Beyond the False Signal: Strategies to Mitigate Ancestral Polymorphism in Introgression Detection
Accurately identifying true introgression is critical for evolutionary studies and biomedical research, yet it is frequently confounded by false positives from ancestral polymorphism and selection.
Beyond the False Signal: Strategies to Mitigate Ancestral Polymorphism in Introgression Detection
Abstract
Accurately identifying true introgression is critical for evolutionary studies and biomedical research, yet it is frequently confounded by false positives from ancestral polymorphism and selection. This article provides a comprehensive framework for researchers and drug development professionals to navigate these challenges. We explore the foundational causes of spurious signals, from lineage-specific rate variation to the pervasive effects of background selection. The content details robust methodological approaches, including alignment-based pipelines and site-pattern methods, and offers practical troubleshooting for dataset optimization. Finally, we present a rigorous validation protocol incorporating statistical benchmarking and case studies from human and plant genomics to ensure the reliability of introgression inferences in clinical and evolutionary contexts.
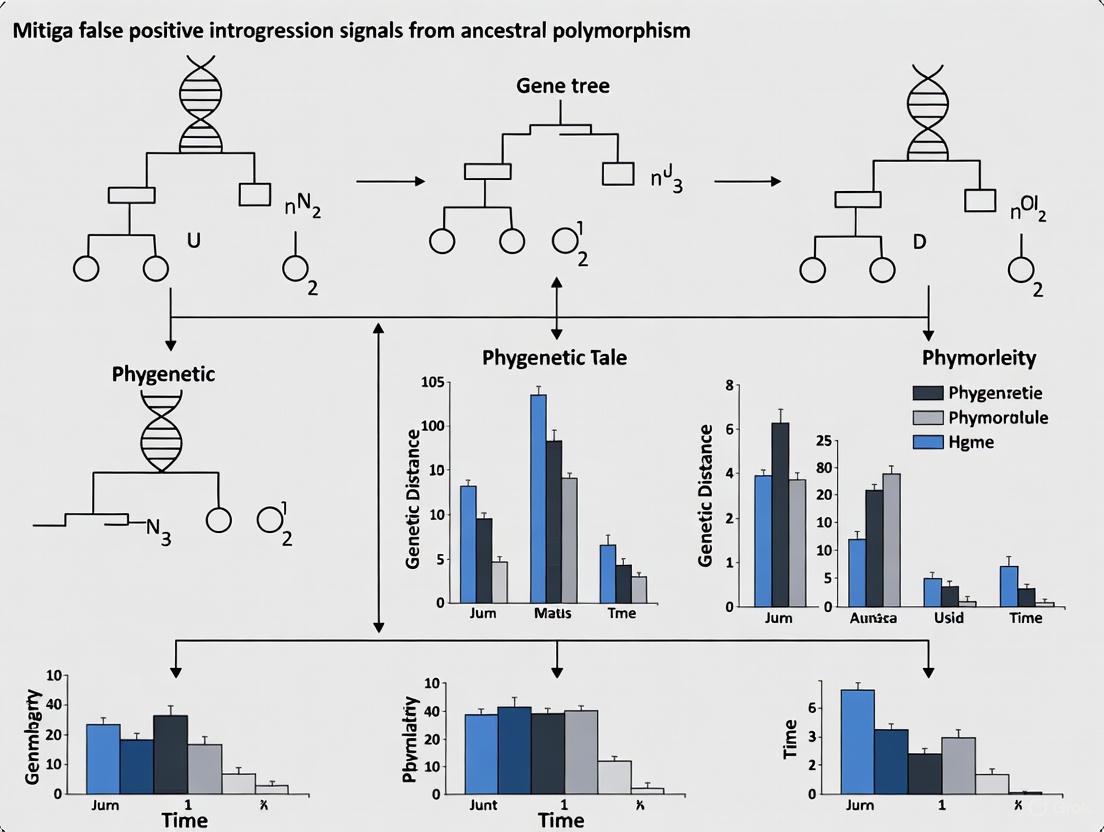
Unmasking the Imposters: How Ancestral Polymorphism and Selection Generate False Introgression Signals
Frequently Asked Questions (FAQs)
1. What is the fundamental difference between introgression and incomplete lineage sorting (ILS)? A: Introgression and ILS both create discordance between gene trees and species trees, but through different mechanisms. Introgression is the movement of genetic material from one species into the gene pool of another through hybridization and repeated backcrossing [1]. Incomplete Lineage Sorting (ILS), also called hemiplasy or deep coalescence, is the failure of ancestral genetic polymorphisms to sort out (or coalesce) into distinct lineages during successive speciation events [2]. This means a gene variant in one species might be more closely related to a variant in a distant species than to its closer relative, not because of gene flow, but simply by chance retention of an ancient polymorphism [2].
2. What are the primary analytical challenges in distinguishing these two signals? A: The core challenge is that both processes produce similar patterns of shared genetic variation, making them difficult to separate using simple tree-based methods [1] [3]. Specific challenges include:
- Similar Gene Tree Discordance: Both ILS and introgression can cause a gene tree to show a different evolutionary relationship than the species tree.
- Model Violations: Statistical methods for distinguishing them often rely on simplifying assumptions (e.g., constant population size, neutral markers) that may be violated in real-world scenarios, complicating interpretation [3].
- Confounding with Other Forces: Variation in mutation rates across the genome can create patterns that mimic recent introgression [4]. Furthermore, natural selection can skew allele frequencies and complicate the picture [4].
3. What kind of data is most powerful for making this distinction? A: Using multiple, independent lines of evidence is crucial. No single locus can provide a definitive answer. Key data includes:
- Genome-Wide Data: Sequencing many unlinked loci from across the nuclear genome provides the statistical power to discern the underlying demographic history [1].
- Population-Level Sampling: Sampling multiple individuals from allopatric (geographically separated) and parapatric (adjacent) populations of the species in question is highly informative. Introgression often leaves a stronger signal in parapatric populations, while ILS is expected to be evenly distributed [1].
- Different Marker Types: Contrasting patterns from biparentally inherited nuclear DNA, maternally inherited mitochondrial DNA (mtDNA), and paternally inherited chloroplast DNA (in plants) can be very revealing. For example, mtDNA can introgress more easily than nuclear genes, creating mito-nuclear discordance [1] [3].
Troubleshooting Guides
Problem 1: Incongruent Phylogenies from Different Genomic Regions
Observation: Gene trees built from different loci show conflicting topologies, and you need to determine if the cause is ILS or introgression.
| Troubleshooting Step | Rationale and Methodology |
|---|---|
| Analyze Geographic Pattern | Compare genetic differentiation between species in allopatry versus parapatry/sympatry. Lower differentiation in parapatry suggests ongoing gene flow (introgression), while similar levels of differentiation in allopatry and parapatry are more consistent with ILS [1]. |
| Utilize Coalescent-Based Model Selection | Use frameworks like Approximate Bayesian Computation (ABC) or the Isolation with Migration (IM) model to compare the statistical support for different speciation scenarios (e.g., strict isolation vs. isolation-with-migration). These methods can estimate parameters like population divergence times and migration rates [1]. |
| Apply Summary Statistics for Introgression | Calculate statistics designed to detect gene flow. RNDmin is a robust method that uses the minimum pairwise sequence distance between two populations relative to an outgroup to detect recently introgressed loci [4]. Gmin (dmin/dXY) is another statistic sensitive to recent migration that is normalized to account for variation in mutation rates [4]. |
The following workflow integrates these steps into a logical decision-making process:
Problem 2: Mito-Nuclear Discordance
Observation: The phylogeny from mitochondrial DNA (mtDNA) does not match the phylogeny from nuclear DNA, a common form of cytonuclear discordance.
| Troubleshooting Step | Rationale and Methodology |
|---|---|
| Test for Pervasive Nuclear Introgression | If mtDNA has introgressed, it is often part of a broader, but potentially weaker, signal of nuclear introgression. Scan the nuclear genome for regions with exceptionally high similarity between the two species, using methods like RNDmin or FST outliers [4] [3]. |
| Evaluate the Role of Selection | Consider if natural selection could be driving the mtDNA capture. For example, an adaptive mitochondrial haplotype might sweep through a population, even in the absence of significant nuclear gene flow [3]. |
| Assess Reproductive Biology | Investigate the known reproductive mode of the species and their hybrids. In some systems, clonal reproduction of hybrids (e.g., gynogenesis) can prevent nuclear introgression while allowing for the complete fixation of allospecific mtDNA, pointing to an ancient hybridization event [3]. |
The following table summarizes empirical findings from published research that successfully differentiated between ILS and introgression.
