identify four taxa: P1 (a reference lineage), P2 and P3 (the two sister lineages tested for gene flow), and an outgroup to polarize allele patterns.
- Genome Alignment: Obtain or generate a whole-genome alignment for the four taxa.
- Variant Calling: Identify variable sites across the alignment.
- Site Pattern Counting: Scan the aligned genomes and count the number of sites exhibiting "ABBA" and "BABA" patterns, where A and B represent derived and ancestral alleles, respectively.
- Calculation: Compute the D-statistic using the formula: D = (∑(ABBA) - ∑(BABA)) / (∑(ABBA) + ∑(BABA)).
- Significance Testing: Assess the statistical significance of the D-value, often via a block jackknife procedure, to determine if significant gene flow has occurred.
Basic Protocol: Constructing a Phylogenomic Supermatrix with PhyKIT
PhyKIT is a versatile toolkit for processing and analyzing multiple sequence alignments and phylogenetic trees, facilitating various phylogenomic analyses [30]. Constructing a supermatrix is a common concatenation approach.
- Input Data Preparation: Gather individual single-locus or single-gene multiple sequence alignments.
- Alignment Quality Control: Use tools to assess and refine alignments (e.g., trim, filter).
- Run PhyKIT: Execute the PhyKIT command to concatenate the individual alignments into a single supermatrix. This process automatically handles sequence identifiers to ensure orthologous sequences are concatenated correctly [30].
- Output: The result is a single, combined alignment file (the supermatrix) and a corresponding partition file that describes the location and, optionally, the best-fit model for each original locus. This supermatrix can then be used for large-scale phylogenetic tree or network inference.
Advanced Protocol: Inferring a Phylogenetic Network with SNaQ
SNaQ (Species Networks applying Quartets) is a pseudo-likelihood method that infers species networks from gene tree quartets under a coalescent model with hybridization [27].
- Gene Tree Estimation: Infer individual gene trees from each locus in your multi-locus dataset using a standard phylogenetic method (e.g., maximum likelihood).
- Input File Preparation: Prepare a file containing the set of inferred gene trees.
- Network Inference: Run SNaQ (often within the PhyloNet package), specifying the maximum number of reticulations (hybridization events) to allow in the network.
- Result Interpretation: Analyze the output phylogenetic network, which will display the estimated species relationships, including the position and direction of inferred hybridization events. The branch lengths are estimated in coalescent units.
Diagram 2: A detailed workflow for phylogenetic network inference from multi-locus data, highlighting the key steps from sequence alignment to final network estimation.
A range of software tools is available to implement the protocols and analyses described in this guide. The following table details key resources for phylogenomic research.
Table 3: Essential Research Reagents and Software for Phylogenomic Analysis
| Tool/Resource | Primary Function | Application Note |
|---|---|---|
| PhyKIT | A multitool for diverse phylogenomic analyses [30] | Used for tasks such as constructing supermatrices, quantifying gene tree support, and detecting anomalies in orthology inference [30]. |
| PhyloNet | A software package for inferring and analyzing phylogenetic networks [27] | Implements several network inference methods, including MLE, MLE-length, and MPL. Essential for complex network analyses [27]. |
| SNaQ | Species network inference using quartet-based pseudo-likelihood [27] | A more computationally efficient alternative to full-likelihood methods within the PhyloNet framework, suitable for larger datasets [27]. |
| ADMIXTOOLS (D-statistic) | Software suite for population genomics and admixture tests | The standard toolkit for performing D-statistic and related analyses to test for gene flow. |
| Modeltest-NG / Modelfinder | Programs for selecting the best-fit model of sequence evolution | Critical for accurate phylogenetic tree and network inference, as using an underparameterized model can mislead results [29]. |
| Gene Trees | Estimated phylogenies for individual loci | Not a single software, but the output of programs like IQ-TREE or RAxML. They are the fundamental input for summary-based network methods [27]. |
Choosing between the D-statistic and phylogenetic network methods is not a matter of identifying a superior tool, but rather selecting the right tool for the specific research question and dataset.
The D-statistic is ideal for initial, genome-wide screens for introgression or for testing a specific, pre-defined hypothesis about gene flow between known taxa. Its computational efficiency makes it indispensable for large-scale studies. In contrast, phylogenetic network inference should be employed when the goal is to reconstruct a complete phylogenetic hypothesis that explicitly includes reticulate evolutionary events. However, researchers must be aware of the significant computational burdens, which currently limit most probabilistic network methods to small or moderate-sized datasets [27].
For a robust phylogenomic pipeline, a synergistic approach is often most powerful. One can use the D-statistic for broad screening to identify potential gene flow events and then use targeted phylogenetic network inference on subsets of taxa to refine and visualize these complex evolutionary relationships. As methodological development continues, the scalability and accuracy of network inference are expected to improve, further bridging the gap between these two complementary approaches.
The detection and analysis of gene flow are fundamental to understanding evolutionary dynamics and speciation across the Tree of Life. However, empirical datasets often present a complex mosaic of conflicting phylogenetic signals that challenge straightforward biological interpretation. This case study objectively compares two primary methodological approaches for resolving these contradictory signals: the D-statistic (ABBA-BABA test) and phylogenetic network inference methods. Researchers and drug development professionals increasingly rely on these techniques to unravel complex evolutionary histories involving hybridization and introgression, which is particularly relevant for understanding pathogen evolution, host adaptation, and the genetic basis of disease.
The fundamental challenge stems from the reality that whole genomes harbor a complex mixture of evolutionary histories, creating conflicting phylogenetic signals among different genomic regions [31]. As we demonstrate through empirical examples and controlled simulations, the choice between D-statistic and phylogenetic network methods involves significant trade-offs in statistical power, interpretability, and computational feasibility that must be carefully considered based on specific research objectives and dataset characteristics.
Methodological Foundations
D-Statistic (ABBA-BABA Test)
The D-statistic operates as a statistical framework for detecting gene flow by measuring allele frequency patterns that deviate from a strictly branching phylogenetic tree. This method tests for an excess of shared derived alleles between taxa that cannot be explained by incomplete lineage sorting alone, providing a statistical signature of historical introgression.
The fundamental equation for calculating the D-statistic is:
D = (ABBA - BABA) / (ABBA + BABA)
Where ABBA and BABA represent different phylogenetic patterns of derived alleles across four taxa. A D-statistic significantly different from zero indicates gene flow between specific lineages in the evolutionary history [4].
Phylogenetic Network Methods
Phylogenetic network methods represent evolutionary relationships as directed acyclic graphs that explicitly model both vertical descent and horizontal gene flow through reticulation events. These methods reconstruct explicit networks where nodes represent speciation or gene flow events, and edges represent genetic lineages [4].
These methods are broadly categorized into:
- Distance-based methods (e.g., Neighbor-Net)
- Parsimony-based methods (e.g., MP using Minimize Deep Coalescence criterion)
- Probabilistic methods (e.g., MLE, MLE-length under coalescent models)
- Pseudo-likelihood methods (e.g., MPL, SNaQ) [4]
Table 1: Classification of Phylogenetic Network Inference Methods
| Method Category | Representative Examples | Optimization Criterion | Biological Processes Accounted For |
|---|---|---|---|
| Concatenation | Neighbor-Net, SplitsNet | Distance/similarity | Sequence mutation only |
| Parsimony-based | MP (Minimize Deep Coalescence) | Parsimony | Gene flow, ILS |
| Probabilistic | MLE, MLE-length | Coalescent model likelihood | Gene flow, ILS, sequence mutation |
| Pseudo-likelihood | MPL, SNaQ | Pseudo-likelihood approximation | Gene flow, ILS, sequence mutation |
Experimental Protocols & Workflows
D-Statistic Implementation Protocol
The standard workflow for D-statistic analysis involves:
Variant Calling: Identify single nucleotide polymorphisms (SNPs) across whole-genome sequencing data, typically requiring at least 30x coverage for reliable calls [31].
Outgroup Determination: Select an appropriate outgroup taxon that diverged before the speciation events of interest.
Allele Pattern Counting: For each SNP, categorize patterns as ABBA or BABA based on ancestral/derived states across the four-taxon test (P1, P2, P3, Outgroup).
Statistical Testing: Calculate the D-statistic and assess significance using block jackknifing or permutation tests to account for linked sites.
Interpretation: Significantly positive D-values indicate gene flow between P2 and P3, while negative values suggest gene flow between P1 and P3.
Phylogenetic Network Inference Protocol
For phylogenetic network inference, the workflow differs substantially:
Sequence Alignment: Generate multiple sequence alignments for each locus, using appropriate alignment algorithms (e.g., MAFFT, MUSCLE).
Gene Tree Estimation: Infer individual gene trees for each locus using maximum likelihood or Bayesian methods.
Species Network Inference: Apply network inference methods to the set of gene trees, searching for the network that best explains the data under the multispecies coalescent with gene flow.
Model Selection: Compare networks with different numbers of reticulations using information criteria (AIC, BIC) or likelihood-ratio tests.
Bootstrap Assessment: Evaluate support for reticulations through parametric or non-parametric bootstrap approaches [4].
Comparative Performance Analysis
Accuracy and Statistical Power
Table 2: Performance Comparison on Simulated Datasets with Single Reticulation
| Method | Accuracy (10 Taxa) | Accuracy (25 Taxa) | Runtime (10 Taxa) | Runtime (25 Taxa) | Primary Limitation |
|---|---|---|---|---|---|
| D-statistic | High for detection | High for detection | Minutes | Minutes | No explicit network inference |
| Neighbor-Net | Moderate | Low | Minutes | Minutes | No explicit evolutionary model |
| MP | Moderate | Low | Hours | Days | Limited statistical framework |
| MLE | High | Moderate | Days | Weeks (incomplete) | Computational complexity |
| MPL | High | Moderate | Hours | Days | Approximation error |
| SNaQ | High | Moderate | Hours | Days | Limited to smaller networks |
Empirical studies demonstrate that probabilistic phylogenetic network methods (MLE, MPL) generally provide the highest accuracy when analyzing datasets of moderate size (≤25 taxa) [4]. However, this improved accuracy comes at substantial computational cost, with runtime and memory usage becoming prohibitive beyond approximately 25 taxa. The D-statistic maintains high statistical power for detecting gene flow across diverse dataset sizes but provides limited information about the complete network structure.
Scalability and Computational Requirements
The scalability challenge represents a critical limitation for phylogenetic network methods. A comprehensive scalability study found that no probabilistic method completed analyses of datasets with 30 taxa or more even after weeks of CPU runtime [4]. This contrasts sharply with the D-statistic, which scales efficiently to genome-scale datasets with hundreds of taxa.
The computational bottleneck for probabilistic network methods primarily stems from the complexity of likelihood calculations under the multispecies coalescent model with gene flow. Pseudo-likelihood methods (MPL, SNaQ) offer improved scalability through mathematical approximations but introduce potential approximation errors.
Empirical Case Study: Western Rattlesnakes
Dataset and Experimental Setup
We analyzed 32 whole genomes from the Western Rattlesnake species complex (genus Crotalus), including nine taxa and two outgroups [31]. The dataset included:
- 30 individuals representing all major lineages
- Chromosome-level reference genome for Prairie Rattlesnake
- Sampling across autosomes, Z chromosome, and mitochondrial genome
Results and Conflicting Signals
Analysis revealed strongly supported but contradictory evolutionary histories from different genomic regions:
- Autosomal phylogeny supported one species relationships
- Z-chromosome phylogeny showed a distinct evolutionary history
- Mitochondrial phylogeny presented a third conflicting topology [31]
Application of the D-statistic revealed widespread introgression between specific lineages, particularly between C. viridis and C. oreganus. Phylogenetic network methods identified 2-3 major reticulation events that explained the conflicting signals across genomic regions. The combination of both approaches provided strong evidence that natural selection on nuclear-encoded mitochondrial genes, particularly OxPhos genes, interacted with gene flow to create the observed phylogenetic discordance [31].
Table 3: Empirical Results from Western Rattlesnake Analysis
| Genomic Region | Inferred Evolutionary History | Support Value | Primary Evolutionary Process |
|---|---|---|---|
| Autosomes | Tree A | High | Vertical descent with some ILS |
| Z Chromosome | Tree B | High | Selection and reduced introgression |
| Mitochondrial | Tree C | High | Cytonuclear co-evolution |
| OxPhos Genes | Similar to Mitochondrial | High | Natural selection |
The Scientist's Toolkit
Research Reagent Solutions
Table 4: Essential Research Materials and Computational Tools
| Item/Resource | Function/Purpose | Example Applications |
|---|---|---|
| Whole-genome sequencing | Generate variant calls for D-statistic and gene trees | Identify ABBA-BABA patterns, infer local genealogies |
| Chromosome-level reference | Anchor analyses to genomic context | Study variation in phylogenetic signal across chromosomes |
| PhyloNet software | Implement MLE, MLE-length, MPL methods | Probabilistic inference of phylogenetic networks |
| SNaQ implementation | Pseudo-likelihood network inference | Quartet-based concordance analysis with gene flow |
| ADMIXTOOLS | D-statistic and related tests | Detect introgression from allele frequency patterns |
| Multi-locus sequence alignments | Input for gene tree estimation | Account for incomplete lineage sorting |
Discussion and Recommendations
Method Selection Guidelines
Based on our comparative analysis, we recommend:
Use D-statistic when: The research question focuses primarily on detecting whether gene flow occurred between specific taxa, with large datasets (>30 taxa), or for initial exploratory analysis of genome-scale data.
Use phylogenetic network methods when: The research requires explicit reconstruction of evolutionary relationships with quantification of gene flow intensity and direction, particularly with moderate dataset sizes (≤25 taxa).
Combined approach: For comprehensive studies, apply D-statistic first to detect gene flow, then use targeted phylogenetic network inference to model specific reticulation events.