Table 1: Empirical Data from Case Studies on ILS vs. Introgression
| Study System | Key Analytical Methods | Evidence for Introgression | Evidence Against ILS | Citation |
|---|---|---|---|---|
| Two pine species (Pinus massoniana and P. hwangshanensis) | Population structure analysis, Comparison of allopatric vs. parapatric FST, Approximate Bayesian Computation (ABC), Ecological Niche Modeling | More admixture in parapatric populations; Lower interspecific differentiation in parapatry; ABC supported a model of long isolation followed by secondary contact. | ILS would predict even sharing of polymorphisms across allopatric and parapatric populations, which was not observed. | [1] |
| Spined loach fish (Cobitis complex) | Multi-locus sequencing (nuclear and mtDNA), Coalescent-based analyses, Knowledge of hybrid clonal reproduction | Mito-nuclear mosaic genome (C. tanaitica nuclear DNA clusters with C. taenia, but its mtDNA clusters with C. elongatoides). Statistical rejection of ILS models. | Contemporary hybrids are clonal and cannot mediate nuclear introgression, implying an ancient hybridization event. | [3] |
| Mosquitoes (Anopheles quadriannulatus and A. arabiensis) | RNDmin statistic to detect introgressed regions | Identification of three novel candidate regions for introgression, including one on the X chromosome. | The RNDmin method is designed to be robust to ILS and variation in mutation rates, helping to pinpoint true introgression. | [4] |
The Scientist's Toolkit: Key Reagents and Computational Methods
Table 2: Essential Reagents and Computational Tools for Differentiation Studies
| Item / Method | Function / Purpose in Analysis |
|---|---|
| Multiple, Independent Intron Markers | Neutral, non-coding regions distributed across the genome are ideal for inferring demographic history without the confounding effects of natural selection [1]. |
| Outgroup Sequences | Essential for rooting phylogenetic trees and for normalizing statistics like RND (Relative Node Depth) and RNDmin to account for variation in mutation rates [4]. |
| Approximate Bayesian Computation (ABC) | A statistical framework for comparing complex demographic models (e.g., with and without gene flow) to identify the scenario best supported by the observed genetic data [1]. |
| Isolation with Migration (IM) Model | A coalescent-based model that jointly estimates population sizes, divergence times, and migration rates from multi-locus sequence data [1]. |
| RNDmin Statistic | A summary statistic used to test for introgression by examining the minimum pairwise sequence distance between populations relative to divergence to an outgroup. It is powerful for detecting recent and strong introgression [4]. |
| Phased Haplotypes | Knowing the phase of alleles (which alleles are linked on the same chromosome) is required for several powerful detection methods, including those based on minimum sequence distances (dmin) [4]. |
| Ecological Niche Modeling (ENM) | Used to infer historical species range shifts (e.g., during past climate oscillations), which can provide independent evidence for potential secondary contact zones that would facilitate introgression [1]. |
Technical Support Center
Troubleshooting Guides
Guide 1: Addressing False Positive Introgression Signals in Genomic Windows
- Problem: Your analysis detects local introgression signals that are not biologically plausible. These spurious signals often appear in genomic regions with low nucleotide diversity.
- Explanation: The widely used Patterson's D statistic can produce unreliable inferences in small genomic windows. Its variance increases in regions of low diversity, leading to false positives [5]. Furthermore, the stochastic nature of the substitution process means that even under a molecular clock, estimated substitution rates can vary erroneously between lineages, creating a pattern that mimics true rate variation [6].
- Solution:
- Implement the D+ Statistic: Use the D+ statistic, which incorporates both shared derived alleles (ABBA/BABA patterns) and shared ancestral alleles (BAAA/ABAA patterns). This increases the number of informative sites per region, improving precision and reducing the false positive rate in local analyses [5].
- Simulate Your Data: Use coalescent simulations under a model without introgression to establish a baseline for expected rate variation and D statistic variance due to stochastic error alone [5] [6].
- Compare Clade Averages: When checking for rate variation, compare average substitution rates between different clades rather than individual lineages, as this approach is less susceptible to erroneous inference from estimation error [6].
Guide 2: Managing Alert Fatigue from Statistical False Positives
- Problem: A high volume of false positive alerts from your detection pipelines is causing "alert fatigue," where real signals may be overlooked.
- Explanation: This is a common operational challenge. False positives waste time and can desensitize researchers to alerts, increasing the risk of missing genuine findings [7] [8].
- Solution:
- Clearly Define Detections: For every statistical test or alert, precisely define the specific evolutionary event or pattern it is designed to catch and the action to be taken if it fires [8].
- Tune Your Thresholds: Set accurate thresholds for your statistics by first baselining normal variation in your dataset. Avoid using default thresholds without adjusting them to your specific data and research question [9] [8].
- Implement a Feedback Loop: When a false positive is identified, use it as feedback to adjust your analytical parameters or detection rules. This continuous tuning prevents the same false positive from recurring [9].
Frequently Asked Questions (FAQs)
Q1: What is the primary cause of false positive introgression signals in local genomic regions? The primary cause is the application of genome-wide statistics, like Patterson's D, to small local regions where low nucleotide diversity maximizes the statistic's variance. This can create spurious signals that are not due to actual gene flow [5].
Q2: How does the D+ statistic improve upon the classic D statistic? The D statistic only uses shared derived alleles (ABBA and BABA site patterns). The D+ statistic increases analytical power by also leveraging shared ancestral alleles (BAAA and ABAA patterns). This provides more information per genomic region, leading to better precision and a lower false positive rate when detecting local introgression [5].
Q3: Can substitution rate variation be inferred even when the molecular clock holds true? Yes. Error in rate estimation, stemming from the stochastic (Poisson) nature of the substitution process and the underestimation of multiple substitutions, can lead to the erroneous inference of rate variation among lineages, even when the underlying substitution process is clock-like [6].
Q4: What is a key best practice for maintaining the reliability of a detection pipeline over time? Treat your detections like code. This "Detection as Code" (DaC) approach involves continuous maintenance, including testing new detections against historical data, tuning thresholds, and disabling or refining rules that become noisy as data and methods evolve [8].
Experimental Protocols & Methodologies
Protocol: Implementing the D+ Statistic for Introgression Detection
This protocol outlines the steps to compute the D+ statistic for a four-population system (P1, P2, P3, Outgroup).
- Data Preparation: Obtain genotype or allele frequency data for at least one individual from each of the four populations across your genomic regions of interest (e.g., 5kb windows) [5].
- Variant Site Identification: Identify biallelic Single Nucleotide Polymorphisms (SNPs). Use the outgroup (P4) to polarize alleles, determining the ancestral (A) and derived (B) states [5].
- Site Pattern Classification: For each SNP in a window, classify it into one of four key patterns based on the alleles present in P1, P2, and P3 (assuming the outgroup is fixed for the ancestral allele):
- ABBA: Derived allele in P3 and P2, ancestral in P1.
- BABA: Derived allele in P3 and P1, ancestral in P2.
- BAAA: Ancestral allele in P3 and P2, derived in P1.
- ABAA: Ancestral allele in P3 and P1, derived in P2 [5].
Calculate the D+ Statistic: Use the following formula, summing over all L sites in the genomic window [5]:
D+ = Σ(ABBA - BABA + BAAA - ABAA) / Σ(ABBA + BABA + BAAA + ABAA)
For sample sizes greater than one, the calculation can be expressed in terms of derived allele frequencies (p̂ᵢⱼ for population j at site i).
Interpretation: A significant positive D+ value indicates introgression between P3 and P2, while a significant negative value indicates introgression between P3 and P1.
Signaling Pathways and Workflows
Diagram 1: D vs. D+ Site Pattern Logic
Diagram 2: False Positive Mitigation Workflow
Data Presentation
Table 1: Comparison of Introgression Detection Statistics
| Feature | Patterson's D Statistic | D+ Statistic |
|---|---|---|
| Core Principle | Measures excess of shared derived alleles (ABBA vs. BABA) [5] | Measures excess of shared derived AND shared ancestral alleles [5] |
| Informative Sites Used | Shared derived alleles only (ABBA, BABA) [5] | Shared derived and shared ancestral alleles (ABBA, BABA, BAAA, ABAA) [5] |
| Optimal Use Case | Detecting genome-wide introgression [5] | Detecting local, targeted introgression in genomic windows [5] |
| Performance in Low-Diversity Regions | High variance, prone to spurious results (false positives) [5] | Improved precision and lower false positive rate due to more informative sites [5] |
Table 2: Common Sources of Error in Substitution Rate Estimation
| Source of Error | Impact on Analysis | Mitigation Strategy |
|---|---|---|
| Stochastic Substitution Process | Even with a molecular clock, the number of substitutions per branch varies, creating erroneous rate variation [6]. | Use coalescent simulations to establish a null distribution; compare average rates between clades [6]. |
| Multiple Hit / Saturation | Underestimation of true substitutions on longer branches, leading to underestimation of rates and erroneous variation [6]. | Employ appropriate substitution models that correct for multiple hits; be aware of the node-density effect [6]. |
| Imperfect Model Fit | No model perfectly captures the substitution process, leading to residual error in rate estimation [6]. | Use model selection tools to choose the best-fitting model; consider model averaging. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools and Resources
| Item | Function / Description | Relevance to Mitigating False Positives |
|---|---|---|
| Coalescent Simulation Software (e.g., ms, SLiM) | Simulates genetic data under evolutionary models without introgression. | Creates a null distribution to test whether observed rate variation or D statistics exceed expectations by chance alone [5] [6]. |
| Population Genomic Analysis Toolkit (e.g., BCFtools, PLINK, admixtools) | Suites for processing VCF files, calculating basic statistics, and implementing tests like D and f4. | The foundational environment for data preparation and initial analysis. |
| D+ Statistic Script | Custom or published script/software to calculate the D+ statistic from aligned sequence or genotype data. | Directly implements the improved method for local introgression detection, reducing false positives [5]. |
| High-Quality Reference Genome & Annotated Outgroup | A well-assembled genome for mapping and an evolutionarily close outgroup for accurate allele polarization. | Critical for correctly identifying ancestral and derived alleles, which is the basis for both D and D+ statistics [5]. |
| High-Performance Computing (HPC) Cluster | Computing resources for large-scale genomic simulations and genome-scans. | Enables the computationally intensive simulations and bootstrapping required for robust statistical testing. |
Troubleshooting Guides
Issue 1: Distinguishing True Introgression from Ancestral Polymorphism
Problem: D-statistics (ABBA-BABA test) yield significant signals, but it is unclear if this results from true introgression or incomplete lineage sorting (ILS) of ancestral variation.
Diagnosis and Solution:
| Test/Metric | Calculation Formula | Interpretation | Key Thresholds |
|---|---|---|---|
| D-Statistic | D = (NABBA - NBABA) / (NABBA + NBABA) | Measures allele frequency asymmetry; significant D ≠ 0. | |D| > 3-4 standard errors suggests significance. |
| f-branch (fb) | Estimated via f4-ratio estimation: fb = (f4(P1, O; A, B) / f4(P1, P2; A, B)) | Estimates the proportion of introgressed ancestry in a genome. | fb ≈ 0: No introgression. fb > 0: Proportion of ancestry from sister lineage. |
| Site Frequency Spectrum (SFS) | — | Compare the SFS in the test population to neutral expectations; an excess of intermediate-frequency alleles may suggest balancing selection. | — |
Experimental Protocol:
- Variant Calling: Generate a whole-genome, multi-individual SNP dataset for P1, P2, P3, and an outgroup (O).