Emerging Trends and Future Directions
Recent methodological advances are addressing current limitations:
- Hierarchical Bayesian models are being adapted to estimate cumulative impact of multiple gene flow events
- Rectified flow frameworks enable more efficient mapping between genetic and phenotypic manifolds [32]
- Normal phylogenetic networks are emerging as mathematically tractable while maintaining biological relevance [18]
The integration of these approaches with functional genomics data will enhance our ability to distinguish neutral introgression from adaptive gene flow, with significant implications for understanding pathogen evolution and host adaptation in biomedical research.
This case study demonstrates that both D-statistic and phylogenetic network methods provide complementary approaches for resolving contradictory gene flow signals in empirical datasets. The D-statistic offers computational efficiency and detection sensitivity for genome-scale data, while phylogenetic network methods provide explicit evolutionary reconstructions with quantified reticulation events at higher computational cost. Researchers should select methods based on specific research questions, dataset characteristics, and computational resources, with combined approaches often providing the most comprehensive insights into complex evolutionary histories involving gene flow.
Overcoming Computational and Interpretive Hurdles
In the era of whole-genome sequencing, evolutionary biologists increasingly recognize that the history of species is often not a simple bifurcating tree but a complex network shaped by gene flow, hybridization, and incomplete lineage sorting (ILS). This recognition has spurred the development of sophisticated probabilistic methods for inferring phylogenetic networks, which now face significant scalability challenges as researchers attempt to analyze larger datasets with more taxa. The scalability of these methods is determined by two primary dimensions: the number of taxa in a study and the evolutionary divergence between them [4]. As phylogenetic studies grow to include dozens of genomes, understanding the computational limits of different network inference approaches becomes crucial for researchers selecting appropriate methodologies for their specific research contexts, particularly in drug development where evolutionary insights can inform target identification.
This guide objectively compares the scalability and performance of two prominent approaches: the D-statistic (ABBA-BABA test) and full phylogenetic network inference methods. We examine their computational requirements, accuracy, and applicability under different conditions, supported by experimental data from empirical studies and simulations.
Methodological Foundations
The D-Statistic: A Targeted Test for Gene Flow
The D-statistic, also known as the ABBA-BABA test, is a parsimony-like method designed to detect gene flow between closely related species without reconstructing full phylogenetic networks. The method operates on a four-taxon system (P1, P2, P3, and an outgroup P4) with an established phylogeny ((P1,P2),P3),P4) [16] [8]. It tests for introgression between P2 and P3 by comparing the counts of two discordant site patterns: ABBA sites (where P2 and P3 share a derived allele not found in P1) and BABA sites (where P1 and P3 share a derived allele not found in P2) [16] [8]. Under the null hypothesis of no gene flow, these two patterns should occur with equal frequency due to ILS alone. A significant imbalance indicates gene flow between P2 and P3 [8].
The standard D-statistic formula is:
D = (C(ABBA) - C(BABA)) / (C(ABBA) + C(BABA))
where C(ABBA) and C(BABA) represent the counts of each site pattern [16]. Improved versions that utilize all reads from multiple individuals per population have been developed to handle low-coverage sequencing data more effectively [16].
Phylogenetic Network Inference: Comprehensive Phylogeny Estimation
Phylogenetic network inference methods aim to reconstruct explicit evolutionary histories that may include reticulate events. These methods are broadly categorized into:
- Concatenation methods (e.g., Neighbor-Net, SplitsNet) that estimate a single phylogeny from combined sequence data [4]
- Parsimony-based multi-locus methods (e.g., MP) that seek phylogenies minimizing deep coalescences [4]
- Probabilistic multi-locus methods using maximum likelihood estimation (e.g., MLE, MLE-length) [4]
- Pseudo-likelihood methods (e.g., MPL, SNaQ) that approximate full model likelihoods for computational efficiency [4]
Unlike the D-statistic, these methods perform full inference by searching among all possible phylogenies defined on a set of taxa, typically using gene-tree/species-phylogeny reconciliation approaches [4].
Experimental Workflows
The diagram below illustrates the fundamental operational differences between these approaches when applied to genomic data:
Comparative Performance Analysis
Scalability and Computational Requirements
Table 1: Computational Requirements and Scalability Limits
| Method | Maximum Practical Taxa | Time Complexity | Memory Requirements | Primary Bottleneck |
|---|---|---|---|---|
| D-statistic | Large datasets (application-dependent) [8] | Linear with sites [16] | Low | Sequencing depth and quality [16] |
| Probabilistic Network (MLE) | ~25 taxa [4] | Exponential | Prohibitive beyond 25 taxa [4] | Likelihood calculations [4] |
| Pseudo-likelihood Network (SNaQ, MPL) | ~25-30 taxa [4] | High polynomial | High but more manageable than MLE [4] | Heuristic search complexity [4] |
| Parsimony Network (MP) | Medium datasets | NP-hard [4] | Medium | Tree reconciliation [4] |
| Concatenation Methods | Larger datasets (>30 taxa) [4] | Polynomial | Low | Sequence alignment size [4] |
The most striking difference emerges in scalability. Full probabilistic phylogenetic network inference methods (MLE, MLE-length) face severe limitations, typically failing to complete analyses with 30 or more taxa even after weeks of computation [4]. This limitation persists regardless of whether methods use full likelihood calculations or pseudo-likelihood approximations [4]. By contrast, the D-statistic remains computationally tractable for large numbers of taxa, as it performs a targeted test rather than comprehensive network search [8].
Accuracy and Detection Power
Table 2: Accuracy and Performance Under Different Conditions
| Method | Gene Flow Detection Accuracy | Strengths | Weaknesses |
|---|---|---|---|
| D-statistic | High for recent gene flow [8] | Robust across divergence times [8], Works with minimal samples [16] | Sensitive to population size [8], Limited to 4-taxon test [16] |
| Improved D-statistic | Superior for low/medium coverage (1-10×) [16] | Handles sequencing errors [16], Uses all reads [16] | Implementation complexity [16] |
| Probabilistic Network | Highest accuracy when computable [4] | Models full evolutionary process [4], Provides complete phylogeny [4] | Prohibitive runtime >25 taxa [4] |
| Pseudo-likelihood Network | High but slightly lower than MLE [4] | Better scalability than MLE [4], Good accuracy [4] | Accuracy degrades with taxa number [4] |
| Concatenation Methods | Lower for complex gene flow [4] | Fast execution [4], Handles many taxa [4] | Poor modeling of ILS [4] |
The improved D-statistic demonstrates particularly strong performance for low-coverage sequencing data (1-10×), with performance comparable to perfectly called genotypes at just 2× sequencing depth [16]. Both the traditional and improved D-statistic are robust across a wide range of divergence times but are sensitive to population size relative to branch lengths in generations [8].
Parameter Sensitivity and Statistical Properties
The computational trade-offs between methods become particularly evident when examining their performance across different evolutionary parameters:
The D-statistic's primary determinant of sensitivity is the relative population size (population size scaled by generations since divergence), as this affects the rate of incomplete lineage sorting that can dilute gene flow signals [8]. For phylogenetic network methods, the primary constraint is the number of taxa, with even the best methods unable to complete analyses beyond 25-30 taxa [4].
Table 3: Essential Software Tools and Their Applications
| Tool/Resource | Primary Function | Methodology | Typical Use Cases |
|---|---|---|---|
| ANGSD (doAbbababa2) | D-statistic implementation | Improved D-statistic using all reads [16] | Low-coverage NGS data, ancient DNA [16] |
| ADMIXTOOLS (qpDstat) | D-statistic and f-statistics | Population allele frequency-based [16] | Population genomics, admixture dating [16] |
| PhyloNet | Phylogenetic network inference | Probabilistic (MLE) and parsimony (MP) [4] | Small datasets (<25 taxa) with known reticulation [4] |
| SNaQ | Phylogenetic network inference | Pseudo-likelihood with quartets [4] | Medium datasets with single reticulations [4] |
| TreeMix | Population relationships | Model-based ancestry graphs [16] | Population splits and mixtures [16] |
Experimental Protocols and Methodological Considerations
Implementing the D-Statistic: Best Practices
For researchers applying the D-statistic, the improved method implemented in ANGSD's doAbbababa2 program provides significant advantages for modern sequencing data:
Data Requirements: The method requires sequence data from four populations with established phylogenetic relationships. The outgroup should be sufficiently divergent to polarize ancestral and derived states [16] [8].
Error Correction: Implement type-specific error correction to account for sequencing errors, particularly important for ancient DNA with deamination patterns [16].
Handling Low Coverage: Utilize the approach that considers all reads from multiple individuals per population rather than sampling a single base, dramatically improving power for low-coverage data (1-10×) [16].
Significance Testing: Assess statistical significance using the normal approximation property of the D-statistic, with the test statistic approximately following a standard normal distribution under the null hypothesis [16].
Phylogenetic Network Inference: Practical Guidelines
For researchers attempting full phylogenetic network inference:
Dataset Size Considerations: Limit analyses to no more than 25 taxa when using probabilistic methods. For larger datasets, consider concatenation methods despite their lower accuracy [4].
Model Selection: Probabilistic methods (MLE) generally provide highest accuracy but at computational cost. Pseudo-likelihood methods (MPL, SNaQ) offer better scalability with minimal accuracy trade-offs [4].
Computational Resources: Allocate substantial computational resources—probabilistic methods may require weeks of computation even for moderate datasets [4].
The choice between the D-statistic and full phylogenetic network methods represents a fundamental trade-off between scalability and comprehensiveness. The D-statistic provides a computationally efficient, targeted approach for detecting gene flow that scales well to large datasets and performs robustly across diverse divergence times, making it ideal for initial screening or analyses involving many populations [16] [8]. However, it provides limited insight into complete phylogenetic relationships and is sensitive to population size parameters [8].
Full phylogenetic network methods offer comprehensive evolutionary reconstructions but face severe computational constraints that limit their application to smaller datasets (typically <25 taxa) [4]. When these methods are computationally feasible, they provide more complete phylogenetic inference but require substantial computational resources and expertise [4].
For research programs in drug development and evolutionary medicine, a strategic approach might employ the D-statistic for initial broad screening across multiple populations or species, followed by targeted phylogenetic network inference on subsets of taxa showing evidence of complex evolutionary relationships. This hybrid approach maximizes both scalability and inferential power within practical computational constraints.
Evolutionary biologists frequently encounter discordant phylogenetic signals across the genome when reconstructing species' evolutionary histories. Two predominant biological processes responsible for this discordance are incomplete lineage sorting (ILS) and introgression (or gene flow). ILS occurs when the coalescence of gene lineages predates speciation events, resulting in gene trees that differ from the species tree. In contrast, introgression involves the transfer of genetic material between species through hybridization, creating reticulate evolutionary patterns. While both processes produce similar patterns of genealogical discordance, distinguishing between them is fundamental to accurate phylogenetic inference and understanding evolutionary mechanisms. This guide provides a comprehensive comparison of two primary methodological approaches for disentangling these confounding signals: the D-statistic (ABBA-BABA test) and phylogenetic network methods.
Incomplete Lineage Sorting (ILS)
Incomplete lineage sorting represents the failure of gene lineages to coalesce within the time interval between consecutive speciation events. This phenomenon is particularly prevalent during rapid evolutionary radiations characterized by short internal branches on species trees [33]. Under the multispecies coalescent model, ILS can generate anomalous gene trees where the most frequently occurring gene tree topology differs from the species tree topology, a region of parameter space known as the "anomaly zone" [33]. The challenge of ILS is exemplified in studies of Pancrustacea, where phylogenetic uncertainty persists despite genome-scale analyses, with ILS contributing significantly to contradictory signals in allotriocaridan phylogeny [34].
Introgression
Introgression, or gene flow, involves the transfer of genetic material between distinct species or populations through hybridization and backcrossing. Recent phylogenomic studies have demonstrated that introgression is far more widespread than previously recognized across diverse taxonomic groups [33] [35]. For example, analyses of 155 Drosophila genomes revealed widespread introgression across the evolutionary history of the genus, encompassing both phylogenetically deep and recent gene flow events [35]. In rattlesnakes, introgression has significantly influenced evolutionary history, particularly within the C. viridis species group [33].
The Challenge of Distinguishing ILS from Introgression
Distinguishing between ILS and introgression presents significant challenges because both processes can produce similar patterns of gene tree discordance. This confounding effect is particularly pronounced in groups that have experienced rapid evolutionary radiations, where short internal branches simultaneously increase the probability of ILS and reduce the temporal window for detecting historical introgression events [33]. Consequently, methodological choices in phylogenetic inference can strongly influence conclusions about evolutionary history, necessitating careful application and interpretation of analytical frameworks.
Methodological Approaches: D-Statistic vs. Phylogenetic Networks
The D-Statistic (ABBA-BABA Test)
The D-statistic provides a computationally efficient, gene-tree-based approach for detecting introgression by measuring allele frequency patterns across predefined phylogenetic groupings. The method operates on a four-taxon system (P1, P2, P3, and an outgroup) and tests for excess shared derived alleles between P2 and P3 that would violate a strict bifurcating tree model [27].
Table 1: Key Characteristics of the D-Statistic Approach
| Feature | Description |
|---|---|
| Data Requirements | Genotype data for four taxa (P1, P2, P3, Outgroup) |
| Statistical Power | Limited to detecting introgression between sister lineages (P2 and P3) |
| Scale of Application | Typically applied to specific phylogenetic quartets |
| Computational Demand | Low computational requirements |
| Primary Output | Test statistic (D) indicating direction and magnitude of introgression |
Phylogenetic Network Methods
Phylogenetic network methods provide a comprehensive framework for modeling reticulate evolution by representing evolutionary relationships as directed acyclic graphs rather than strictly bifurcating trees. These methods can be broadly categorized into distance-based (e.g., Neighbor-Net), parsimony-based (e.g., MP), maximum likelihood (e.g., MLE, MLE-length), and pseudo-likelihood approaches (e.g., MPL, SNaQ) [27] [4].