- D-statistic Calculation: Use packages like
Dsuiteto calculate D-statistics in sliding windows across the genome. Identify regions with significant D-values. - f-branch Estimation: Apply f4-ratio estimation to significant regions to quantify the proportion of ancestry from an introgressed lineage.
- Coalescent Simulation: Simulate genomic data under a model of strict isolation (no introgession) with parameters (e.g., population size, divergence time) estimated from your data.
- Comparison: Compare the distribution and magnitude of significant D-values from your empirical data to the distribution obtained from simulations where no introgression is possible. If empirical signals are stronger/more frequent, true introgression is a more likely cause than ILS alone.
Issue 2: Confounding Effects of Background Selection
Problem: Background Selection (BGS) reduces genetic diversity in regions of low recombination, creating "valleys" of diversity that can be mistaken for selective sweeps or local reductions in gene flow, potentially creating spurious introgression signals.
Diagnosis and Solution:
| Analysis Method | Application | Interpretation of Results |
|---|---|---|
| Diversity (π) vs. Recombination Rate Correlation | Calculate π in non-overlapping windows and plot against the local recombination rate. | A strong positive correlation is a hallmark signature of BGS. |
| B-value Calculation | B = πobserved / πneutral expectation. The B-value estimates the reduction in diversity due to BGS. | B ≈ 1: No BGS effect. B < 1: Diversity is reduced by BGS. |
| Comparison to Neutral Model | Use software (e.g., SLiM, msprime) to simulate genomes with and without BGS. |
If patterns in empirical data (e.g., diversity valleys) are replicated in BGS simulations, BGS is a sufficient explanation. |
Experimental Protocol:
- Estimate Recombination Rates: Generate a genetic map for your organism or use a published one.
- Calculate Genomic Diversity: Compute nucleotide diversity (π) in windows across the genome.
- Correlation Analysis: Perform a statistical test (e.g., Spearman's correlation) between π and recombination rate.
- Modeling and Simulation:
- Estimate the density of functional elements (e.g., using annotation files) across the genome.
- Input the recombination map and functional density into a simulator like
SLiMto generate expected diversity patterns under a BGS model.
- Identify Outliers: Compare empirical diversity values to the simulated BGS distribution. Genomic windows where π is significantly lower than the BGS expectation are candidate regions for bona fide selective sweeps or other processes.
Frequently Asked Questions (FAQs)
Q1: My D-statistic is significant, and the f-branch estimate is high in a specific region. Can I conclusively say this is introgression? A1: Not yet. A high f-branch estimate suggests gene flow but does not rule out the retention of ancestral polymorphism in that specific region due to linked selection. You must test whether the region is also subject to BGS (low recombination, gene-dense) or balancing selection. If it is, the signal may be a false positive driven by Selection Past.
Q2: How can I tell if balancing selection is maintaining ancestral polymorphism in my data? A2: Look for specific genomic signatures: (1) an excess of intermediate-frequency alleles in the Site Frequency Spectrum (SFS); (2) higher than expected genetic diversity (e.g., π, θw) for long, deeply divergent haplotypes; and (3) trans-species polymorphism, where alleles are shared between species that diverged a long time ago.
Q3: What is the most critical negative control when performing these tests? A3: The most critical control is to analyze genomic regions you believe a priori to be evolving neutrally—areas far from genes, with high recombination rates, and devoid of conserved elements. Establish the null distribution of your test statistics (like D) in these regions. Any inference from putatively selected regions should be compared against this neutral baseline.
Experimental Workflow for Mitigating False Positives
The following diagram outlines the core decision-making workflow for determining the source of an apparent introgression signal.
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Resource | Type | Primary Function in Analysis |
|---|---|---|
| Reference Genome Assembly | Dataset | Serves as the coordinate system for mapping sequence reads and calling genetic variants. A high-quality assembly is crucial. |
| Population SNP Dataset | Dataset | A VCF file containing genotype calls for multiple individuals across populations. The fundamental input for most population genetic analyses. |
| Genetic Map | Dataset | Provides estimates of recombination rates across the genome. Essential for diagnosing the effects of Background Selection. |
| Dsuite | Software Package | Efficiently calculates D-statistics (ABBA-BABA) and related metrics (f-branch) for genome-wide data to test for introgression. |
| ANGSD | Software Package | Used for estimating the Site Frequency Spectrum (SFS) and other summary statistics from next-generation sequencing data, even without full genotype calls. |
| SLiM | Software Package | A powerful simulation framework for forward-time, individual-based population genetic simulations. Ideal for modeling complex scenarios involving selection and demography. |
| msprime / stdpopsim | Software Package | Tools for coalescent simulations. Efficient for generating large genomic datasets under complex demographic models with neutral evolution or background selection. |
Accurate detection of introgression—the exchange of genetic material between species—is fundamental to understanding evolutionary processes. However, in shallow phylogenies (involving closely related taxa), commonly used methods like the D-statistic can produce false positive signals. These spurious signals are not due to actual gene flow but are often artifacts of substitution rate variation across lineages. This guide provides troubleshooting and methodologies to help researchers distinguish true introgression from these false positives, a critical consideration for research in areas like drug development where genetic insights can inform target identification.
Troubleshooting Guide: False Positive Introgression Signals
Problem: Your analysis using site-pattern methods (e.g., D-statistic, HyDe) on a shallow phylogeny indicates significant introgression, but you suspect the signal might be false.
Solution: Investigate the following potential causes and solutions.
| Symptom | Potential Cause | Diagnostic Steps | Recommended Solution |
|---|---|---|---|
| Significant D-statistic or HyDe signal in young phylogenies (< 300,000 generations) with small population sizes [10]. | Lineage-specific substitution rate variation (rate heterogeneity) [10]. | Perform a relative rate test to quantify rate differences between sister lineages [10]. | Use methods robust to rate variation, such as full-likelihood tests that incorporate branch length information [10]. |
| Inflated false-positive rates (up to 35% for weak, 100% for moderate rate variation) with a 500 Mb genome [10]. | Violation of the molecular clock assumption, leading to homoplasy that creates ABBA/BABA asymmetry [10]. | Simulate data under a null model of no gene flow but with estimated rate variation to establish a baseline false-positive rate [10]. | Employ the D+ statistic, which leverages both shared derived and ancestral alleles, as it has been shown to have better precision [5]. |
| Signal intensifies when a more distant outgroup is used [10]. | Increased potential for multiple hits with distant outgroups, violating the "no multiple hits" assumption [10]. | Re-run the analysis with a closer outgroup, if available, and compare the results. | Ensure the outgroup is not excessively distant from the ingroup taxa under study. |
| Unreliable local introgression signals in genomic windows, especially in regions of low nucleotide diversity [5]. | High variance of the D-statistic in small genomic windows with low diversity [5]. | Calculate the D+ statistic in these windows, as it incorporates more informative sites (BAAA and ABAA patterns) [5]. | Switch from the standard D-statistic to the D+ statistic for local inference of introgressed regions [5]. |
| Significant D-statistic but no known historical opportunity for gene flow. | Ghost introgression or other demographic complexities not accounted for in the model [10]. | Test complex demographic models that include population size changes and structure. | Consider using supervised learning or probabilistic modeling approaches designed for complex scenarios [11]. |
Frequently Asked Questions (FAQs)
1. What are the primary methods for detecting introgression, and how do they work?
Introgression detection methods generally fall into two categories:
- Site-pattern or gene-tree topology methods: These methods, like the D-statistic (ABBA-BABA test) and HyDe, detect gene flow by identifying significant asymmetries between discordant gene trees or site patterns. They operate on the principle that without introgression, two discordant site patterns (ABBA and BABA) occur with equal frequency due to Incomplete Lineage Sorting (ILS). An excess of one pattern suggests gene flow [10] [5].
- Gene-tree branch length methods: Methods like D3 and QuIBL use information from gene-tree branch lengths, examining whether their distributions deviate from expectations under ILS alone [10].
2. Why are "shallow phylogenies" particularly vulnerable to false positives?
Shallow phylogenies, involving closely related taxa, are often assumed to adhere to a molecular clock. However, empirical studies show that even within genera, species can exhibit substitution rate disparities of 10% to over 50% [10]. Methods like the D-statistic assume no multiple hits, but rate variation causes homoplasy (independent mutations), which creates an asymmetry in ABBA and BABA site patterns that mimics the signal of introgression [10].
3. What is the D+ statistic, and how does it improve upon the D-statistic?
The D+ statistic is an extension of the D-statistic that leverages both shared derived alleles (like ABBA and BABA) and shared ancestral alleles (BAAA and ABAA) [5]. Introgression reintroduces chunks of DNA containing both types of alleles into the recipient population. By using this additional information, D+ increases the number of informative sites per genomic region. This improves the precision of local introgression detection and reduces the false positive rate in small genomic windows compared to the standard D-statistic [5].
D+ = [ Σ(ABBA - BABA) + Σ(BAAA - ABAA) ] / [ Σ(ABBA + BABA) + Σ(BAAA + ABAA) ] [5]
4. Besides rate variation, what other factors can cause spurious introgression signals?
Other factors include:
- Ghost Introgression: Gene flow from an unsampled (ghost) lineage can produce a significant D-statistic signal that is misinterpreted [10].
- Inaccurate Branch Lengths: The use of "pseudo-chronograms" with suboptimal branch lengths (e.g., estimated via algorithms like BLADJ) can lead to strong overestimation of phylogenetic signal in other comparative metrics, highlighting the general sensitivity of evolutionary analyses to branch length quality [12].
- Purifying Selection: Selection against deleterious introgressed variants can create patterns that mimic adaptive introgression [5].
Experimental Protocols
Protocol 1: Quantifying Lineage-Specific Rate Variation
Objective: To test the molecular clock assumption and quantify the degree of substitution rate variation between sister lineages.
Materials:
- Genomic sequence data for the ingroup taxa and a suitable outgroup.
- Software for multiple sequence alignment (e.g., MAFFT, MUSCLE).
- Phylogenetic software capable of performing relative rate tests (e.g., HYPHY, MEGA).
Methodology:
- Sequence Alignment: Generate a whole-genome or multi-locus alignment for the taxa of interest, including the outgroup.
- Tree Estimation: Infer a time-calibrated species tree under a strict molecular clock model.
- Relative Rate Test: Apply the Relative Rate Test (RRT) to pairs of sister lineages. This test checks if the genetic distance from each sister lineage to the outgroup is equal [10].
- Interpretation: A significant RRT result indicates a violation of the molecular clock. The percentage difference in distances estimates the magnitude of rate variation.
Protocol 2: Implementing the D+ Statistic for Local Introgression Detection
Objective: To identify specific introgressed regions in a genome with improved precision.
Materials:
- Genotype or allele frequency data for four populations: P1 and P2 (sister taxa), P3 (putative donor), and P4 (outgroup).
- Scripts or software to calculate the D+ statistic (implementation may require custom scripts based on the published formula [5]).
Methodology:
- Data Preparation: Organize genomic data into non-overlapping or sliding windows.
- Site Pattern Counts: For each window, count the occurrences of the four key site patterns:
- ABBA: Derived allele in P2 and P3, ancestral in P1 and P4.
- BABA: Derived allele in P1 and P3, ancestral in P2 and P4.
- BAAA: Derived allele only in P1, ancestral in P2, P3, P4.
- ABAA: Derived allele only in P2, ancestral in P1, P3, P4 [5].
- Calculate D+: Compute the D+ value for each window using the formula provided in the FAQ section.
- Significance Testing: Determine the statistical significance of the D+ value, typically achieved through a block jackknife or bootstrap approach across windows or sites.
Signaling Pathways and Workflows
Diagram: Site Patterns in Introgression Detection
Research Reagent Solutions
Essential computational tools and conceptual "reagents" for investigating introgression.
| Research Reagent | Function / Description | Example Use Case |
|---|---|---|
| D-statistic (ABBA-BABA) | A summary statistic that tests for an excess of shared derived alleles between populations to detect genome-wide introgression [10] [5]. | Initial screening for the presence of gene flow in a four-taxon system. |
| HyDe | A site-pattern method for detecting and characterizing hybrid speciation events by testing if the two least frequent site patterns occur at comparable frequencies [10]. | Identifying if a taxon is of hybrid origin and determining its parental species. |
| D+ Statistic | An extension of the D-statistic that incorporates shared ancestral alleles to improve precision in detecting local introgressed regions [5]. | Pinpointing the exact genomic locations of introgressed tracts in windows of low diversity. |
| Relative Rate Test | A phylogenetic method used to test the molecular clock hypothesis by comparing rates of evolution between two sister lineages using an outgroup [10]. | Diagnosing potential false positive drivers by quantifying lineage-specific rate variation. |
| Coalescent Simulator (e.g., MSci) | Software that implements the multispecies coalescent model with introgression to simulate genomic data under user-defined demographic parameters [10]. | Generating null distributions to estimate false-positive rates and validate findings. |
Frequently Asked Questions
Q1: What is the primary statistical limitation of Patterson's D statistic when analyzing local genomic regions, and how can it lead to false positives? The primary limitation is its high variance and spurious inferences in genomic windows with low nucleotide diversity. The D statistic becomes unreliable in small regions because it measures an excess of shared derived alleles (ABBA or BABA sites) but does not leverage shared ancestral variation. This can produce false positive signals of introgression in areas with low diversity, as the statistic is sensitive to the limited number of informative sites in these windows [5].
Q2: How does the D+ statistic improve upon the D statistic for detecting local introgression? The D+ statistic incorporates both shared derived alleles (like the D statistic) and shared ancestral alleles. This increases the number of informative sites per genomic region, which improves precision and reduces the false positive rate when identifying local introgressed segments. By using more data, D+ provides a more robust measure for pinpointing the exact location of introgression in a genome [5].
Q3: My analysis of local genomic windows shows high D statistic values, but I suspect they are false positives. What is a likely cause? A likely cause is analyzing regions with low nucleotide diversity. In such windows, the D statistic has high variance and can produce strong but spurious signals of gene flow. It is recommended to use statistics like D+ that are designed for local analysis and to validate findings with additional demographic context and independent methods [5].
Q4: Why are small sequence fragments (like 15-mers) problematic in genomic studies and patent claims? Short sequences are highly non-specific. A 15-nucleotide sequence from one gene can perfectly match sequences in hundreds of other genes. This creates extensive "cross-matches," leading to ambiguous results in sequence-based assays and uncertain infringement liability in patent claims. For example, a 15mer from the human BRCA1 gene matches at least 689 other genes [13].
Q5: What are the common challenges in detecting microexons, and how do they relate to broader issues in genomic analysis? Microexons (≤15 nucleotides) are frequently misannotated or missed entirely in genome annotations due to their small size, which challenges standard RNA-seq read mapping and statistical models for gene prediction. This reflects a broader pervasiveness of detection problems for small genomic features, similar to the challenges of identifying short, informative sequences for introgression analysis [14].
Troubleshooting Guides
| Observation | Potential Cause | Solution |
|---|---|---|
| High D statistic in small genomic windows | Low nucleotide diversity inflating variance | Use the D+ statistic to incorporate shared ancestral alleles and increase informative sites [5]. |
| Spurious local introgression signals | Incomplete Lineage Sorting (ILS) mimicking gene flow | Apply the D+ statistic; simulate data under a null model of no gene flow to establish a baseline [5]. |
| Inability to detect local introgression | Low number of informative derived alleles in the region | Switch from D to D+ statistic, which uses both derived and ancestral alleles to increase power [5]. |
| Primer dimers (40-80 bp peaks) in library prep | Self-annealing of primers during PCR | Perform a bead purification using the same ratio as the final library purification to remove the contaminant [15]. |
| Adaptor dimers (120-160 bp peaks) in library prep | Ligation of adaptors to each other instead of target DNA | Perform an additional bead purification; ensure fresh ethanol is used and beads are fully dry before elution [15]. |
| Low library yields from SPRI bead purifications | Beads not at room temperature, improper mixing, or old ethanol | Allow beads to reach room temperature; mix bead suspension and sample thoroughly; use fresh 70% ethanol [15]. |
Experimental Protocols
Protocol 1: Detecting Local Introgression with the D+ Statistic
- Define Populations: Select four populations for analysis: two closely related populations (P1 and P2) that are potential recipients of gene flow, a donor population (P3), and an outgroup (P4).
- Sequence and Call Variants: Obtain high-coverage genome sequencing data for all populations. Perform variant calling to identify biallelic Single Nucleotide Polymorphisms (SNPs).
- Determine Ancestral Alleles: Use the outgroup (P4) to determine the ancestral (A) and derived (B) allele at each SNP site.
- Calculate Site Patterns: For a given genomic window, tabulate the counts of the following site patterns across all SNPs:
- ABBA: Derived allele in P3 and P2, ancestral in P1.
- BABA: Derived allele in P3 and P1, ancestral in P2.
- BAAA: Ancestral allele in P3 and P2, derived only in P1.
- ABAA: Ancestral allele in P3 and P1, derived only in P2.
- Compute D+: Calculate the D+ statistic for the window using the formula:
- Formula: D+ = [Σ(ABBA - BABA) + Σ(BAAA - ABAA)] / [Σ(ABBA + BABA) + Σ(BAAA + ABAA)]
- Interpretation: A significant positive D+ value indicates introgression from P3 into P2. A significant negative value indicates introgression from P3 into P1 [5].
Protocol 2: A Combined Pipeline for Plant Microexon Discovery
- Data Preparation: Collect RNA-seq datasets from multiple tissues and conditions.
- Read Mapping (Combined Approach):
- Perform ab initio spliced read mapping using OLego to identify novel splice junctions without reference to existing annotations.
- Perform a second mapping with STAR, using a splice junction database that combines existing genome annotations and the novel junctions found by OLego.
- Microexon Identification: Identify candidate internal microexons (1-15 nt) supported by junction reads. Require a minimum of 5 reads spanning each splice junction.
- Filtering and Validation: Filter candidates and experimentally validate a subset via RT-PCR followed by sequencing [14].
Methodology Visualization
Four-Population Tree for Introgression Detection
D+ Statistic Calculation Workflow
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Tool | Function in Analysis |
|---|---|
| D+ Statistic | A refined population genetic statistic that leverages both shared derived and ancestral alleles to detect local genomic introgression with higher precision than the D statistic [5]. |
| OLego | A de novo spliced read mapping tool that is highly effective for the initial discovery of novel splice junctions, including those bordering microexons, without prior annotation [14]. |
| STAR | A fast and accurate RNA-seq aligner that is optimal for annotation-guided mapping. Used in conjunction with OLego for comprehensive microexon discovery [14]. |
| SPRI Beads | Magnetic beads used for the size-selective purification of DNA fragments (e.g., in NGS library preparation) to remove unwanted products like primer and adaptor dimers [15]. |
| EvaGreen | A fluorescent DNA dye that binds double-stranded DNA with high specificity. Recommended for optional qPCR steps to accurately estimate the cycle number needed for NGS library amplification [15]. |
Building a Robust Toolkit: Methodological Approaches to Minimize False Inferences
IntroMap is a bioinformatics pipeline that employs signal analysis techniques on next-generation sequencing (NGS) data to detect genomic introgressions—the transfer of genetic material between species through hybridization and backcrossing [16]. Designed primarily for plant breeding programs, it offers an automated approach to screen large populations, potentially replacing more labor-intensive marker-assisted assays. Its key innovation is the accurate identification of introgressed genomic regions using alignment to a reference genome without requiring a variant calling step or de novo assembly of the read data [16]. This article provides a technical support center for researchers using IntroMap, framed within the critical context of mitigating false positive signals in introgression research.
Troubleshooting Guides
Installation and Environment Setup
Problem: Conda environment creation fails.
- Solution: Ensure your conda installation is up to date. If the provided YAML file with specific package versions fails, try using the version-agnostic YAML file available on the IntroMap GitHub repository, which offers more flexibility [17].