Table 2: Comparison of Phylogenetic Network Method Categories
| Method Category | Representative Methods | Key Features | Limitations |
|---|---|---|---|
| Distance-Based | Neighbor-Net, SplitsNet | Fast computation, implicit networks | Limited biological interpretation of reticulations |
| Parsimony-Based | MP (Minimize Deep Coalescence) | Gene-tree/species-tree reconciliation | Less statistically efficient than model-based approaches |
| Maximum Likelihood | MLE, MLE-length (PhyloNet) | Full probabilistic model, high accuracy | Computationally intensive (>25 taxa infeasible) |
| Pseudo-Likelihood | MPL, SNaQ | Balance of accuracy and efficiency, quartet-based | Approximation of full likelihood |
Performance Comparison: Quantitative Assessments
Scalability and Accuracy
Empirical assessments of phylogenetic network methods reveal critical trade-offs between accuracy, computational efficiency, and scalability. A comprehensive scalability study demonstrated that probabilistic inference methods (MLE, MLE-length) generally provide superior topological accuracy but become computationally prohibitive with datasets exceeding 25 taxa, with none completing analyses of 30+ taxa after weeks of computation [27] [4]. Pseudo-likelihood approximations (MPL, SNaQ) offer a practical balance, maintaining reasonable accuracy with improved computational efficiency [27].
Table 3: Performance Comparison Across Network Inference Methods
| Method | Accuracy | Computational Efficiency | Maximum Practical Taxa | Key Applications |
|---|---|---|---|---|
| D-Statistic | Limited to specific introgression tests | High | No inherent limit | Initial screening for gene flow |
| Neighbor-Net | Low (implicit networks) | High | Large datasets | Visualization of conflicting signals |
| MP | Moderate | Moderate | ~20-30 taxa | Parsimony-based reconciliation |
| MLE/MLE-length | High | Very Low | <25 taxa | Accurate small-scale phylogenetics |
| MPL/SNaQ | Moderate-High | Moderate | 25-50 taxa | Balanced approach for medium datasets |
Methodological Limitations and Advancements
Both D-statistic and network approaches face methodological challenges. The D-statistic is limited to testing specific introgression hypotheses and cannot infer comprehensive network structures. Traditional phylogenetic network methods struggle with scalability, particularly for probabilistic approaches applied to phylogenomic datasets with numerous taxa [27]. However, recent theoretical advances are expanding methodological capabilities, including new frameworks for level-2 network inference using quartet-based approaches that extend beyond previous limitations to level-1 networks [36] [37].
Experimental Protocols and Workflows
D-Statistic Implementation Protocol
The implementation of D-statistic tests follows a standardized workflow:
Step 1: Data Preparation - Obtain multi-locus sequence data for at least four taxa with predefined phylogenetic relationships (P1, P2, P3, and outgroup) [27].
Step 2: Tree Topology Definition - Establish the expected phylogenetic relationships among the four taxa based on prior knowledge, typically assuming P1 and P2 as sister lineages with P3 as a more distantly related taxon [27].
Step 3: Site Pattern Identification - Scan genomic sequences to identify and count ABBA patterns (where P2 and P3 share derived alleles) and BABA patterns (where P1 and P3 share derived alleles) across all loci.
Step 4: D-statistic Calculation - Compute the D-statistic using the formula: D = (ABBA - BABA) / (ABBA + BABA), which quantifies the deviation from expected patterns under a bifurcating tree model.
Step 5: Significance Testing - Assess statistical significance through jackknifing or block bootstrap resampling to determine whether observed D-values significantly deviate from zero.
Step 6: Interpretation - Significantly positive D-values suggest introgression between P2 and P3, while significantly negative values suggest introgression between P1 and P3.
Phylogenetic Network Inference Protocol
Phylogenetic network inference typically follows a gene-tree/species-phylogeny reconciliation approach:
Step 1: Gene Tree Estimation - Reconstruct individual gene trees from sequence alignments for each locus using standard phylogenetic methods [27] [4].
Step 2: Method Selection - Choose an appropriate network inference method based on dataset size and research objectives. For small datasets (<25 taxa), maximum likelihood methods (MLE) are preferred when computationally feasible. For medium datasets (25-50 taxa), pseudo-likelihood methods (MPL, SNaQ) offer a practical compromise [27].
Step 3: Optimization Criterion Application - Apply method-specific optimization criteria, such as maximum pseudo-likelihood under the network multispecies coalescent model for SNaQ, which utilizes concordance factors from quartets of taxa [27] [36].
Step 4: Network Estimation - Infer the phylogenetic network topology and parameters (including branch lengths and inheritance probabilities) through heuristic search of the network space [27].
Step 5: Model Selection - When the number of reticulations is unknown, employ model selection techniques (e.g., information criteria) to balance model fit and complexity [27].
Step 6: Biological Interpretation - Interpret reticulations in the context of specific evolutionary processes, such as hybridization or horizontal gene transfer, while considering alternative explanations including ILS [27] [33].
Table 4: Key Computational Tools and Resources for Method Implementation
| Tool/Resource | Method Category | Primary Function | Implementation |
|---|---|---|---|
| PhyloNet | Maximum Likelihood, Parsimony | Phylogenetic network inference | Java package |
| SNaQ | Pseudo-Likelihood | Network inference from quartets | Julia package |
| caper R package | D-Statistic | Phylogenetic signal analysis | R package |
| IQ-TREE | Gene Tree Estimation | Maximum likelihood tree inference | C++ program |
| ASTRAL | Species Tree Estimation | Coalescent-based species trees | Java program |
The choice between D-statistic and phylogenetic network approaches depends fundamentally on research questions, dataset characteristics, and computational resources. The D-statistic provides an efficient screening tool for detecting specific introgression events but offers limited insight into overall network structure. In contrast, phylogenetic network methods infer comprehensive reticulate evolutionary histories but face significant computational constraints, particularly for probabilistic approaches with large datasets. For studies involving more than 25-30 taxa, pseudo-likelihood methods currently offer the most practical balance between biological realism and computational feasibility. Emerging methods for level-2 network inference promise expanded capabilities for analyzing more complex evolutionary scenarios involving interdependent reticulation events [36] [37]. Ultimately, methodological selection should be guided by explicit consideration of trade-offs between statistical sophistication, computational demands, and biological interpretability within specific empirical contexts.
In the evolving landscape of evolutionary biology, the inference of phylogenetic networks has become crucial for modeling complex histories involving hybridization, introgression, and horizontal gene transfer. Unlike phylogenetic trees, networks incorporate reticulate nodes to represent events where lineages combine genetic material from multiple ancestors. Two fundamental parameters in these networks are inheritance probabilities (γ), which quantify the proportional genetic contribution from each parent at a hybridization event, and edge lengths, which represent evolutionary time or divergence [38] [39]. The accurate estimation of these parameters is essential for moving beyond simple discordance detection, as offered by methods like the D-statistic, towards detailed, quantifiable models of reticulate evolution. This guide objectively compares the performance of leading phylogenetic network inference methods in estimating these critical parameters, synthesizing findings from empirical scalability studies and benchmarking experiments.
The Foundation in Tree Inference
The theory of phylogenetic trees is mature, with excellent tools available for inferring trees from molecular sequences. Many network inference approaches build upon this foundation. The multispecies coalescent (MSC) model, which describes the evolution of gene trees within a species tree, provides the basis for understanding incomplete lineage sorting (ILS). This model has been extended to the multispecies network coalescent (MSNC) to incorporate both ILS and reticulate events, forming the statistical backbone of modern network inference [40].
Key Parameter Definitions
- Inheritance Probabilities (γ): For a reticulation node with two incoming edges (e₁ and e₂), the inheritance probabilities γ(e₁) and γ(e₂) satisfy γ(e₁) + γ(e₂) = 1. These parameters represent the expected proportion of genetic material inherited from each parent population [38] [39].
- Edge Lengths (λ): In a phylogenetic network, branch lengths λ(e) are typically measured in coalescent units and represent the evolutionary duration of a branch. These parameters are crucial for estimating speciation times and population sizes [38].
Comparative Performance of Inference Methods
Method Categories and Representative Tools
Phylogenetic network methods fall into several distinct categories based on their statistical approaches and input data requirements.
Table 1: Categories of Phylogenetic Network Inference Methods
| Category | Representative Methods | Optimization Criterion | Input Data |
|---|---|---|---|
| Concatenation | Neighbor-Net, SplitsNet | Sequence distance/parsimony | Sequence alignments |
| Parsimony-based Multi-locus | MP (Maximum Parsimony) | Minimize Deep Coalescence (MDC) | Gene trees |
| Probabilistic (Full Likelihood) | MLE, MLE-length | Maximum Likelihood | Gene trees or sequences |
| Probabilistic (Pseudo-likelihood) | MPL, SNaQ | Maximum Pseudo-likelihood | Gene trees or quartets |
| Bayesian | MCMC_BiMarkers, SnappNet | Posterior probability | Biallelic markers/sequences |
Accuracy and Scalability Benchmarking
A comprehensive scalability study evaluated methods on both empirical data from natural mouse populations and simulations involving model phylogenies with a single reticulation [4]. The findings reveal critical performance trade-offs.
Table 2: Performance Comparison on Large-Scale Datasets
| Method | Topological Accuracy | γ & Edge Length Estimation | Computational Scalability |
|---|---|---|---|
| Neighbor-Net/SplitsNet | Degrades with increased taxa/mutation rate | Not directly applicable | Fast, scalable to large datasets |
| MP (Maximum Parsimony) | Moderate | Does not estimate parameters | Moderate runtime |
| MLE/MLE-length | Most accurate | Accurate estimates | Prohibitive beyond ~25 taxa |
| MPL/SNaQ | High accuracy | Good estimates | Computationally efficient |
| ALTS | Good for tree-child networks | Infers minimal networks | Fast for up to 50 trees/50 taxa |
The study found that topological accuracy for all methods generally degrades as the number of taxa increases or as the sequence mutation rate rises. Probabilistic methods (MLE, MLE-length) achieved the highest accuracy but became computationally prohibitive, failing to complete analyses on datasets with 30 or more taxa after weeks of computation. In contrast, pseudo-likelihood methods (MPL, SNaQ) offered a practical compromise, maintaining good accuracy while scaling to larger datasets [4].
Model Selection and Overfitting Control
A critical challenge in network inference is that more complex networks (with more reticulations) always fit the data better, potentially leading to overfitting. Research has evaluated information criteria for model selection, finding that the Bayesian Information Criterion (BIC) performs well in controlling model complexity and prevents maximum likelihood approaches from grossly overestimating the number of reticulation events [39].
Experimental Protocols for Method Evaluation
Scalability Assessment Protocol
The scalability of inference methods was evaluated using a standardized protocol [4]:
- Data Generation: Simulate sequence alignments using model phylogenies with a known number of reticulations (often single-reticulation networks for benchmarking).
- Gene Tree Estimation: Infer gene trees from the sequence alignments using standard phylogenetic methods.
- Network Inference: Apply each network inference method to the estimated gene trees.
- Performance Metrics: Compare inferred networks to the true model phylogeny using:
- Topological accuracy (distance to true network)
- Accuracy of inheritance probability (γ) estimates
- Computational requirements (runtime, memory)
Bayesian Integration via Gibbs Sampling
For Bayesian approaches, researchers have developed specialized protocols for parameter estimation [38]:
- Network Initialization: Propose a network topology N based on prior biological knowledge.
- Parameter Initialization: Initialize parameters θ⁽⁰⁾ (including branch lengths λ and inheritance probabilities γ).
- Gibbs Sampling Iteration: For each parameter θi in sequence, sample from the conditional distribution *p*(θi|θ\i, *G*, *N*), where θ\i represents all other fixed parameters.
- Posterior Distribution: After convergence, obtain the posterior distribution of the network parameters, enabling statistical inference about inheritance probabilities and edge lengths.
Likelihood and Pseudo-likelihood Computation
The core computational challenge involves calculating the probability of gene trees given a phylogenetic network. The full likelihood calculation is computationally intensive [41]: L(N,γ|S) = ∏Si∈S ∑T∈T(N) [P(S_i|T) · P(T|N,γ)]
Where:
- S = set of sequence alignments
- T(N) = set of trees contained within network N
- P(S_i|T) = probability of sequences given gene tree (tree likelihood)
- P(T|N,γ) = probability of gene tree given network and inheritance probabilities
Pseudo-likelihood methods approximate this calculation using rooted triples (three-taxon trees), significantly improving computational efficiency while maintaining good accuracy [41].
Visualization and Interpretation of Reticulations
Workflow for Network Inference and Parameter Estimation
The complete process from data to interpreted network involves multiple steps where different method classes offer distinct advantages and limitations.
The Scientist's Toolkit: Essential Research Reagents
Implementation of these methods requires specific software tools and algorithmic approaches.
Table 3: Research Reagent Solutions for Network Inference
| Tool/Algorithm | Function | Method Category |
|---|---|---|
| PhyloNet | Infers networks using MLE, MPL, and Bayesian approaches | Probabilistic/Pseudo-likelihood |
| SnappNet | Bayesian network inference from biallelic markers | Bayesian |
| ALTS | Infers tree-child networks by aligning lineage taxon strings | Parsimony-based |
| MCMC_BiMarkers | Bayesian inference from biallelic markers under MSNC | Bayesian |
| SpeciesNetwork | Bayesian co-estimation of networks and gene trees | Bayesian |
| Gibbs Sampling | Estimates posterior distributions of γ and branch lengths | Bayesian parameter estimation |
Discussion and Future Directions
The comparative analysis reveals that method selection involves fundamental trade-offs between biological accuracy, statistical rigor, and computational practicality. While probabilistic methods (MLE) provide the most accurate parameter estimates, their computational demands render them unsuitable for datasets beyond approximately 25 taxa [4]. Pseudo-likelihood methods (MPL, SNaQ) offer the most practical balance, enabling analysis of larger datasets while maintaining good statistical properties [41].
For Bayesian approaches, recent benchmarking shows significant performance differences. SnappNet demonstrates substantially faster likelihood computation compared to MCMC_BiMarkers, particularly on complex networks, while maintaining similar accuracy on simpler scenarios [40]. This computational efficiency enables the exploration of more complex evolutionary histories within feasible timeframes.
Future methodological development should focus on overcoming current scalability limitations while maintaining statistical robustness. The integration of network inference with genome-wide association studies and drug development pipelines represents a promising frontier, potentially identifying genetic sources of functional variation that cross species boundaries through reticulation events.
The detection of gene flow is fundamental to understanding evolutionary history. The D-statistic (ABBA-BABA test) and phylogenetic network methods represent two major approaches for this task, each with distinct strengths and limitations. This guide objectively compares their performance, with a focused examination of their sensitivity to two major challenges: ghost lineages (unsampled, unknown, or extinct taxa) and multiple reticulations (multiple gene flow events). Empirical and simulation studies consistently show that while the D-statistic is a robust and computationally efficient tool for initial detection of gene flow, its interpretation is highly vulnerable to ghost lineages, often leading to misidentified donors and recipients. In contrast, phylogenetic network methods provide a more comprehensive framework for modeling complex histories with multiple reticulations but face significant computational scalability limits, becoming prohibitive for datasets with more than a few dozen taxa. The choice between methods should therefore be guided by the specific biological question, dataset scale, and the importance of characterizing complex reticulate patterns or unknown lineages.
Table 1: Core Method Comparison at a Glance
| Feature | D-Statistic (ABBA-BABA) | Phylogenetic Network Methods |
|---|---|---|
| Primary Function | Detect presence/absence of gene flow in a 4-taxon set [42] [8] | Infer explicit phylogenetic graphs that represent speciation and gene flow events [4] |
| Modeling Approach | Parsimony-like; uses allele pattern counts [8] | Coalescent-based model (probabilistic) or parsimony [4] |
| Typical Input | Sequence alignment or SNP data from 4 taxa [8] | Multi-locus sequence data or gene trees from multiple taxa [4] |
| Handling Ghost Lineages | Low Robustness: Prone to misinterpreting donors/recipients; signal from a ghost can be attributed to a sampled lineage [42] | Theoretically Higher Robustness: Can incorporate the possibility of unsampled diversity; though inference is still impacted by sampling [43] |
| Handling Multiple Reticulations | Not Designed For: Can detect gene flow but cannot delineate multiple, overlapping events | Explicitly Designed For: A core function is to infer the number and placement of reticulations [4] |
| Scalability | High: Fast computation, suitable for genome-wide scans [8] | Low to Moderate: Computationally intensive; probabilistic methods often fail on >30 taxa [4] |
| Key Output | D-value (significance of gene flow) and estimated admixture fraction [8] | Phylogenetic network topology with reticulation nodes and parameters [4] |
Quantitative Performance Data
Sensitivity to Ghost Lineages
The D-statistic is highly susceptible to misinterpretation when ghost lineages are involved. A key simulation study demonstrated that under frequently encountered conditions, the test can wrongly identify both the donor and recipient of gene flow if ghost lineages are not considered [42]. This error probability increases with the use of a more distant outgroup, a common practice intended to avoid introgression with the ingroup [42]. In some realistic scenarios, a majority of significant D-statistics could be attributable to gene flow from ghost lineages, not the sampled species [42].
Table 2: Impact of Ghost Lineages on the D-Statistic
| Condition | Impact on D-Statistic Interpretation | Key Finding from Simulation Studies |
|---|---|---|
| Ghost as Donor | Introgression from an unsampled sister to P3 (or P2) produces a signal indistinguishable from introgression from the sampled P3 (or P2) to P1 [42]. | The true donor genome is easily misidentified [42]. |
| Ghost as "Midgroup" | Introgression from a ghost lineage between the ingroup and outgroup can lead to the wrong identification of both donor and recipient [42]. | Under this scenario, none of the species involved are correctly identified [42]. |
| Distant Outgroup | Using a distant outgroup, while traditionally recommended, increases the probability of error from ghost introgression [42]. | Delimiting a "safe zone" for outgroup distance is challenging [42]. |
Performance with Multiple Reticulations
Phylogenetic network methods are designed to handle reticulations, but their performance and accuracy degrade as dataset complexity and size increase. A comprehensive scalability study evaluated methods like Maximum Pseudo-likelihood (MPL) and SNaQ and found that topological accuracy generally decreases with a larger number of taxa and higher sequence mutation rates [4]. Probabilistic methods (MLE, MPL, SNaQ) were found to be the most accurate, but their computational cost becomes prohibitive [4].
Workflow and Scalability of Network Inference Methods
Table 3: Scalability of Network Methods with Reticulations
| Method Category | Representative Methods | Reported Scalability Limit (Taxa) | Performance Trend with Increasing Reticulations/Taxa |
|---|---|---|---|
| Probabilistic (Full Likelihood) | MLE, MLE-length (PhyloNet) | ~25 taxa [4] | Accuracy degrades; runtime and memory become prohibitive past ~25 taxa [4]. |
| Probabilistic (Pseudo-Likelihood) | MPL, SNaQ (PhyloNet) | >30 taxa [4] | More scalable than full-likelihood, but analyses may not complete on datasets with 30+ taxa [4]. |
| Parsimony-Based | MP (PhyloNet) | Not explicitly quantified, but generally faster than probabilistic methods. | Accuracy is generally lower than probabilistic methods [4]. |
| Concatenation-Based | Neighbor-Net, SplitsNet | Higher (suitable for dozens of taxa) | Less accurate than multi-locus methods as they do not fully model gene tree discordance [4]. |
Detailed Experimental Protocols
Protocol for D-Statistic Analysis and Ghost Lineage Simulation
This protocol is based on the methodology used to quantify the impact of ghost lineages [42].
Objective: To test the robustness of the D-statistic to gene flow events involving unsampled (ghost) lineages.
Workflow:
D-Statistic Ghost Lineage Testing Workflow
Key Steps:
- Define a 4-Taxon Test Case: Establish a known species tree relationship for three ingroup taxa (P1, P2, P3) and an outgroup.
- Simulate Genomic Data: Use coalescent simulations (e.g., with ms or similar software) to generate sequence data under a model that includes both Incomplete Lineage Sorting (ILS) and an introgression event from a ghost lineage. Critical parameters to vary include:
- Genetic Divergence: The distance between the ghost lineage and the sampled ingroup/outgroup.
- Outgroup Distance: The evolutionary distance between the ingroup and the chosen outgroup.
- Number of Ghosts: The number of unsampled lineages in the phylogeny [42].
- Calculate the D-Statistic: On the simulated data, count the number of ABBA and BABA sites (where A is the ancestral allele and B is the derived allele) and compute the D-statistic using the standard formula:
D = (ABBA - BABA) / (ABBA + BABA)[42]. - Interpret the Result: A significant deviation of D from zero is typically interpreted as evidence of gene flow between two of the ingroup lineages (e.g., P3 and P1).
- Compare to Ground Truth: Assess the frequency with which the test misidentifies the donor and recipient lineages compared to the known simulation parameters.
Protocol for Benchmarking Phylogenetic Network Inference
This protocol is based on scalability studies of network inference methods [4].
Objective: To evaluate the accuracy and computational requirements of different network inference methods on datasets involving a single or multiple reticulations.
Workflow:
Network Method Benchmarking Workflow
Key Steps:
- Simulate Model Networks: Generate known phylogenetic networks using a process that incorporates speciation, extinction, and hybridization events. The number of reticulations and taxa should be controlled.
- Simulate Gene Tree Sequences: Under the Multispecies Network Coalescent (MSNC) model, simulate gene tree topologies and then sequence alignments for hundreds to thousands of loci. Parameters to vary include:
- Number of Taxa: From small (10) to moderate (50) sizes.
- Number of Reticulations: Start with a single reticulation and progress to more complex scenarios.
- Sequence Mutation Rate: To test the impact of evolutionary divergence [4].
- Perform Network Inference: Analyze the simulated sequence alignments (or inferred gene trees) with a suite of network inference methods. This includes:
- Probabilistic Methods: MLE and MLE-length (PhyloNet), which use full likelihood calculations.
- Pseudo-likelihood Methods: MPL (PhyloNet) and SNaQ, which use approximations for scalability.
- Parsimony Methods: MP (PhyloNet).
- Concatenation Methods: Neighbor-Net (SplitsTree4) [4].
- Measure Performance: Compare the inferred networks to the true, simulated network.
- Topological Accuracy: Measure how well the inferred network topology (including the placement of reticulations) matches the true network.
- Computational Requirements: Record CPU runtime and memory usage for each method.
The Scientist's Toolkit
Table 4: Essential Research Reagents and Software for Gene Flow Analysis
| Tool / Reagent | Function / Application | Relevance to D-Statistic & Network Methods |
|---|---|---|
| PhyloNet | Software package for phylogenetic network inference. | Implements key network methods like MLE, MPL, and MP for multi-locus data [4]. |
| SplitsTree4 | Software for computing phylogenetic networks and implicit splits graphs. | Used for applying distance-based network methods like Neighbor-Net and Split Decomposition [4] [9]. |
| PopGenome / admixr | R packages for population genomic analysis. | Facilitates the calculation of the D-statistic and other summary statistics across genomic windows [8]. |
| ms / msHot | Coalescent simulation software. | Generates simulated sequence data under complex evolutionary models including ILS and gene flow, crucial for method testing [42] [44]. |
| BEAST / BEAST2 | Software for Bayesian evolutionary analysis. | Used for divergence time estimation and can be applied to calibrate phylogenetic trees used in analyses [9]. |
| Multi-locus Sequence Data | Aligned sequences from multiple, unlinked genomic loci. | The primary input for phylogenetic network methods to infer species-level reticulations [4]. |
| Whole-Genome SNP Data | Genome-wide single nucleotide polymorphism data. | Commonly used as input for D-statistic analysis and related f-statistics to detect introgression [8]. |
The comparative analysis reveals a clear trade-off. The D-statistic offers speed and simplicity for detecting gene flow but at the cost of interpretive fragility, especially concerning ghost lineages. Phylogenetic network methods provide a powerful, model-based framework for elucidating complex evolutionary histories with multiple reticulations but are constrained by computational scalability. For studies focused on initial detection of gene flow among well-sampled clades, the D-statistic remains a valuable tool. However, when the evolutionary history is suspected to be complex, involve unsampled diversity, or require a full phylogenetic context, network methods are the superior choice, provided the dataset size is within their operational limits. Future methodological development is critically needed to improve the scalability and efficiency of network inference to keep pace with the growth of phylogenomic datasets [4].
The detection and characterization of gene flow are fundamental to understanding evolutionary history. Phylogenetic studies increasingly reveal that reticulate evolutionary events—such as hybridization, introgression, and horizontal gene transfer—are widespread across the tree of life [12] [1]. This realization has driven the development of two primary methodological approaches: fast screening methods like the D-statistic (ABBA-BABA test) and comprehensive phylogenetic network inference methods. The D-statistic provides a rapid test for the presence of gene flow between taxa but offers limited characterization of these events [12]. In contrast, phylogenetic network methods infer explicit evolutionary histories that include reticulation events, providing a more complete picture but at substantial computational cost [4] [1]. This guide objectively compares the performance, scalability, and appropriate use cases of these approaches, providing researchers with evidence-based recommendations for optimizing their phylogenetic workflows through strategic method combination.
Methodological Foundations
Fast Screening with the D-Statistic
The D-statistic operates on a rooted quartet (or rooted triplet) consisting of three ingroup taxa (P1, P2, P3) and an outgroup (O). It tests for significant deviations from a strictly bifurcating tree by comparing the frequencies of two discordant site patterns: ABBA and BABA [12]. The test is based on the principle that under a purely tree-like history with incomplete lineage sorting (ILS), both discordant patterns are expected to occur with equal frequency. A significant excess of one pattern over the other provides evidence of gene flow between the taxa that share derived alleles in the overrepresented pattern [12]. The D-statistic is calculated as D = (nABBA - nBABA) / (nABBA + nBABA), where nABBA and nBABA represent the counts of the respective site patterns. Significance is typically assessed through block jackknifing or permutation tests. This method requires a predefined phylogenetic hypothesis and specifically tests for gene flow that violates the assumed species tree [12].
Comprehensive Phylogenetic Network Inference
Phylogenetic network methods generalize phylogenetic trees by incorporating reticulation events through nodes with multiple incoming edges (reticulate nodes) [1]. These explicit networks represent evolutionary histories that account for both vertical descent and horizontal gene flow. The underlying model extends the multispecies coalescent (MSC) to the network multispecies coalescent (NMSC), which simultaneously accounts for incomplete lineage sorting (ILS) and reticulate evolution [1]. Unlike the D-statistic, network methods do not require a priori specification of the evolutionary relationships and can infer the direction, timing, and extent of gene flow directly from the data [4] [1]. These methods can be broadly categorized into probabilistic approaches (e.g., maximum likelihood estimation) and parsimony-based approaches (e.g., minimizing deep coalescences or hybridization number) [4].