Problem: Unable to execute the IntroMap.py script.
- Solution: Verify that the script has executable permissions. Run
chmod 755 IntroMap.pyin your terminal to grant the necessary permissions [17].
Runtime and Input Errors
Problem: The pipeline terminates with an error related to input files.
- Solution: Confirm that your input BAM file is properly aligned to the reference genome you provide using the
-roption. The reference genome must share homology with the recurrent parental cultivar used in the experiment [16] [17].
Problem: No output files are generated, or the output is empty.
- Solution: Check the command-line arguments. The
-oparameter specifies the output filename format. For example, using-o lowesswould create files likechr1.lowess. Ensure you have write permissions in the output directory [17].
Performance and Interpretation
Problem: The pipeline runs successfully, but no introgressed regions are reported.
- Solution: Adjust the classification threshold using the
-tparameter (default is 0.90) and the-bflag, which controls whether to report regions below (True) or at/above (False) the threshold [17]. The optimal threshold may vary based on your specific data and the expected signal strength.
Problem: Results are inconsistent with marker-based assays or other methods.
- Solution: Be aware that methods for detecting introgression, including the popular D-statistic, can produce false-positive signals when evolutionary rates vary between the divergent taxa being analyzed [18]. These rate variations lead to homoplasies (independent substitutions at the same site) that can be misinterpreted as evidence of introgression [18]. Always validate key findings with independent methods.
Frequently Asked Questions (FAQs)
Q1: What is the primary advantage of IntroMap over variant-calling methods? IntroMap identifies introgressed regions directly from alignment data, bypassing the variant calling step. This can reduce complexity and improve accuracy compared to some marker-based approaches [16].
Q2: What input data does IntroMap require? The pipeline requires an aligned BAM file from your sample and the reference genome sequence (in FASTA format) to which the reads were aligned. Genome annotation is not required [16] [17].
Q3: How does IntroMap mitigate issues with false positives? While the core method relies on alignment and signal analysis, users should be cautious of general limitations in introgression detection. A significant source of false positives in many methods, including the D-statistic, is variation in evolutionary rates among lineages [18]. IntroMap's signal-based approach may offer a different pathway, but it is crucial to tune parameters and validate results experimentally.
Q4: Can IntroMap be used for ancient introgression events? The pipeline was demonstrated on plant breeding data. Methods developed for recent introgression (like the D-statistic) can be unreliable for ancient events due to accumulated homoplasies and evolutionary rate variation [18]. IntroMap's applicability to very old hybridization events would require further validation.
Q5: Where can I find the software and its documentation? The IntroMap software is freely available on GitHub at https://github.com/danshea/IntroMap, which includes a Jupyter notebook with usage examples [17].
Essential Research Reagent Solutions
The following table details key materials and resources required for a successful IntroMap experiment.
Table 1: Key Research Reagents and Resources for IntroMap
| Item Name | Function / Description | Critical Parameters |
|---|---|---|
| Reference Genome | A genome sequence sharing homology with the recurrent parental line. | Required format: FASTA. Genome annotation is not necessary [16] [17]. |
| Aligned Sequencing Data | The sample data to be screened for introgressed regions. | Must be provided as a BAM file, aligned to the specified reference genome [17]. |
| Diagnostic SNP Sets | For mapping alien introgressions from specific donor species. | In Arachis, for example, pre-compiled sets for five diploid species are available. The pipeline can also generate new diagnostic SNPs [19]. |
| Conda Environment | A reproducible software environment to run IntroMap. | Resolves dependencies (e.g., Python, NumPy, SciPy). Use the YAML file from the GitHub repository [17]. |
Experimental Workflow and Signaling Pathways
The following diagram illustrates the logical workflow of the IntroMap pipeline, from data input to final output.
Diagram 1: IntroMap analysis workflow
Methodological Protocols
Core Introgression Detection Protocol using IntroMap
- Data Input Preparation: Generate a BAM file by aligning your NGS reads to a reference genome that is closely related to the recurrent parent in your hybridization experiment [16] [17].
- Software Activation: Activate the IntroMap conda environment using the command
source activate IntroMap[17]. - Pipeline Execution: Run the IntroMap.py script from the command line. A sample invocation is:
where
-iis the input BAM,-ris the reference,-ois the output prefix,-tis the classification threshold,-b Truereports regions below the threshold, and-fsets the LOWESS smoothing window as a fraction of the chromosome length [17]. - Output Analysis: The pipeline generates output files for each chromosome containing the predicted introgressed regions. It also creates visualizations (PNG files) of the predictions for manual inspection [17].
- Validation: As with any computational prediction, it is critical to validate introgressed regions identified by IntroMap using independent methods, such as targeted marker-based assays or PCR, to confirm their biological validity [16].
Protocol for Mitigating False Positives from Rate Variation
- Awareness of Limitations: Understand that all introgression detection methods, including tree-based and site-based (D-statistic) approaches, are susceptible to false positives caused by homoplasies when evolutionary rates differ between lineages [18].
- Method Selection Consideration: For analyses involving highly divergent taxa, where rate variation is more likely, be cautious in interpreting results. A tree-based equivalent of the D-statistic (
Dtree) has been used in some studies, but it may also be affected by rate variation [18]. - Independent Verification: Employ a new test based on the expected clustering of introgressed sites along the genome, as implemented in the program Dsuite, to help distinguish genuine introgression from patterns caused by rate variation [18]. Using multiple, methodologically distinct detection approaches strengthens the evidence for true introgression.
Frequently Asked Questions (FAQs)
Q1: What is the fundamental difference between the D-statistic and the HyDe method? Both the D-statistic and HyDe use site pattern frequencies to detect hybridization but differ in their specific approach and output. The D-statistic (or ABBA-BABA test) calculates a normalized difference in the frequencies of two site patterns (ABBA and BABA) that are expected to be equal under a null model of no gene flow [20]. A significant deviation from zero is evidence of introgression. In contrast, HyDe uses a ratio of these same site pattern frequencies to not only test for the presence of hybridization but also to directly estimate the admixture proportion, gamma (γ) [20].
Q2: My analysis with HyDe shows significant hybridization, but I suspect it might be a false positive due to ancestral polymorphism. How can I investigate this? Ancestral polymorphism can create gene tree patterns that mimic hybridization. To mitigate this, you can:
- Incorporate an Outgroup: Always use a carefully selected outgroup population that diverged before the speciation event of your ingroup taxa. This helps polarize the site patterns (e.g., determine ancestral vs. derived states) and is a requirement for both D-statistic and HyDe analyses [20].
- Use a Network-Based Method: Consider using phylogenetic network inference software (e.g., like those referenced in [20]) as a complementary approach. These methods can explicitly model both incomplete lineage sorting and hybridization, helping to distinguish between the two processes.
- Validate with Simulations: Perform coalescent simulations under a model without hybridization but with ancestral population structure. If your empirical data shows significantly stronger signals than these simulations, it strengthens the case for true hybridization.
Q3: When running HyDe with multiple individuals per population, what is the software actually doing? HyDe leverages all available individuals by calculating site patterns across all possible quartets formed by individuals from the four populations (Outgroup, P1, Hybrid, P2) [20]. This approach increases the effective sample size for estimating site pattern probabilities, which can lead to more accurate parameter estimates and greater statistical power for detecting hybridization.
Q4: Can HyDe tell me if only some individuals in a population are hybrids?
Yes, this is a key feature of HyDe. The standard test assumes all individuals in the putative hybrid population are admixed. However, HyDe provides the individual_hyde.py script to test each individual within the hybrid population separately [21] [22]. Furthermore, the bootstrap_hyde.py script can be used to assess heterogeneity in the admixture process through resampling [20] [21]. Non-uniform bootstrap support distributions can indicate that not all individuals are hybrids.
Q5: What are the common sources of false positives in D-statistic and HyDe analyses?
- Incorrect Rooting: Using an outgroup that does not properly root the tree can lead to incorrect interpretation of site patterns.
- Ancestral Population Structure: Population structure in the ancestral population of P1, P2, and the hybrid can create spurious signals of introgression.
- Genome Assembly Biases: Reference genome biases or alignment errors can create systematic artifacts that mimic introgression signals.
- Uneven Missing Data: If missing data is not random and is correlated with population relationships, it can bias site pattern counts.
Troubleshooting Guides
Issue 1: Weak or Non-Significant Introgression Signal
Problem: Your analysis yields a non-significant D-statistic or HyDe p-value, but you have a biological reason to expect hybridization.
Solution:
- Verify Data Quality:
- Check for an adequate number of informative sites (SNPs). Genome-scale data is typically required for sufficient power [20].
- Ensure high-quality alignments and variant calls to minimize noise.
- Check Population Labels:
- Confirm that you have correctly specified the parental populations (P1 and P2). Mis-specification will severely reduce power. Use population structure analyses (e.g., PCA, ADMIXTURE) to validate your assignments.
- Explore Different Triplets:
- Consider the Direction of Gene Flow: The signal might be stronger if you swap your hypotheses for P1 and P2.
Issue 2: Suspected False Positive Due to Ancestral Polymorphism
Problem: You detect a strong and significant signal of introgression, but you are concerned it is caused by incomplete lineage sorting (ILS) rather than true hybridization.
Solution:
- Re-run Analysis with a Closer Outgroup: If possible, use an outgroup that is more closely related to the ingroup. A more distant outgroup has a higher probability of containing ancestral polymorphisms.
- Conduct a D-statistic Test with Multiple Outgroups: Compare the D-statistic results across different outgroups. A consistent signal across multiple outgroups is more robust.
- Use the
bootstrap_hyde.pyScript: Bootstrap resampling of individuals can provide a distribution of the estimated admixture parameter (γ). A wide and unstable distribution might indicate a weak or false signal [20] [22]. - Compare with Other Methods: Corroborate your findings using an independent method. For example, use a phylogenetic network tool or a supervised machine learning approach like FILET, which combines multiple summary statistics to identify introgressed loci [23].
Issue 3: Interpreting HyDe's Output for Individual Tests
Problem: You have run individual_hyde.py and get varied p-values and γ estimates for different individuals within the same population.
Solution:
- Identify Pure Hybrids and Non-Hybrids: Individuals with significant p-values and intermediate γ estimates are strong hybrid candidates. Individuals with non-significant p-values are likely non-hybrids.
- Check for Backcrossing: Individuals with a significant signal but a γ estimate very close to 0 or 1 might be backcrosses to one parental population (F1 hybrid crossed with a pure parent).
- Visualize the Results: Create a plot of the γ estimate for each individual (or its bootstrap distribution) to easily visualize the variation in admixture proportions within the population.
The table below summarizes the core quantitative relationships for the D-statistic and HyDe methods.
Table 1: Key Formulas and Thresholds for Site-Pattern Methods
| Method | Core Formula / Statistic | Null Hypothesis Value | Key Output |
|---|---|---|---|
| D-Statistic | ( D = \frac{(N{ABBA} - N{BABA})}{(N{ABBA} + N{BABA})} ) [20] | D = 0 (ABBA = BABA) | Test statistic (Z-score) for introgression [20] |
| HyDe | ( \gamma = 1 - \frac{f{BABA}}{f{ABBA}} ) (estimated from data) [20] | γ = 0 (Ratio test) | P-value for hybridization test and γ, the admixture proportion [20] |
Table 2: Essential Research Reagent Solutions for HyDe Analysis
| Item / Reagent | Function in Analysis | Implementation Note |
|---|---|---|
| Genomic SNP Data | The raw input data used to calculate site pattern counts (ABBA, BABA, etc.). | Data should be unlinked SNPs, ideally from genome-wide sequencing [20]. Can be in Phylip, Plink, or VCF format. |
| Population Map File | A text file specifying which individuals belong to which population. | Critical for correctly assigning individuals to Outgroup, P1, Hybrid, and P2 roles. |
| HyDe Software (phyde) | The Python package that performs the phylogenetic invariants-based test [22]. | Installed via pip install phyde. Contains the core scripts: run_hyde.py, individual_hyde.py, and bootstrap_hyde.py [21] [22]. |
| Triples File | A text file specifying the combinations of populations (P1, Hybrid, P2) to test. | Required for individual_hyde.py and bootstrap_hyde.py. Can be generated from the output of run_hyde.py [21]. |
Experimental Protocols
Protocol 1: Standard Genome-Wide Hybridization Detection with HyDe
This protocol describes a standard workflow for running a HyDe analysis on a genomic dataset.
1. Input Data Preparation:
- Obtain a aligned genomic sequence data or a called SNP dataset in a supported format (e.g., Phylip, VCF).
- Create a population map file, a simple text file with two columns:
individual_nameandpopulation_name.
2. Software Installation:
- Install the HyDe package using pip:
pip install phyde[22].
3. Running the Analysis:
- Execute the main analysis script from the command line:
- This script will test all possible triplets and produce a filtered results file showing significant hybridization events.
4. Results Interpretation:
- Examine the output file. Key columns include:
P1,Hybrid,P2: The tested populations.Gamma: The estimated admixture proportion.Z_scoreandP_value: The test statistic and its significance.
Protocol 2: Testing for Heterogeneous Introgression within a Population
This protocol is used when you suspect that not all individuals in a "hybrid" population are actually admixed.
1. Generate a Triples File:
- Use the significant results from your
run_hyde.pyanalysis or create a custom file listing the specific (P1, Hybrid, P2) triplets you wish to investigate.
2. Run Individual-Level Tests:
- Use the
individual_hyde.pyscript to test each individual in the hybrid population:
3. Run Bootstrap Resampling:
- To assess the stability of the admixture signal, run:
4. Interpret Individual Variation:
- The individual test output will provide a γ and p-value for each specimen. The bootstrap output allows you to visualize the distribution of γ, helping you identify whether admixture is a uniform property of the population.
Workflow and Conceptual Diagrams
HyDe Analysis and Troubleshooting Workflow
Conceptual Model of Signals and False Positives
In genomic research, accurately distinguishing somatic mutations from germline variants is a critical challenge. Somatic variants arise in non-germline tissues and are not inherited, playing key roles in diseases like cancer, while germline variants are present in all cells and are inherited from parents. This guide provides troubleshooting and best practices for leveraging allele frequency spectra to differentiate these variant types, a methodology crucial for avoiding false positives in introgression and ancestry analysis.
## Key Concepts and Definitions
Allele Frequency Spectrum (AFS): The distribution of allele frequencies across multiple genomic sites in a population. Somatic Variants: DNA alterations acquired in somatic (non-germline) cells, not present in all cells, and not inherited by offspring. Germline Variants: DNA variations present in germ cells, inherited from parents, and found in all cells of an organism. Variant Allele Frequency (VAF): The proportion of sequencing reads at a genomic locus that support a specific variant.
## Allele Frequency Characteristics: Somatic vs. Germline
Table 1: Key Characteristics of Somatic and Germline Variants Based on Allele Frequency Spectra
| Characteristic | Germline Variants | Somatic Variants |
|---|---|---|
| Expected VAF in Heterozygous State | ~50% (or 100% for homozygous) [24] [25] | Highly variable (5-100%), often subclonal [24] |
| Distribution in Population | Follows Hardy-Weinberg equilibrium in large cohorts [26] | Population-specific, tissue-specific |
| Presence in Matched Normal Tissue | Present in all tissues [25] | Absent in matched normal tissue [24] [25] |
| Supporting Evidence | Population databases (gnomAD), clinical databases (ClinVar) [27] | Somatic databases (COSMIC), tumor-only evidence [27] [28] |
## Experimental Protocols
### Protocol 1: Tumor-Normal Paired Analysis
Purpose: To identify somatic variants by comparing tumor tissue to matched normal tissue.
Materials:
- DNA from tumor tissue
- DNA from matched normal tissue (e.g., blood, saliva)
- Next-generation sequencing platform
- Computational resources
Method:
- Sequencing: Perform whole-exome or whole-genome sequencing on both tumor and normal samples to a minimum coverage of 30x for normal and 60x for tumor samples [24].
- Variant Calling: Use somatic variant callers such as:
- Variant Filtering: Apply filters to remove artifacts:
- Remove variants with VAF < 5% in tumor unless ultra-deep sequencing
- Exclude variants present in matched normal at VAF > 2%
- Filter against population databases (gnomAD) to remove common germline variants [27]
- Annotation: Use annotation tools such as:
### Protocol 2: Tumor-Only Analysis with Germline Contamination Assessment
Purpose: To identify somatic variants when matched normal tissue is unavailable.
Materials:
- DNA from tumor tissue
- Population frequency databases (gnomAD)
- Computational resources
Method:
- Sequencing: Sequence tumor sample to high coverage (≥100x recommended).
- Variant Calling: Use tools capable of tumor-only analysis:
- GATK Mutect2's tumor-only mode [28]
- Implement additional filtering for germline contamination
- Germline Filtering:
- Validation: Confirm putative somatic variants with orthogonal methods (e.g., digital PCR, Sanger sequencing).
### Protocol 3: RNA-Seq Based Variant Calling
Purpose: To identify expressed variants and assess allele-specific expression.
Materials:
- RNA from tissue of interest
- RNA-Seq library preparation kit
- Computational resources
Method:
- Library Preparation and Sequencing: Perform RNA sequencing with a minimum of 70 million reads [24].
- Alignment: Use STAR two-pass alignment to GRCh38 [24].
- Variant Calling:
- Use GATK best practices for RNA-seq short variant discovery [24]
- Apply base quality score recalibration and split reads with N in CIGAR string
- Classification:
- Implement VarRNA or similar tool to classify variants as germline, somatic, or artifact [24]
- Compare VAFs between DNA and RNA to identify allele-specific expression
Diagram 1: Experimental Workflow for Variant Differentiation
## Troubleshooting FAQs
### FAQ 1: How do I handle variants with intermediate VAF (30-70%) in tumor-only samples?
Issue: Variants with VAF between 30-70% could be either germline heterozygous variants or clonal somatic variants, creating ambiguity in tumor-only analyses.
Solution:
- Database Filtering: Check population frequency databases (gnomAD). Variants with population frequency >0.1% are likely germline [27] [25].
- B-allele Frequency: Analyze B-allele frequency across the genomic region to identify copy number changes that might alter VAF expectations.
- Machine Learning Classification: Implement tools like VarRNA that use XGBoost models trained on multiple features to classify variants [24].
- Pathogenicity Assessment: Evaluate variant pathogenicity using ClinVar and other clinical databases - pathogenic germline variants in cancer predisposition genes may be present at ~50% VAF but are clinically significant [29] [25].
### FAQ 2: What are the best practices for minimizing false positives in somatic variant calling?
Issue: High false positive rates in somatic variant calling due to sequencing artifacts, mapping errors, and germline contamination.
Solution:
- Sequencing Depth: Ensure adequate sequencing depth (≥100x for tumor, ≥30x for normal) [24].
- Duplicate Removal: Remove PCR duplicates to avoid artifacts [28].
- Base Quality Recalibration: Implement base quality score recalibration (BQSR) as in GATK best practices [24].
- Strand Bias Filtering: Filter variants with significant strand bias (Fisher's exact test p-value < 0.05).
- Annotation Filtering: Use databases of common sequencing artifacts and cross-replicate validation.
### FAQ 3: How can I differentiate true somatic variants from RNA editing events in RNA-Seq data?
Issue: RNA-seq variant calling may detect RNA editing events rather than true DNA variants, leading to misinterpretation.
Solution:
- Database Comparison: Cross-reference with known RNA editing databases (e.g., REDIportal).
- DNA-RNA Concordance: Require DNA validation for variants called from RNA-seq alone [24].
- Editing Context: Identify characteristic sequence contexts of common RNA editing (e.g., A-to-I editing in Alu elements).
- VAF Comparison: Compare VAF between DNA and RNA - RNA editing events typically show sub-50% VAF and may be tissue-specific.
### FAQ 4: What thresholds should I use for allele frequency in population filtering?
Issue: Inappropriate allele frequency thresholds can either remove true somatic variants or retain common germline variants.