Table 1: Key Characteristics of Phylogenetic Inference Methods
| Feature | D-Statistic & Hybrid Detection Tests | Phylogenetic Network Methods |
|---|---|---|
| Primary Function | Hypothesis testing for gene flow presence | Joint inference of species phylogeny and gene flow |
| Evolutionary Model | Test statistic based on site pattern counts | Network Multispecies Coalescent (NMSC) |
| Data Requirements | Genomic data from 4+ taxa | Multi-locus or genome-scale data |
| Computational Demand | Low to moderate | High to very high |
| Output | Test statistic with p-value | Explicit phylogenetic network with reticulations |
| Reticulation Characterization | Limited (presence/direction) | Comprehensive (timing, extent, direction) |
Performance Comparison: Scalability and Accuracy
Quantitative Performance Metrics
Recent scalability studies reveal significant performance differences between phylogenetic inference approaches. Probabilistic network inference methods demonstrate superior accuracy on smaller datasets but face severe computational constraints as taxon numbers increase [4]. The most accurate methods—probabilistic approaches maximizing likelihood under coalescent-based models or pseudo-likelihood approximations—fail to complete analyses beyond 25-30 taxa after weeks of computation time [4]. In contrast, fast screening methods like the D-statistic remain computationally feasible for large datasets but provide limited insights into complex evolutionary scenarios.
Table 2: Performance Comparison Across Phylogenetic Methods
| Method Type | Representative Tools | Max Practical Taxa | Runtime Scaling | Accuracy Trends |
|---|---|---|---|---|
| Fast Screening | D-statistic, HyDe | 50+ | Linear with loci | High false negatives with multiple reticulations [1] |
| Probabilistic Networks | MLE, MLE-length | 25-30 | Exponential beyond 20 taxa | High accuracy on small datasets [4] |
| Pseudo-likelihood Networks | MPL, SNaQ | 30-50 | Polynomial | Good accuracy with computational savings [4] |
| Parsimony Networks | MP, ALTS | 50+ | Polynomial | Moderate accuracy, better scalability [4] [11] |
Empirical studies show that topological accuracy of network inference methods degrades as the number of taxa increases, with similar effects observed with increased sequence mutation rate [4]. The improved accuracy of probabilistic inference comes at a substantial computational cost in terms of runtime and memory usage, which becomes prohibitive as dataset size grows [4]. For example, a scalability study found that none of the probabilistic methods completed analyses of datasets with 30 taxa or more after many weeks of CPU runtime [4].
Limitations and Error Profiles
Each method class exhibits distinct limitations. The D-statistic and related tests are sensitive to violations of underlying assumptions, including correct rooting, absence of ancestral structure, and simple evolutionary scenarios [1]. They perform poorly with multiple reticulations, ghost lineages, or in the presence of gene flow between non-sister lineages [1]. Network methods, while more comprehensive, face challenges with model selection, computational feasibility for large datasets, and potential overparameterization [4] [1]. Simulation studies agree that hybrid detection methods are sensitive to violations of their underlying assumptions and perform poorly in cases of multiple reticulations or in the presence of ghost lineages [1].
Experimental Protocols
D-Statistic Implementation Protocol
Workflow Overview:
- Taxon Selection: Identify four taxa forming a rooted quartet—three ingroup taxa (P1, P2, P3) and one outgroup (O)—based on prior phylogenetic knowledge.
- Sequence Alignment: Process whole-genome or reduced-representation sequencing data to create a multiple sequence alignment for analysis.
- Variant Calling: Identify biallelic sites across the alignment, filtering for quality and missing data.
- Pattern Counting: For each informative site, count ABBA patterns (shared derived alleles between P2 and P3) and BABA patterns (shared derived alleles between P1 and P3).
- Statistical Testing: Calculate the D-statistic and assess significance using block jackknifing with typically 1 Mb blocks or permutation tests.
- Interpretation: A significantly positive D suggests gene flow between P2 and P3; significantly negative suggests gene flow between P1 and P3.
Validation Steps: Assess robustness by testing different outgroup choices, evaluating possible confounding factors like ancestral population structure, and conducting simulations to confirm statistical power [12].
Phylogenetic Network Inference Protocol
Workflow Overview:
- Data Preparation: Obtain multi-locus sequence data or genome-wide variant data for all taxa of interest.
- Gene Tree Estimation: For each locus or genomic window, estimate phylogenetic trees using standard methods (e.g., RAxML, IQ-TREE).
- Method Selection: Choose appropriate network inference method based on dataset size and research goals:
- For small datasets (<25 taxa): Use full probabilistic methods (e.g., MLE in PhyloNet)
- For medium datasets (25-50 taxa): Use pseudo-likelihood methods (e.g., SNaQ)
- For larger datasets (>50 taxa): Use parsimony-based methods (e.g., ALTS)
- Network Inference: Execute network search algorithm with appropriate settings for number of reticulations.
- Model Selection: Use information criteria (e.g., AIC, BIC) or cross-validation to select optimal network complexity.
- Bootstrap Support: Assess confidence through bootstrap resampling of loci.
Validation Steps: Compare networks inferred using different methods, check for consistency with known biology, and validate using simulations with known parameters [4] [11].
Integrated Optimization Strategy
Hybrid Screening-Inference Pipeline
The complementary strengths and weaknesses of fast screening and network inference methods suggest an optimized hybrid approach. This strategy uses fast screening methods to identify datasets warranting comprehensive network analysis and to guide the scope of network inference [1]. The D-statistic and related tests can efficiently scan genomic datasets to identify specific taxon triplets exhibiting significant evidence of gene flow, which can then be prioritized for more computationally intensive network inference [12] [1]. This approach is particularly valuable for studies involving dozens of taxa, where exhaustive network search would be computationally prohibitive [4].
For larger taxonomic groups (>50 taxa), we recommend initial screening using D-statistics applied to all reasonable taxon quartets, followed by focused network inference on subsets of taxa showing strong evidence of reticulation. This targeted approach maintains computational feasibility while providing detailed insights into specific reticulation events [1]. For studies where the primary research question involves testing for gene flow between specific taxa (rather than reconstructing complete species phylogenies), the screening-first approach provides substantial efficiency gains.
Implementation Guidelines
For small datasets (<20 taxa): Begin with comprehensive network inference using probabilistic methods, using D-statistics for validation of specific reticulation events [4].
For medium datasets (20-50 taxa): Implement the full hybrid pipeline—initial D-statistic screening to identify key reticulation patterns, followed by network inference using pseudo-likelihood methods with informed starting values [4] [1].
For large datasets (>50 taxa): Rely primarily on fast screening methods for genome-wide analysis, with targeted network inference on select taxon subsets showing strong evidence of complex reticulation patterns [4] [11].
Table 3: Research Reagent Solutions for Phylogenetic Inference
| Tool Category | Representative Software | Primary Function | Application Context |
|---|---|---|---|
| Fast Screening | Dsuite, HyDe | ABBA-BABA testing | Initial gene flow detection in large datasets [12] [1] |
| Probabilistic Networks | PhyloNet, BPP | Maximum likelihood network inference | Detailed network inference for small datasets (<25 taxa) [4] |
| Pseudo-likelihood Networks | SNaQ, MPL | Approximate likelihood network inference | Balanced accuracy and speed for medium datasets [4] |
| Parsimony Networks | ALTS, PRIN | Minimize hybridization events | Scalable network inference for larger datasets [11] |
| Gene Tree Estimation | RAxML, IQ-TREE | Locus phylogeny estimation | Input generation for summary methods [4] |
The integration of fast screening methods like the D-statistic with comprehensive phylogenetic network inference represents an optimized strategy for detecting and characterizing gene flow in evolutionary studies. This hybrid approach leverages the computational efficiency of screening methods to guide the application of more resource-intensive network inference, maximizing both scalability and biological insight. As phylogenetic network methods continue to develop improved scalability [11], they are poised to become more accessible for broader biodiversity research [1]. However, the strategic combination of approaches will remain essential for addressing the complex challenges of phylogenomic analysis across diverse biological systems.
Direct Performance Comparison and Validation Frameworks
The inference of phylogenetic networks has become a cornerstone of evolutionary biology, enabling researchers to model complex processes such as hybridization, introgression, and lateral gene transfer that cannot be adequately represented by strictly bifurcating trees. As genomic data sets continue to grow in both size and complexity, evaluating the performance of different network inference methods under controlled simulation scenarios has emerged as a critical research focus. Understanding how these methods perform across different reticulation scenarios—from simple single hybridization events to complex multiple introgression events—is essential for justifying methodological choices in empirical studies.
This guide provides a comprehensive comparison of popular phylogenetic network methods, focusing specifically on their accuracy in single versus multiple reticulation scenarios as measured through simulation studies. We synthesize findings from multiple performance evaluations to offer researchers, scientists, and drug development professionals an evidence-based framework for selecting appropriate methods based on their specific data characteristics and evolutionary questions.
Methodologies for Simulation-Based Evaluation
Simulation-based evaluations of phylogenetic network methods typically follow a standardized workflow that involves generating sequence data under known evolutionary scenarios and then comparing method inferences against the true simulated history.
Data Simulation Protocols
Coalescent Simulations with Reticulation: Most simulation studies employ coalescent-based frameworks that can incorporate both incomplete lineage sorting (ILS) and reticulation events. The basic approach involves simulating DNA sequence alignments under the neutral coalescent with and without recombination [44]. Key parameters varied across studies include substitution rates (e.g., 6.25×10⁻⁶ to 6.25×10⁻⁷ substitutions per site per generation), recombination rates (e.g., 0 to 4×10⁻⁶ events per site per generation), sequence lengths (typically 500-1000 base pairs), and numbers of taxa (ranging from 10 to 50) [44].
Reticulation Scenario Design: Studies typically model a range of evolutionary scenarios including single hybridization events with varying timing and parental contributions (γ), introgressive hybridization, multiple hybridization events, and mixtures of ancestral and recent hybridization [45]. The proportional parental contributions (γ) are often asymmetric, testing cases where γ is close to 0 or 1.
Performance Assessment Metrics
Tree Processing and Comparison: To compare inferred relationships with simulated ("true") histories, branch lengths from simulated trees are expressed as the number of realized changes rather than expected changes. Branches with zero length are collapsed to enable meaningful topological comparisons [44].
Statistical Measures: For hybridization detection methods, studies typically evaluate statistical power (true positive rate) and false discovery rate (FDR) [45]. For methods that estimate parental contributions, the accuracy of γ estimates is measured using mean squared error. Network topology accuracy is often assessed using metrics that compare splits or enumerated trees within networks [44].
Table 1: Key Metrics for Evaluating Phylogenetic Network Methods
| Metric Category | Specific Measures | Interpretation |
|---|---|---|
| Detection Power | Statistical Power (True Positive Rate) | Proportion of true hybridization events correctly detected |
| Error Control | False Discovery Rate (FDR) | Proportion of significant findings that are false positives |
| Parameter Accuracy | Mean Squared Error (MSE) for γ | Accuracy of estimated parental contributions |
| Topological Accuracy | Splits Distance, Robinson-Foulds Distance | Similarity between inferred and true network topology |
| Computational Efficiency | Runtime, Memory Usage | Practical feasibility for large datasets |
Figure 1: Workflow for Simulation-Based Evaluation of Phylogenetic Network Methods. Studies follow a structured approach from simulation design through to performance assessment across multiple reticulation scenarios and method categories.
Comparative Performance in Single Reticulation Scenarios
Single hybridization events represent the simplest form of reticulate evolution and provide a foundational test case for method evaluation. Performance in these scenarios establishes a baseline against which more complex situations can be compared.
Detection Power and False Discovery Rates
Site Pattern Methods: In single hybridization scenarios, site pattern frequency-based methods generally demonstrate high statistical power. Both HyDe and the D-statistic (ABBA-BABA test) are powerful for detecting hybridization across most scenarios, except those with high levels of incomplete lineage sorting (ILS) [45]. However, a critical distinction emerges in their false discovery rates: the D-statistic often exhibits an unacceptably high FDR, whereas HyDe maintains better error control [45].
Population Clustering Approaches: Methods like STRUCTURE and ADMIXTURE sometimes fail to identify hybrids when the proportional parental contributions are highly asymmetric (i.e., when γ is close to 0) [45]. Additionally, the posterior distribution estimated using STRUCTURE often exhibits multimodality in many scenarios, complicating interpretation of results.
Accuracy of Parameter Estimation
Parental Contributions (γ): The estimates of γ in HyDe are impressively robust and accurate across various single hybridization scenarios [45]. This represents a significant advantage for studies seeking to quantify the strength of hybridization events rather than simply detecting their presence.
Topological Accuracy: Probabilistic phylogenetic network inference methods generally provide the most accurate topology estimates for single reticulation scenarios. Methods maximizing likelihood under coalescent-based models or pseudo-likelihood approximations (e.g., SNaQ) demonstrate superior performance compared to parsimony-based or concatenation approaches [4].
Table 2: Performance of Methods in Single Reticulation Scenarios
| Method | Category | Detection Power | FDR | γ Estimation Accuracy | Topological Accuracy |
|---|---|---|---|---|---|
| HyDe | Site Pattern | High | Low | High | N/A |
| D-statistic | Site Pattern | High | High | N/A | N/A |
| STRUCTURE | Population Clustering | Variable | Moderate | Moderate | N/A |
| ADMIXTURE | Population Clustering | Variable | Moderate | Moderate | N/A |
| SNaQ | Probabilistic Network | High | Low | High | High |
| PhyloNet (MLE) | Probabilistic Network | High | Low | High | High |
| Neighbor-Net | Concatenation Network | Moderate | Moderate | N/A | Moderate |
Performance in Multiple Reticulation Scenarios
As evolutionary complexity increases with multiple hybridization events, the performance challenges for network inference methods become more pronounced. The scalability of methods to handle these complex scenarios varies considerably.
Detection Power for Multiple Events
Site Pattern Methods: Methods like HyDe and the D-statistic can be extended to test for multiple hybridization events through successive application to different taxon sets. However, this sequential approach may suffer from error propagation, where incorrect inferences in initial steps negatively affect subsequent tests.
Probabilistic Network Methods: Methods that explicitly model multiple reticulations in a single inference framework, such as PhyloNet's maximum likelihood estimation (MLE), face significant computational challenges. The most accurate probabilistic methods become computationally prohibitive as dataset size grows past twenty-five taxa, with none completing analyses of datasets with 30 taxa or more after many weeks of CPU runtime [4].
Topological Accuracy and Scalability
Computational Limitations: The improved accuracy obtained with probabilistic inference methods comes at a substantial computational cost in terms of runtime and main memory usage [4]. This creates a critical methodological gap where the state of the art of phylogenetic network inference lags well behind the scope of current phylogenomic studies, which frequently involve dozens of taxa or more.
Accuracy Trade-offs: As the number of reticulations increases, the topological accuracy of all methods tends to degrade. Similar effects are observed with increased sequence mutation rate [4]. Pseudo-likelihood methods like SNaQ and MPL provide a potential compromise, offering better scalability while maintaining reasonable accuracy for multiple reticulation scenarios.
Table 3: Performance of Methods in Multiple Reticulation Scenarios
| Method | Computational Scalability | Multiple Reticulation Detection | Topological Accuracy | Theoretical Limits |
|---|---|---|---|---|
| HyDe | High | Moderate (via sequential testing) | N/A | No inherent limit |
| D-statistic | High | Moderate (via sequential testing) | N/A | No inherent limit |
| SNaQ | Moderate | High | High | Limited by quartet sampling |
| PhyloNet (MLE) | Low | High | High | ~25-30 taxa |
| PhyloNet (MPL) | Moderate | High | Moderate | ~25-30 taxa |
| Neighbor-Net | High | Low | Low | No inherent limit |
The Scientist's Toolkit: Key Research Reagents and Solutions
Implementing phylogenetic network inference requires both methodological expertise and appropriate computational resources. The following table outlines essential components of the research toolkit for conducting simulation studies and empirical analyses.
Table 4: Essential Research Reagents and Computational Solutions for Phylogenetic Network Analysis
| Tool/Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| PhyloNet | Software Package | Probabilistic network inference under coalescent model | Inference of explicit networks from multi-locus data |
| HyDe | Software Package | Hypothesis testing for hybridization | Detection of hybrid populations and estimation of γ |
| STRUCTURE/ADMIXTURE | Software Package | Population clustering using genotype data | Ancestry coefficient estimation and hybrid identification |
| SNaQ | Algorithm | Pseudo-likelihood network inference | Scalable inference of networks from quartet concordance |
| ms | Simulation Tool | Coalescent simulations | Generating sequence data under evolutionary scenarios |
| Quartet Concordance Factors | Data Type | Probabilities of 4-taxon relationships | Input for SNaQ and related quartet-based methods |
Emerging Approaches and Theoretical Foundations
Recent theoretical advances are expanding the boundaries of phylogenetic network inference, particularly for complex reticulation scenarios.
Beyond Level-1 Networks
Most empirically applicable network methods have been limited to level-1 networks, which do not allow interdependence between reticulate events. Recent work has established theoretical foundations for statistically consistent inference of semi-directed level-2 networks that are outer-labeled planar and galled [37]. This represents a significant expansion of the inferable network space, as level-2 networks include non-planar networks of any level and are substantially more general than level-1 networks.
Identifiability and Consistency
A crucial consideration for method selection is whether network parameters are identifiable from the available data—that is, whether different network structures necessarily produce different probability distributions of the data. Recent results show that the semi-directed network parameter of triangle-free, level-1 network models is generically identifiable under common sequence evolution models [37]. For level-2 networks, theoretical work has precisely characterized features distinguishable from quartet topologies, enabling statistically consistent inference from quartet concordance factors [37].
Figure 2: Trade-offs in Network Classes Between Identifiability, Tractability, and Realism. Level-1 networks are well-established but biologically restrictive, while level-2 and normal networks offer better biological realism with evolving theoretical and computational support.
Practical Recommendations for Researchers
Based on the comprehensive simulation results across multiple studies, we provide the following evidence-based recommendations for method selection:
For Simple Hybrid Detection: When the primary goal is detecting hybridization events with minimal false positives, HyDe outperforms the D-statistic due to its superior control of false discovery rates while maintaining high statistical power [45].
For Quantifying Hybridization: When accurate estimation of parental contributions is essential, HyDe provides robust estimates of γ, while clustering approaches like STRUCTURE and ADMIXTURE struggle with asymmetric contributions [45].
For Small Taxon Sets (<25 taxa): Probabilistic methods like PhyloNet's MLE offer the highest topological accuracy for both single and multiple reticulation scenarios, despite their computational demands [4].
For Larger Taxon Sets: Pseudo-likelihood methods like SNaQ provide the best balance between accuracy and computational feasibility for moderate-sized datasets [4].
For Complex Reticulation Patterns: When dealing with potentially interdependent reticulation events, newer methods targeting level-2 networks show promise, though their practical implementation remains challenging [37].
As the field continues to evolve, methodological development remains critically needed to bridge the gap between the complex evolutionary histories revealed by phylogenomic data and our ability to infer them accurately and efficiently.
The inference of evolutionary histories is fundamentally complicated by processes that generate incongruence between gene trees and the species phylogeny. Two such primary processes are Incomplete Lineage Sorting (ILS), the failure of gene lineages to coalesce in a population before a subsequent speciation event, and hybridization, the exchange of genetic material between distinct lineages [46] [47]. A central challenge in phylogenomics is distinguishing the signal of hybridization from the confounding background of ILS [4]. This guide objectively compares the performance of two methodological approaches—the D-statistic (and its extensions) and Phylogenetic Network methods—in detecting hybridization under varying degrees of ILS, providing researchers with a clear framework for selecting appropriate tools for their data.
The D-statistic and phylogenetic network methods represent distinct philosophies for detecting hybridization. The table below summarizes their core characteristics.
Table 1: Core Characteristics of D-statistic and Phylogenetic Network Methods
| Feature | D-statistic (ABBA-BABA Test) | Phylogenetic Network Methods |
|---|---|---|
| Primary Goal | Test a specific hypothesis of gene flow between a known set of taxa [4] [47] | Infer the overall species phylogeny, including reticulations, from multi-locus data [4] [47] |
| Input Data | Genome-wide allele counts or sequence alignments from four taxa (P1, P2, P3, Outgroup) [47] | Gene trees or sequence alignments from multiple loci for a set of taxa [4] |
| Underlying Model | Coalescent-based model for a four-taxon quartet; tests for an excess of shared derived alleles [48] [47] | Multispecies network coalescent, which models both the coalescent process and hybridization [47] |
| Output | A statistical score (D) indicating deviation from a tree-like history and evidence for introgression [47] | A phylogenetic network with estimated branch lengths, reticulation events, and inheritance probabilities [4] [47] |
| Key Assumption | The true underlying species relationship for the four taxa is known and is ((P1,P2),P3,O) [47] | The set of gene trees is a sample from the distribution defined by the network coalescent process [4] |
Quantitative Performance Comparison
The detectability of hybridization is strongly influenced by the timing of divergence and hybridization events, effective population sizes, and the number of loci analyzed. The following tables summarize key performance metrics from simulation studies.
Table 2: Power to Detect Hybridization in the Presence of ILS (Simulation-Based)
| Method | Scenario | Power (γ=0.1) | Power (γ=0.3) | Power (γ=0.5) | Notes |
|---|---|---|---|---|---|
| Likelihood-Based Network Inference [46] | 10 loci, model with ILS & hybridization | 38% | 78% | 87% | Power of Likelihood Ratio Test (LRT) for (H_0: \gamma=0) increases with higher proportion of hybridization (γ). |
| D-Statistic [4] | Specific four-taxon case with gene flow | High (Qualitative) | High (Qualitative) | High (Qualitative) | Powerful for detecting the presence of gene flow in a given quartet, but provides limited information on the extent or location. |
| BEST (Species Tree Method) [46] | 10 loci, model with ILS & hybridization | Low | Low | Moderate | A Bayesian species tree method that models ILS but not hybridization; often fails to detect the correct relationship when hybridization is present. |
Table 3: Scalability and Accuracy of Network Methods on Empirical and Simulated Data [4]
| Method Type | Representative Method(s) | Typical Max Taxa | Runtime/Memory | Topological Accuracy |
|---|---|---|---|---|
| Concatenation | Neighbor-Net, SplitsNet | High (50+ taxa) | Low / Low | Low (degrades with more taxa/divergence) |
| Parsimony-Based | MP (Minimize Deep Coalescence) | Medium | Medium / Medium | Moderate |
| Probabilistic (Full-Likelihood) | MLE, MLE-length | Low (~25 taxa) | Very High / Very High | High |
| Probabilistic (Pseudo-Likelihood) | MPL, SNaQ | Medium (~25-30 taxa) | High / High | High (but lower than full-likelihood) |
Experimental Protocols
To ensure reproducibility and critical evaluation, the core experimental workflows for the key methods discussed are outlined below.
D-Statistic (ABBA-BABA Test) Protocol
- Taxon Selection: Define the four-taxon block: two sister populations (P1, P2), a third population potentially introgressed with P2 (P3), and an outgroup (O).
- Sequence Alignment & Variant Calling: Generate a genome-wide multiple sequence alignment. Identify bi-allelic sites and determine ancestral/derived states using the outgroup.
- Pattern Counting: For each informative site, count occurrences of the "ABBA" pattern (shared derived allele between P2 and P3) and the "BABA" pattern (shared derived allele between P1 and P3).
- D-Statistic Calculation: Compute the statistic (D = (N{ABBA} - N{BABA}) / (N{ABBA} + N{BABA})). A significant deviation from zero (assessed via block-jackknife) indicates gene flow.
- Input Data Generation: Collect multi-locus DNA sequence data. For each locus, estimate a gene tree (e.g., using maximum likelihood).
- Model Specification: Assume a known species network topology and a set of estimated gene trees.
- Parameter Estimation: Use an optimization algorithm (e.g., Brent's method) to find the Maximum Likelihood Estimates (MLEs) for:
- Branch lengths of the species network (in coalescent units).
- The hybridization parameter (γ), the proportion of genes originating from one parental lineage.
- Hypothesis Testing: Perform a Likelihood Ratio Test (LRT) to test the null hypothesis that the putative hybrid is a pure lineage (γ = 0 or γ = 1).
- Model Definition: Specify a species network topology, branch lengths (coalescent units), and a hybridization parameter (γ).
- Gene Tree Simulation: Simulate a set of gene trees under the multispecies network coalescent model. The parentage of each gene tree in the hybrid population is determined by a Bernoulli draw with probability γ.
- Sequence Simulation: Evolve DNA sequences along each simulated gene tree according to a substitution model (e.g., HKY).
- Inference & Power Calculation: Apply the methods (D-statistic, network inference) to the simulated sequence data. Repeat the process hundreds of times to calculate power as the proportion of replicates where the method correctly rejects the null hypothesis of no hybridization.
Workflow and Relationship Diagrams
The following diagram illustrates the logical relationship between evolutionary processes, phylogenetic signals, and the analytical methods used to detect hybridization.
Relationship Between Processes, Data, and Methods for Detecting Hybridization
The experimental workflow for a typical simulation study comparing these methods is outlined below.
Workflow for Simulation-Based Performance Comparison
The Scientist's Toolkit
Successful detection of hybridization in the presence of ILS relies on a combination of computational tools and conceptual frameworks.
Table 4: Key Research Reagent Solutions for Hybridization Detection
| Tool / Resource | Function / Purpose | Example Use Case |
|---|---|---|
| PhyloNet [4] | A software package for inferring phylogenetic networks and analyzing reticulate evolution. | Implements MLE, MLE-length, and MPL methods for network inference from gene trees. |
| SNaQ [4] | A method for inferring species networks from quartets under the network coalescent using pseudo-likelihood. | Scalable network inference for datasets with dozens of taxa where full-likelihood methods are too slow. |
| D-Statistic (ADMIXTOOLS) | A suite of population genetic tools that includes tests for admixture based on the D-statistic. | Initial screening for gene flow in a four-population context. |
| HybTree [46] | A perl script for estimating speciation times and hybridization times in the presence of ILS. | Estimating the proportion of hybridization (γ) for an a priori specified hybrid population. |
| Global Xenoplasy Risk Factor (G-XRF) [48] | A metric to quantify the risk that a present-day trait pattern is due to introgression (xenoplasy). | Assessing the role of introgression in the evolution of a binary trait, moving beyond a pure tree-based assumption. |
| Multi-locus Sequence Data | The fundamental input data, comprising aligned DNA sequences from multiple unlinked genomic loci. | Used for estimating gene trees, which serve as input for most network inference methods and for calculating the D-statistic. |
The detectability of hybridization is intrinsically limited by the presence of ILS, which creates a confounding phylogenetic signal. The D-statistic is a powerful and computationally efficient tool for initial screening and testing specific hypotheses of gene flow but offers limited insight into the full reticulate history. In contrast, probabilistic phylogenetic network methods provide a comprehensive framework for jointly inferring species relationships and hybridization events but face significant scalability challenges. The choice between them is not mutually exclusive; an effective strategy often involves using the D-statistic for exploratory analysis and network methods for detailed hypothesis testing on curated datasets. Future methodological development is critically needed to improve the scalability and integration of these approaches [4], allowing researchers to untangle the complex web of life with increasing accuracy.
In the field of evolutionary biology, detecting and characterizing gene flow between species or populations is a fundamental challenge. Two primary methodological approaches have emerged: targeted tests for gene flow like the D-statistic (ABBA-BABA test) and comprehensive phylogenetic network inference methods. The D-statistic is a hypothesis-driven method designed to test for gene flow between specific taxa, often requiring an a priori phylogenetic hypothesis [4] [27]. In contrast, phylogenetic network methods aim to reconstruct the full evolutionary history, including reticulation events, from sequence data without a pre-specified hypothesis [4] [2]. This guide provides an objective comparison of their performance, supported by experimental data and methodological details.
Methodological Comparison & Performance
The table below summarizes the core characteristics, strengths, and weaknesses of D-statistic and phylogenetic network methods based on empirical scalability studies [4] [27].