Solution: Table 2: Recommended Allele Frequency Thresholds for Variant Filtering
| Application | Recommended Threshold | Rationale |
|---|---|---|
| Common Germline Filter | MAF < 0.1% in gnomAD [29] [27] | Removes polymorphic germline variants while retaining rare disease-associated variants |
| Tumor-Only Somatic Calling | VAF 10-90% with supporting evidence [25] | Allows for subclonal populations and aneuploidy effects |
| Low-Frequency Somatic | VAF ≥ 5% (≥1% with ultra-deep sequencing) | Balances sensitivity with false positive rates |
| Germline Contamination | VAF 40-60% in tumor-only plus population frequency >1% [25] | Identifies likely heterozygous germline variants |
## Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for Variant Analysis
| Tool/Reagent | Function | Application Context |
|---|---|---|
| GATK Mutect2 [24] [28] | Somatic variant calling | Tumor-normal paired and tumor-only analysis |
| VarRNA [24] | Machine learning classification of RNA variants | Differentiation of germline/somatic variants from RNA-seq data |
| Ensembl VEP [28] | Variant effect prediction | Functional annotation of coding and non-coding variants |
| gnomAD [27] | Population frequency database | Filtering of common germline polymorphisms |
| COSMIC [27] [28] | Catalog of somatic mutations | Evidence for somatic variant classification |
| AutoGVP [29] | Automated germline variant pathogenicity classification | Standardized variant interpretation per ACMG/AMP guidelines |
| ANNOVAR [28] | Functional annotation of genetic variants | Comprehensive variant annotation including dbNSFP, ClinVar |
## Advanced Analysis Techniques
### Allele Frequency Spectrum Analysis
Purpose: To distinguish somatic from germline variants based on population-level allele frequency distributions.
Method:
- Cohort AFS Construction: Calculate allele frequencies across your study cohort for all putative variants.
- Distribution Analysis: Germline variants will show peaks at ~50% (heterozygous) and 100% (homozygous), while somatic variants show a continuous distribution skewed toward lower frequencies [26].
- Statistical Modeling: Implement binomial mixture models to identify different variant classes based on their AFS.
- Validation: Use known germline and somatic variants from public databases to validate the AFS-based classification.
Diagram 2: Allele Frequency Spectrum Analysis Workflow
### Machine Learning Approaches
Purpose: To improve classification accuracy using multiple variant features beyond allele frequency.
Method:
- Feature Selection: Include VAF, read depth, base quality, mapping quality, population frequency, and functional impact.
- Model Training: Train XGBoost or random forest classifiers using validated variant sets [24].
- Cross-Validation: Use k-fold cross-validation to assess model performance.
- Implementation: Apply trained models to novel datasets for variant classification.
## Quality Control and Validation
### Internal QC Metrics
- Sequenceing Depth: Minimum 30x for germline, 60x for somatic variants
- VAF Concordance: Technical replicates should show VAF correlation R² > 0.98
- Positive Controls: Include known variants with expected VAFs in each run
### External Validation
- Orthogonal Methods: Validate a subset of variants using digital PCR or Sanger sequencing
- Database Comparison: Compare variant calls with public datasets (TCGA, ICGC)
- Proficiency Testing: Participate in external quality assessment programs [28]
Frequently Asked Questions
Q1: Why should I use a Bayesian approach over traditional methods for estimating allele frequencies?
Traditional methods, like simply counting observed alleles, can be unreliable with modern sequencing data, which often has high error rates and low coverage [26]. A Bayesian framework allows you to formally incorporate prior knowledge, such as allele frequency distributions from evolutionarily related populations, to improve estimation accuracy. Crucially, it avoids the need for a "hard threshold" on whether to combine data from different samples, which can introduce bias. Instead, it uses a continuous "affinity measure" to adaptively borrow strength from related samples, typically resulting in a lower mean squared error for the frequency estimates [30].
Q2: How can I mitigate false positive signals of introgression that are actually caused by ancestral genetic polymorphism?
Ancestral polymorphism can be misidentified as recent introgression because both processes can create similar genetic patterns. To mitigate these false positives, your analysis should:
- Use an Unbiased Root: Employ an outgroup species to root your phylogenetic trees reliably. This helps distinguish shared ancestral alleles (present in the common ancestor) from alleles that have moved between species after divergence (introgression) [23].
- Refine Species Borders: Use patterns of gene flow to define species boundaries (creating BSC-species) rather than relying solely on arbitrary genetic distance thresholds (ANI-species). This can prevent the misclassification of recently diverged lineages within a single species as separate species experiencing high introgression [31].
- Leverage Machine Learning: Use supervised machine learning frameworks like FILET (Finding Introgressed Loci using Extra-Trees Classifiers) that combine multiple population genetic summary statistics. These methods are trained to recognize the complex genomic signatures of true introgression and can achieve higher accuracy and power than single-statistic approaches [23].
Q3: What are the main differences between the STRUCTURE software and the empirical Bayes approach for modeling population structure?
While both methods use Bayesian principles, they are designed for different primary purposes and operate differently, as summarized in the table below.
| Feature | STRUCTURE | Empirical Bayes for Allele Frequencies |
|---|---|---|
| Primary Purpose | Identify populations and assign individuals to them based on genetic similarity [32]. | Improve the estimation of allele frequencies in a target population using data from related populations [30]. |
| Core Methodology | Uses a Bayesian clustering algorithm with MCMC to estimate individual ancestry proportions (the Q-matrix) [32]. | Uses an empirical prior (e.g., a Beta distribution) informed by related samples to compute a posterior estimate for the frequency at each marker [30]. |
| Key Assumptions | Assumes markers are in linkage equilibrium and selectively neutral; some models assume Hardy-Weinberg equilibrium within populations [32]. | Assumes independence between markers and Hardy-Weinberg equilibrium within the target population [30]. |
| Output | Individual membership coefficients to K clusters; inference of the most likely number of populations (K) [32]. | A refined, lower-variance estimate of the allele frequency for each genetic marker in the target population [30]. |
Q4: My Bayesian model for genomic prediction is computationally slow. Are there efficient alternatives to MCMC?
Yes, several efficient alternatives exist. You can use models like Bayes-C0, which is equivalent to Genomic BLUP (GBLUP) and can be solved using efficient linear algebra methods without MCMC [33]. Furthermore, fast, non-MCMC approaches based on the Expectation-Maximization (EM) algorithm have been developed for models like Bayes-A and Bayes-B, providing estimates that are the maximum likelihood equivalent of the Bayesian approaches [33].
Experimental Protocols & Methodologies
Protocol 1: Empirical Bayes Estimation of Allele Frequencies with a Booster Sample
This protocol outlines the method for improving allele frequency estimates in a primary target population by incorporating data from a related "booster" population [30].
- Data Preparation: For a large number of biallelic markers (e.g., SNPs), collect genotype data. The data should include a primary sample of
nalleles from your target population, ( \mathcal{P} ), and a booster sample ofnalleles from a related population, ( \mathcal{S} ). - Calculate Observed Frequencies: For each marker
i, compute the maximum likelihood estimate (MLE) in each population:- ( \hat{q}i = Xi / n ) (Frequency in target population ( \mathcal{P} ))
- ( \hat{p}i = Yi / n ) (Frequency in booster population ( \mathcal{S} ))
- Establish an Empirical Prior:
- For a given marker
iof interest in ( \mathcal{P} ), consider the set of all other markersjwhose observed frequency in ( \mathcal{S} ), ( \hat{p}j ), is within a small window of ( \hat{p}i ). - Examine the distribution of the allele counts ( Xj ) in ( \mathcal{P} ) for this set of markers.
- Fit a Beta distribution to this empirical distribution using maximum likelihood. This fitted Beta distribution, ( \text{Beta}(a, b) ), serves as your empirical prior for the true frequency ( qi ) at marker
i.
- For a given marker
- Compute the Posterior Estimate:
- The prior is ( q_i \sim \text{Beta}(a, b) ).
- The likelihood of observing ( Xi ) successes in
ntrials is Binomial: ( Xi | qi \sim \text{Bin}(n, qi) ). - Thanks to conjugacy, the posterior distribution is also a Beta distribution: ( qi | Xi \sim \text{Beta}(a + Xi, b + n - Xi) ).
- The Bayesian point estimate for the allele frequency is the mean of this posterior distribution: ( \hat{q}i^{\text{Bayes}} = (a + Xi) / (a + b + n) ).
The following diagram illustrates the logical workflow and the relationship between the data, the prior, and the posterior in this empirical Bayes approach.
Protocol 2: A Bayesian Multiple Regression Framework for Genome-Wide Association (GWA)
This protocol uses Bayesian variable selection models for GWA analysis to identify markers associated with a quantitative trait, which helps control for population structure and mitigates false positives [33].
- Model Specification: Define a multiple regression model where the phenotype of an individual is a function of all genotyped markers simultaneously:
( y = \mu + \sum{k=1}^K zk \betak + e )
Here, ( y ) is the vector of phenotypes, ( \mu ) is the overall mean, ( zk ) is the genotype vector for SNP
k, ( \beta_k ) is its effect, and ( e ) is the residual. - Choose a Prior for Marker Effects: Select an appropriate prior distribution from the "Bayesian Alphabet" to match your assumptions about the genetic architecture:
- Bayes-A: Each SNP effect has a normal prior with its own variance, which itself follows a scaled inverse chi-square distribution. This allows for a t-distributed prior for effects.
- Bayes-B: A variable selection model. A pre-specified proportion ( \pi ) of SNPs are assumed to have zero effect, while the non-zero effects have a normal prior with a common variance.
- Bayes-Cπ: Similar to Bayes-B, but the proportion ( \pi ) of zero-effect SNPs is treated as unknown and estimated from the data.
- Model Fitting via MCMC: Use Markov Chain Monte Carlo (MCMC) sampling (e.g., Gibbs sampling) to obtain samples from the joint posterior distribution of all model parameters. This includes the SNP effects (( \beta_k )), the residual variance, and any hyperparameters.