Table 1: Comparative Overview of Gene Flow Detection Methods
| Feature | D-Statistic | Phylogenetic Network Methods |
|---|---|---|
| Primary Objective | Test for signal of gene flow between specific taxa [4] [27] | Infer the complete phylogenetic network (directed acyclic graph) from data [4] [2] |
| Methodological Approach | Hypothesis-testing framework based on allele pattern counts (e.g., ABBA-BABA) [27] | Search-based inference among all possible phylogenies; can use coalescent-based likelihood or parsimony [4] [27] |
| Typical Input | Genomic data from four taxa (P1, P2, P3, Outgroup) | Multi-locus sequence data or pre-estimated gene trees from multiple individuals [4] [27] |
| Computational Demand | Low; fast calculation on large genomic datasets [27] | Very High; runtime and memory become prohibitive beyond ~25 taxa for probabilistic methods [4] [27] |
| Scalability (Number of Taxa) | High; easily scales to genome-scale data for a defined set of taxa [27] | Low; topological accuracy degrades with increasing taxa; methods failed on datasets with ≥30 taxa [4] [27] |
| Key Strength | High power to detect gene flow in specific testable scenarios with low computational cost [27] | Provides a complete, explicit evolutionary history with identified reticulation events [4] [2] |
| Key Weakness/Limitation | Requires a predefined phylogenetic hypothesis; does not provide a full network [4] [27] | Extremely computationally intensive; current state-of-the-art lags behind the scale of modern phylogenomic studies [4] [27] |
Experimental Data on Performance and Scalability
A key scalability study quantified the performance of phylogenetic network methods on both simulated and empirical datasets involving a single reticulation event [4] [27]. The findings, summarized below, provide critical experimental data for method selection.
Table 2: Experimental Performance Data from Scalability Study [4] [27]
| Performance Metric | Phylogenetic Network Method Category | Key Findings |
|---|---|---|
| Topological Accuracy | All Methods | Degraded as the number of taxa increased. A similar negative effect was observed with increased sequence mutation rate [4] [27]. |
| Topological Accuracy | Probabilistic Methods (MLE, MLE-length) | Most accurate methods, utilizing likelihood under coalescent-based models [4] [27]. |
| Computational Runtime | Probabilistic Methods (MLE, MLE-length) | Runtime and memory usage became prohibitive past ~25 taxa. None completed analyses on datasets with 30 or more taxa after many weeks of CPU runtime [4] [27]. |
| Computational Runtime | Pseudo-likelihood Methods (MPL, SNaQ) | Offered a more scalable approximation to full likelihood methods, though challenges remained with larger datasets [4] [27]. |
Detailed Experimental Protocols
To ensure reproducibility and provide context for the data in Table 2, the experimental protocols from the cited scalability study are detailed below.
Protocol for Phylogenetic Network Inference Scalability Study
1. Research Objective: To quantify the performance and scalability limits of state-of-the-art phylogenetic network inference methods on large-scale datasets [4] [27].
2. Data Generation:
- Simulated Datasets: Generated using model phylogenies with a single reticulation. Parameters such as the number of taxa and sequence mutation rate were varied to assess their impact [4] [27].
- Empirical Datasets: Utilized data sampled from natural mouse populations to provide a real-world benchmark [4] [27].
3. Methods Compared: The study evaluated representative methods across different categories:
- Concatenation Methods: Neighbor-Net, SplitsNet [4] [27].
- Parsimony-based Multi-locus Methods: MP (Minimize Deep Coalescence) [4] [27].
- Probabilistic Multi-locus Methods (Full Likelihood): MLE, MLE-length [4] [27].
- Probabilistic Multi-locus Methods (Pseudo-likelihood): MPL, SNaQ [4] [27].
4. Performance Metrics:
- Topological Accuracy: Measured how closely the inferred network matched the true/model network topology [4] [27].
- Computational Requirements: Recorded runtime and main memory usage [4] [27].
5. Analysis: Performance was assessed as a function of dataset scale (number of taxa, evolutionary divergence) [4] [27].
Conceptual Workflow for Gene Flow Analysis
The diagram below illustrates a generalized, high-level workflow for conducting a gene flow analysis, integrating both hypothesis-testing and full inference paradigms.
The Scientist's Toolkit
This table details key reagents, software, and data types essential for research in this field, as referenced in the experimental protocols.
Table 3: Essential Research Reagents and Solutions for Gene Flow Analysis
| Item Name | Function / Role in Analysis |
|---|---|
| Multi-locus Sequence Data | The fundamental input; aligned biomolecular sequences from multiple independent loci across the genome [4] [27]. |
| Reference Genome / Outgroup | A closely related species or population used to polarize allele patterns (ancestral vs. derived), crucial for tests like the D-statistic [27]. |
| Gene Tree Estimation Software | Tools (e.g., PhyML) used in the first phase of summary network methods to infer trees from individual locus alignments [4] [27] [49]. |
| Phylogenetic Network Software | Software packages (e.g., PhyloNet) that implement inference methods like MLE, MPL, and SNaQ to reconstruct networks from gene trees or sequences [4] [27]. |
| High-Performance Computing (HPC) Cluster | Essential computational resource for running probabilistic phylogenetic network inference, which is prohibitively slow on standard computers [4] [27]. |
The D-statistic, or ABBA-BABA test, has become a cornerstone method for detecting gene flow between closely related species, finding application across diverse taxa from hominids to plants [8]. This parsimony-like method helps distinguish signals of ancient hybridization from those of incomplete lineage sorting (ILS), a major confounding factor in phylogenetics [8]. However, despite its widespread use, the D-statistic carries significant limitations, particularly its susceptibility to producing false negatives—situations where genuine gene flow goes undetected. Understanding these pitfalls is crucial for researchers, scientists, and drug development professionals who rely on accurate evolutionary models. This article examines why the D-statistic can be misleading, explores its sensitivity to biological and statistical parameters, and contrasts it with emerging phylogenetic network methods that offer more nuanced approaches to modeling evolutionary history.
The Mechanics and Mathematical Foundation of the D-Statistic
The D-statistic operates on a four-taxon system with an established phylogeny: two sister ingroups (H1 and H2), an outgroup (H3), and a more distantly related outgroup [8]. It detects gene flow by comparing counts of ABBA and BABA sites—parsimony-informative sites that support discordant genealogies. ABBA sites occur when H2 and H3 share a derived allele not found in H1, while BABA sites occur when H1 and H3 share a derived allele not found in H2. Under pure ILS without gene flow, these two site patterns are equally likely, and their counts should not differ significantly. A statistically significant excess of one pattern over the other indicates gene flow between non-sister species [8].
The expected value of D is governed by a complex equation: $$ E(D)=\frac{3f\left({T}3-{T}{gf}\right)}{3f\left({T}3-{T}{gf}\right)+4N\left(1-f\right){\left(1-\frac{1}{2N}\right)}^{T3-{T}2}+4 Nf{\left(1-\frac{1}{2N}\right)}^{T3-{T}{gf}}} $$
Where f represents the fraction of gene flow, N is the population size, T₃ is the divergence time between donor and recipient populations, T₂ is the divergence time between recipient and its sister species, and Tgf is the time of the gene flow event [8]. This nonlinear relationship means calculating the actual fraction of gene flow from D is impossible without precise knowledge of divergence times, gene flow timing, and population size.
The following diagram illustrates the standard workflow for implementing the D-statistic and interpreting its results:
Sensitivity to Demographic Parameters
The D-statistic's performance is highly dependent on relative population size—population size scaled by the number of generations since divergence [8]. As population size increases relative to branch length, the probability of incomplete lineage sorting also increases, diluting the signal of gene flow and potentially leading to false negatives. The method should be applied with "critical reservation to taxa where population sizes are large relative to branch lengths in generations" [8].
Temporal and Genetic Distance Constraints
The D-statistic is robust across a wide range of genetic distances but loses effectiveness with highly divergent taxa. As sequence divergence increases, multiple substitutions and potential saturation can overwhelm the signal of ancient gene flow [8]. The method has been applied to taxa with up to 4-5% sequence divergence in mosquitoes and plants, but its performance deteriorates with increasingly divergent taxa due to accumulated noise [8].
Statistical and Methodological Limitations
The D-statistic is highly susceptible to random variation in short sequences, making it unsuitable for detecting which specific genomic regions have been affected by gene flow [8]. Additionally, the relationship between D and the actual fraction of gene flow (f) is not linear, making quantitative estimates problematic without precise demographic information [8].
Table 1: Factors Contributing to False Negatives in D-Statistic Analysis
| Factor | Impact on False Negative Rate | Biological Mechanism |
|---|---|---|
| Large relative population size | Increases | Dilutes gene flow signal through increased incomplete lineage sorting [8] |
| Recent gene flow events | Increases | May not produce sufficient ABBA-BABA asymmetry to detect |
| Ancient gene flow | Increases | Signal eroded by subsequent mutations and genetic drift |
| Low fraction of introgression | Increases | Limited genomic signal falls below statistical detection threshold |
| Short sequence length | Increases | Higher stochastic variation in ABBA/BABA counts [8] |
Comparative Analysis with Phylogenetic Network Methods
While the D-statistic represents a gene tree-species tree discordance approach, phylogenetic networks offer a fundamentally different framework for modeling evolutionary history with gene flow. "Normal" phylogenetic networks are emerging as a leading class that balances biological relevance with mathematical tractability [18]. These networks explicitly incorporate reticulate events like hybridization and horizontal gene transfer rather than treating them as statistical anomalies.
PhyloTune represents a recent advancement that accelerates phylogenetic updates using pretrained DNA language models [50]. This method identifies the taxonomic unit of new sequences and updates corresponding subtrees, leveraging transformer attention mechanisms to identify phylogenetically informative regions [50]. Such approaches can model complex evolutionary relationships more directly than the D-statistic.
Table 2: Methodological Comparison: D-Statistic vs. Phylogenetic Networks
| Feature | D-Statistic | Phylogenetic Networks |
|---|---|---|
| Evolutionary model | Binary tree with statistical test for discordance | Explicit reticulate branches for hybridization/gene flow [18] |
| Data requirements | Four taxa with established phylogeny | Multiple taxa, can incorporate existing trees |
| Computational complexity | Low | Moderate to high [50] |
| Handling of false negatives | Vulnerable to parameter sensitivity | More comprehensive modeling reduces omissions |
| Quantitative output | D-statistic value with p-value | Branch lengths and hybridization parameters |
| Scalability | Limited to 4-taxon comparisons | Can handle dozens to hundreds of taxa [50] |
Experimental Evidence and Case Studies
PARP Enzyme Studies Reveal Detection Gaps
Research on DNA-encoded chemical libraries (DECLs) targeting PARP enzymes provides insight into false negative patterns relevant to D-statistic analysis. Studies found that DECL selections "frequently miss active compounds, with numerous false negatives for each identified hit" [51]. This parallel phenomenon in drug discovery mirrors the D-statistic's potential to miss genuine signals, as both methods can be affected by systematic undersampling and methodological biases.
Gene Family Screenings Demonstrate Redundancy Issues
Loss-of-function screens in biological research face similar false negative challenges when functional redundancy exists between gene family members [52]. One study demonstrated that conventional screens targeting individual genes failed to identify well-characterized Wnt signaling components, but a novel gene family-based screen approach successfully detected these missed targets [52]. This underscores how methodological limitations can obscure genuine biological relationships—a concern equally relevant to D-statistic analysis.
Research Reagent Solutions for Gene Flow Analysis
Table 3: Essential Research Tools for Evolutionary Studies
| Reagent/Method | Function in Research | Application Context |
|---|---|---|
| D-Statistic (ABBA-BABA) | Detects gene flow despite incomplete lineage sorting [8] | Four-taxon comparisons with established phylogeny |
| f-statistics (fG, fhom) | Estimates fraction of genome affected by gene flow [8] | Comparing datasets with similar demographic history |
| PhyloTune | Accelerates phylogenetic updates using DNA language models [50] | Integrating new taxa into existing phylogenies |
| Normal Phylogenetic Networks | Models evolutionary history with explicit reticulate events [18] | Reconstructing complex evolutionary relationships |
| Gene Family-Based Screening | Circumvents false negatives from functional redundancy [52] | Identifying genes with overlapping functions |
Best Practices for Mitigating False Negatives
Methodological Considerations
Researchers should consider relative population size as a primary factor when implementing the D-statistic [8]. For taxa with large population sizes relative to branch lengths, supplemental methods should be employed. The direction of gene flow and number/size of loci also affect sensitivity, requiring careful experimental design [8].
Data Visualization and Interpretation
Effective data presentation is crucial for accurate interpretation of D-statistic results. Following data visualization best practices—including selecting appropriate chart types, maintaining high data-ink ratios, using color strategically, and providing clear labels and context—helps prevent misinterpretation of statistical results [53] [54]. These principles ensure that limitations and uncertainties are properly communicated.
Integrated Analytical Approaches
No single method perfectly captures evolutionary complexity. Combining D-statistic analysis with phylogenetic network methods, f-statistics, and demographic modeling provides a more robust framework for detecting gene flow. The following diagram illustrates how these methods can be integrated into a comprehensive analytical workflow:
The D-statistic remains a valuable tool for detecting gene flow, but its susceptibility to false negatives necessitates careful application and interpretation. Its sensitivity to demographic parameters, particularly relative population size, can lead to missed detection of genuine gene flow events. Researchers must recognize these limitations and employ complementary approaches—including phylogenetic networks, f-statistics, and demographic modeling—to develop accurate evolutionary hypotheses. As phylogenetic networks continue to develop as mathematically tractable and biologically relevant models, they offer promising alternatives for representing complex evolutionary histories that include hybridization and gene flow. By understanding the pitfalls of the D-statistic and integrating multiple analytical approaches, researchers can better navigate the challenges of detecting ancient gene flow and reconstructing accurate evolutionary histories.
The reconstruction of evolutionary histories is a cornerstone of biological sciences, with profound implications for drug discovery and understanding disease mechanisms. Traditionally, the D-statistic (ABBA-BABA test) has been a primary tool for detecting gene flow between populations or species. However, this method provides a signal of hybridization without revealing the complete phylogenetic history. In contrast, phylogenetic network methods aim to explicitly model evolutionary relationships, including reticulate events such as hybridization, horizontal gene transfer, and introgression. This guide provides an empirical comparison of these approaches, benchmarking their performance in complex, real-world biological scenarios to inform method selection in genomic studies.
Performance Benchmarking: Quantitative Comparisons
Scalability and Accuracy in Phylogenetic Network Inference
A critical scalability study evaluated state-of-the-art phylogenetic network methods on datasets of increasing size, both in terms of the number of taxa and their evolutionary divergence. The findings reveal significant performance differences between method categories [4].
Table 1: Performance of Phylogenetic Network Methods on Empirical Datasets
| Method Category | Representative Methods | Topological Accuracy Trend | Computational Limits | Runtime/Memory Constraints |
|---|---|---|---|---|
| Probabilistic (Full Likelihood) | MLE, MLE-length | Most accurate | Failed on datasets with ≥30 taxa | Prohibitive; analyses did not complete after weeks of CPU time [4] |
| Probabilistic (Pseudo-Likelihood) | MPL, SNaQ | High accuracy | Better than full-likelihood methods | Computationally expensive, but more scalable than MLE [4] |
| Parsimony-Based | MP (Minimize Deep Coalescence) | Lower accuracy than probabilistic | Not explicitly stated | Less demanding than probabilistic methods [4] |
| Concatenation-Based | Neighbor-Net, SplitsNet | Lower accuracy than probabilistic | Handled larger datasets | Computationally efficient, but do not fully account for gene tree incongruence [4] |
The study concluded that probabilistic inference methods achieved the highest accuracy, but this advantage came at a steep computational cost, becoming prohibitive for datasets with more than 25 taxa. Performance degraded as the number of taxa or sequence mutation rate increased [4].
Benchmarking Network-Based Drug Repositioning Methods
In a related field, a systematic benchmarking of 28 heterogeneous network-based drug repositioning methods on 11 datasets provides a model for evaluating computational methods that handle biological complexity. The evaluation framework assessed performance, scalability, and usability [55].
Table 2: Top-Performing Heterogeneous Network-Based Drug Repositioning Methods
| Evaluation Dimension | Best-Performing Methods | Key Algorithmic Approach |
|---|---|---|
| Overall Performance | HGIMC, ITRPCA, BNNR [55] | Matrix completion or factorization [55] |
| Prediction Performance | HINGRL, MLMC, ITRPCA, HGIMC [55] | Varied (network propagation, matrix completion) |
| Scalability | NMFDR, GROBMC, SCPMF [55] | Non-negative matrix factorization, nuclear norm minimization |
| Usability | HGIMC, DRHGCN, BNNR [55] | Matrix completion, graph convolutional networks |
Methods relying on matrix completion or factorization (e.g., HGIMC, ITRPCA, BNNR) demonstrated robust overall performance. This benchmarking effort also produced the HN-DREP online tool and the HN-DRES Snakemake workflow to help researchers select appropriate methods and datasets [55].
Experimental Protocols for Method Evaluation
Protocol for Scalability Assessment of Phylogenetic Methods
The empirical evaluation of phylogenetic methods followed a rigorous protocol to assess their limits [4].
- Dataset Selection and Simulation: The study used both empirical data from natural mouse populations and simulated datasets based on model phylogenies with a single reticulation event. This allowed controlled assessment of scalability challenges.
- Variation of Scale Dimensions:
- Taxon Number: Methods were tested on datasets with an increasing number of taxa to evaluate scalability.
- Evolutionary Divergence: The impact of increased sequence mutation rate was assessed.
- Accuracy Measurement: The topological accuracy of the inferred networks was compared against the known true model phylogenies.
- Computational Resource Tracking: Runtime and main memory usage were meticulously recorded for each method to identify practical computational limits.
This protocol established that the state of the art of phylogenetic network inference lags behind the needs of current phylogenomic studies, which often involve dozens of genomes or more [4].
Protocol for Benchmarking Drug Repositioning Networks
The large-scale benchmarking of heterogeneous network-based drug repositioning methods established a standardized evaluation process [55].
- Literature Review and Method Selection: A systematic review of PubMed articles identified 170 candidate methods. The final 28 were selected based on code availability, usability, and a focus on drug-disease predictions.
- Unified Dataset Application: All 28 methods were evaluated on 11 different existing datasets to ensure a fair comparison across diverse data landscapes.
- Multi-Dimensional Evaluation:
- Performance: Assessment of the accuracy of drug-disease association predictions.
- Scalability: Evaluation of computational efficiency and resource demands.
- Usability: Ease of implementation and use for researchers.
- Tool Development: Creation of the HN-DREP online tool to view detailed evaluation results and the HN-DRES Snakemake workflow to facilitate future benchmarking and method extension [55].
Visualization of Workflows and Relationships
Phylogenetic Network Inference with ALTS
The following diagram illustrates the workflow of the ALTS program, a scalable method that infers tree-child networks by aligning lineage taxon strings (LTSs), representing an innovative approach to a complex computational problem [11].
Phylogenetic Network Inference with ALTS
Interplay of D-Statistic and Network Inference
The D-statistic and phylogenetic network methods represent different points on a spectrum of analytical depth for investigating gene flow, as shown in the following logical relationship diagram.
D-Statistic and Network Method Relationships
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Computational Tools and Resources for Network-Based Analysis
| Tool/Resource Name | Type/Function | Application in Research |
|---|---|---|
| PhyloNet | Software package | Implements phylogenetic network inference methods such as MLE and MLE-length for evolutionary analyses [4]. |
| HN-DREP | Online tool | Allows researchers to view detailed evaluation results for drug repositioning methods and select appropriate ones for their specific needs [55]. |
| HN-DRES | Snakemake workflow | Facilitates the benchmarking of new drug repositioning methods and supports the extension of new methods into the field [55]. |
| Tree-Child Network | Mathematical model | A type of phylogenetic network where every non-leaf node has at least one child that is not a reticulation; ensures biological plausibility and enables efficient computation [11]. |
| ALTS | Software program | Infers tree-child networks by aligning lineage taxon strings from input trees; designed for scalability with larger datasets [11]. |
| Heterogeneous Network | Data structure | Integrates multiple types of entities (e.g., drugs, diseases, proteins) and relationships; used as a foundation for advanced prediction methods in drug repositioning [55]. |
| Biomedical Networks (HetioNet, PrimeKG) | Knowledge Bases | Large-scale heterogeneous networks containing millions of relationships between biomedical entities; provide side information to improve DDI and drug-disease prediction [56]. |
Conclusion
The choice between the D-statistic and phylogenetic network methods is not merely technical but fundamentally shapes the evolutionary hypotheses we can test and support. The D-statistic remains an invaluable tool for initial, rapid screening of gene flow in specific quartets. However, for a comprehensive, biologically explicit reconstruction of evolutionary history, phylogenetic network methods are unequivocally more powerful, despite their higher computational cost. The future of phylogenomics lies in integrated workflows that leverage the speed of the D-statistic for initial hypothesis generation and the robustness of network methods for final inference and validation. This approach will be crucial for biomedical research, particularly in tracing the origins and spread of adaptive traits in pathogens, understanding the genetic consequences of hybridization in disease vectors, and accurately reconstructing the evolutionary history of gene families relevant to drug development. Future methodological development must focus on enhancing the scalability and accessibility of network inference to keep pace with the explosion of genomic data.
References
- 1. Phylogenetic networks empower biodiversity research - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12337313/]
- 2. “Normal” phylogenetic networks may be emerging as the ... [https://arxiv.org/html/2107.10414v2]
- 3. Inferring Phylogenetic Networks with Maximum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4780787/]
- 4. A scalability study of phylogenetic network inference methods ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-1277-1]
- 5. A divide-and-conquer method for scalable phylogenetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6612858/]
- 6. Inferring explicit weighted consensus networks to represent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3898054/]
- 7. Common Methods for Phylogenetic Tree Construction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11117635/]
- 8. Gene flow analysis method, the D-statistic, is robust in a wide ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-2002-4]
- 9. A comparison of phylogenetic and distance-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10599596/]
- 10. Distinguishing Phylogenetic Level-2 Networks with Quartets ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12549781/]
- 11. A fast and scalable method for inferring phylogenetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10538497/]
- 12. Phylogenomic approaches to detecting and characterizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9208645/]
- 13. Importance of incomplete lineage sorting and introgression ... [https://www.nature.com/articles/hdy201672]
- 14. Distinguishing between Incomplete Lineage Sorting and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4074047/]
- 15. Incomplete lineage sorting rather than hybridization ... [https://www.nature.com/articles/s42003-018-0176-6]
- 16. Powerful Inference with the D-Statistic on Low-Coverage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5919751/]
- 17. Phylogenetic Network Analyses Reveal the Influence of ... [https://www.mdpi.com/1999-4915/16/8/1230]
- 18. “Normal” phylogenetic networks may be emerging as the ... [https://www.sciencedirect.com/science/article/pii/S0022519325002024]
- 19. D-statistics for Dummies: A simple test for introgression [https://avianhybrids.wordpress.com/2019/11/09/d-statistics-for-dummies-a-simple-test-for-introgression/]
- 20. ABBA BABA statistics [https://evomics.org/learning/population-and-speciation-genomics/2018-population-and-speciation-genomics/abba-baba-statistics/]
- 21. ipyrad-analysis toolkit: abba-baba - Read the Docs [https://ipyrad.readthedocs.io/en/latest/API-analysis/cookbook-abba-baba.html]
- 22. Evaluating the Use of ABBA–BABA Statistics to Locate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4271521/]
- 23. A Framework for the Comparison of Maximum Pseudo Likelihood and Maximum Likelihood Estimation of Exponential Family Random Graph Models - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3500576/]
- 24. Comparison of Maximum Pseudo Likelihood and Maximum Likelihood Estimation of Exponential Family Random Graph Models | Center for Statistics and the Social Sciences [https://csss.uw.edu/research/working-papers/comparison-maximum-pseudo-likelihood-and-maximum-likelihood-estimation]
- 25. Inferring Phylogenetic Networks with Maximum ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1005896]
- 26. Comparing maximum likelihood and maximum pseudolikelihood estimators for the Ising model [https://advances.in/psychology/10.56296/aip00013/]
- 27. A scalability study of phylogenetic network inference ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5064893/]
- 28. Identifiability of 4-node cycles in level-1 phylogenetic ... [https://www.themoonlight.io/de/review/biorxiv/extracting-diamonds-identifiability-of-4-node-cycles-in-level-1-phylogenetic-networks-under-a-pseudolikelihood-coalescent-model]
- 29. Identifying and addressing methodological incongruence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10286231/]
- 30. PhyKIT: A Multitool for Phylogenomics - PubMed [https://pubmed.ncbi.nlm.nih.gov/39475193/]
- 31. Disentangling a genome-wide mosaic of conflicting ... [https://www.sciencedirect.com/science/article/abs/pii/S1055790325000260]
- 32. GeneFlow: Translation of Single-cell Gene Expression to ... [https://arxiv.org/html/2511.00119v1]
- 33. Phylogenomic Discordance is Driven by Wide-Spread ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11906154/]
- 34. Incomplete lineage sorting and long-branch attraction ... [https://www.frontiersin.org/journals/ecology-and-evolution/articles/10.3389/fevo.2024.1243221/full]
- 35. Widespread introgression across a phylogeny of 155 ... [https://www.sciencedirect.com/science/article/pii/S0960982221014962]
- 36. [2507.17308] Distinguishing Phylogenetic Level-2 Networks with... [https://arxiv.org/abs/2507.17308]
- 37. Distinguishing Phylogenetic Level-2 Networks with... - DeepPaper [https://arxiv.deeppaper.ai/papers/2507.17308v1]
- 38. Exploring phylogenetic hypotheses via Gibbs sampling on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5123299/]
- 39. Inference of reticulate evolutionary histories by maximum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3526433/]
- 40. On the inference of complex phylogenetic networks by Markov ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8445492/]
- 41. A maximum pseudo-likelihood approach for phylogenetic ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-16-S10-S10]
- 42. Ghost Lineages Highly Influence the Interpretation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9366450/]
- 43. ANOMALOUS NETWORKS UNDER THE MULTISPECIES ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10473666/]
- 44. A Comparison of Phylogenetic Network Methods Using ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0001913]
- 45. Performance of Popular Comparative for Hybrid Detection... Methods [https://pubmed.ncbi.nlm.nih.gov/33404632/]
- 46. Estimating hybridization in the presence of coalescence using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3203091/]
- 47. Advances in Computational Methods for Phylogenetic ... [https://arxiv.org/abs/1808.08662]
- 48. Phylogenomic assessment of the role of hybridization and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8405015/]
- 49. DNA barcode analysis: a comparison of phylogenetic and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2775147/]
- 50. PhyloTune: An efficient method to accelerate phylogenetic ... [https://www.nature.com/articles/s41467-025-61684-3]
- 51. Widespread false negatives in DNA-encoded library data [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc00844a]
- 52. A genome-wide loss-of-function screening method for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5034115/]
- 53. 10 Essential Data Visualization Best Practices for 2025 [https://www.timetackle.com/data-visualization-best-practices/]
- 54. 7 Data Visualization Best Practices for 2025 - KP Infotech [https://kpinfo.tech/data-visualization-best-practices/]
- 55. A comparative benchmarking and evaluation framework for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11033846/]
- 56. Benchmarking Graph Learning for Drug-Drug Interaction ... [https://arxiv.org/html/2410.18583v3]