- Inference for GWA: For GWA, identify SNPs with a significant effect by monitoring the posterior inclusion probability for each SNP (in variable selection models like Bayes-B/Cπ) or the posterior distribution of its effect size. A common approach is to calculate the probability that a SNP's effect is non-zero or exceeds a certain threshold.
The workflow for this Bayesian GWA analysis is visualized below, showing the key stages from data preparation to association inference.
Table 1: Performance Comparison of Allele Frequency Estimation Methods in Low-Coverage Sequencing
A comparison of a maximum likelihood (ML) method that integrates over genotype uncertainty versus traditional methods based on genotype calling, as applied to low/medium-coverage next-generation sequencing data [26].
| Method | Key Feature | Reported Performance |
|---|---|---|
| Maximum Likelihood (ML) | Directly estimates allele frequency from sequencing data without first calling genotypes, integrating over individual genotype uncertainty. | Outperformed genotype calling methods in accuracy of allele frequency estimation and statistical power in association studies [26]. |
| Genotype Calling with Filtering | First calls a genotype for each individual using a confidence threshold (e.g., 10x more likely than next best), then treats called genotypes as correct. | Less accurate for estimating the distribution of allele frequencies and for association mapping compared to the ML method. Filtering based on call confidence can further reduce performance [26]. |
Table 2: Reported Levels of Core Genome Introgression Across Bacterial Genera
Summary of introgression levels found in a systematic analysis of 50 bacterial lineages, highlighting the variation between different taxonomic and analytical definitions [31].
| Lineage / Group | Reported Level of Introgression | Context and Notes |
|---|---|---|
| Average across 50 genera | 8.13% (median 2.76%) | Percentage of core genes inferred as introgressed between ANI-defined species [31]. |
| Escherichia–Shigella | Up to 14% | The genus with the highest observed level of introgression [31]. |
| Streptococcus parasanguinis ANI-sp32 | 33.2% | High introgression with ANI-sp67; however, these were reclassified as a single BSC-species after gene-flow analysis, showing how species definition impacts introgression estimates [31]. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Software and Statistical Tools
| Tool / Reagent | Function in Analysis |
|---|---|
| STRUCTURE | A widely used software for inferring population structure and assigning individuals to populations using a Bayesian clustering algorithm [32]. |
| Bayesian Alphabet Methods (e.g., Bayes-A, B, Cπ) | A family of Bayesian multiple regression models used for genomic prediction and genome-wide association studies (GWA) by fitting all marker effects simultaneously [33]. |
| FILET (Finding Introgressed Loci using Extra-Trees) | A supervised machine learning framework that combines multiple population genetic statistics to identify introgressed genomic regions with high power and infer directionality [23]. |
| Extra-Trees Classifier (Extremely Randomized Trees) | The machine learning algorithm underlying FILET; it creates an ensemble of decision trees for robust classification, here used to distinguish introgressed from non-introgressed loci [23]. |
| ANI (Average Nucleotide Identity) | A genomic metric used to define bacterial species boundaries based on a percentage identity cutoff (e.g., 94-96%), resulting in "ANI-species" [31]. |
| BSC-species (Biological Species Concept) | In bacteria, a species definition based on patterns of gene flow (homologous recombination), refined from ANI-species to reflect genetic cohesiveness [31]. |
Troubleshooting Guides
Issue 1: High False Positive Rates in Introgression Detection
- Problem: A high proportion of candidate introgressed regions are likely false positives, potentially due to ancestral polymorphism or sequencing errors in repetitive regions.
- Solution:
- Apply RAfilter: Use the RAfilter algorithm to identify and remove false-positive alignments in repetitive genomic regions by leveraging rare k-mers that represent copy-specific features. Filter alignments based on the kMAPQ score [34].
- Utilize Ensemble Genotyping: Reduce false positives by integrating multiple variant calling algorithms instead of relying on a single pipeline [35].
- Implement RNDmin: Confirm candidates with the RNDmin statistic, which is robust to variation in mutation rates and inaccurate divergence time estimates [4].
Issue 2: Low Sensitivity to Recent or Rare Introgression
- Problem: Standard methods like
dXYorFSTfail to detect recent or rare introgressed lineages because they are not sensitive to low-frequency migrants [4]. - Solution:
- Use
dminorGmin: Switch to statistics likedmin(the minimum sequence distance between any pair of haplotypes from two taxa) orGmin(which normalizesdminbydXYto account for mutation rate variation). These methods are more powerful for detecting recent and strong migration [4]. - Leverage Phased Data: Ensure you use phased haplotype data, as required by these more sensitive statistics [4].
- Use
Issue 3: Inaccurate Signals Due to Mutation Rate Variation
- Problem: Genomic regions with low neutral mutation rates can be mistaken for loci that have experienced recent introgression [4].
- Solution:
Frequently Asked Questions (FAQs)
Q1: What is the primary cause of false positive introgression signals in my data? False positives often arise from incorrect sequence alignments in repetitive genomic regions (e.g., centromeres, telomeres), which can be mistaken for true evolutionary relationships. Ancestral polymorphism (incomplete lineage sorting) can also lead to shared alleles between species that are not the result of introgression [34].
Q2: How can I differentiate between true introgression and false positives caused by ancestral polymorphism?
Methods like the RNDmin statistic are designed for this purpose. It tests for introgression by using the minimum pairwise sequence distance between two population samples relative to divergence to an outgroup. This makes it robust to some of the confounding effects of ancestral polymorphism and variation in mutation rates [4].
Q3: My research focuses on non-model organisms with less complete genomic resources. Which methods are most suitable?
Summary statistic methods like FST, dXY, dmin, and RNDmin are often more practical for non-model organisms. They do not require a complex demographic model and can be applied with genome assemblies of moderate quality [4].
Q4: Are there specific methods to improve variant calling accuracy and reduce false positives? Yes, two effective approaches are:
- Logistic Regression (LR) Based Filtering: This method uses variant quality measures (like genotype quality, dbSNP status, overlap with repetitive elements) to calculate the probability of a variant being a true positive [35].
- Ensemble Genotyping: This approach integrates the results of multiple variant-calling algorithms, significantly reducing false positives compared to using any single method alone [35].
Experimental Protocols & Data
Table 1: Summary of Key Statistics for Introgression Detection
| Statistic | Formula | Data Requirements | Key Advantage | Key Disadvantage |
|---|---|---|---|---|
| FST | Wright's 1931 formula [4] | Unphased or phased data; does not require an outgroup. | Well-established, quantifies population structure. | Confounded by linked selection; not sensitive to low-frequency migrants [4]. |
| dXY | Average of d_x,y between all sequences in two species [4]. |
Unphased or phased data. | Robust to the effects of linked selection [4]. | Not sensitive to low-frequency migrants; confounded by mutation rate variation [4]. |
| dmin | min<sub>x∈X,y∈Y</sub>{d_x,y} [4] |
Phased haplotypes. | High power to detect rare or recent introgression [4]. | Confounded by mutation rate variation; requires phased data [4]. |
| RND | dXY / d_out where d_out = (d_XO + d_YO)/2 [4] |
Requires an outgroup (O). | Robust to variation in the neutral mutation rate [4]. | Not sensitive to low-frequency migrants [4]. |
| Gmin | dmin / dXY [4] |
Phased haplotypes. | Robust to mutation rate variation; sensitive to recent migration [4]. | Requires phased data [4]. |
| RNDmin | Minimum RND between two populations [4] | Phased haplotypes; requires an outgroup. | Robust to mutation rate and inaccurate divergence times; powerful for detecting introgression [4]. | Requires phased data and an outgroup [4]. |
Table 2: Performance of RAfilter on Simulated Datasets
| Dataset Type | Repeat Type | Sensitivity (HiFi) | Precision (HiFi) | Sensitivity (ONT) | Precision (ONT) |
|---|---|---|---|---|---|
| Simulated (Human Chr 1 & 2) | Tandem Repeats | >60-80% [34] | ~100% [34] | ~50% [34] | ~80% [34] |
| Simulated (Human Chr 18) | Interspersed Repeats | ~90% [34] | ~100% [34] | ~50% [34] | ~80% [34] |
Protocol 1: Detecting Introgressed Loci Using the RNDmin Statistic
- Data Preparation: Obtain whole-genome sequencing data from two sister species and a suitable outgroup. Phase the haplotypes.
- Calculate Minimum Distance: For each locus, calculate
dmin, the minimum pairwise sequence distance between any haplotype in species A and any haplotype in species B [4]. - Calculate Outgroup Distance: Compute the average sequence distance (
d_out) from each species to the outgroup [4]. - Compute RNDmin: Calculate the RNDmin statistic for the locus [4].
- Generate Null Distribution: Simulate a null distribution of RNDmin under a model of no migration using coalescent simulations.
- Identify Candidates: Identify candidate introgressed regions where the observed RNDmin falls in the lower tail of the null distribution (e.g., p < 0.05) [4].
Protocol 2: Filtering False-Positive Alignments with RAfilter
- Read Alignment: Align long reads (HiFi or ONT) to a reference genome using an aligner like minimap2 [34].
- Run RAfilter: Process the alignment file with RAfilter.
- kMAPQ Calculation: RAfilter assigns a kMAPQ score to each alignment, representing the possibility of it being a false positive based on rare k-mer profiles [34].
- Apply Threshold: Filter the alignments by applying a kMAPQ threshold (user-defined) to remove low-confidence alignments [34].
- Downstream Analysis: Use the filtered alignment file for subsequent variant calling or genome assembly tasks [34].
Workflow Visualization
Primary Research Workflow
False Positive Mitigation Strategy
The Scientist's Toolkit
Table 3: Research Reagent Solutions for Genomic Analysis
| Item | Function |
|---|---|
| RAfilter | An algorithm designed to filter false-positive sequence alignments in repetitive genomic regions using rare k-mers, improving the reliability of T2T assembly and variant calling [34]. |
| RNDmin | A summary statistic for detecting introgressed genomic regions that is robust to mutation rate variation and inaccurate species divergence time estimates [4]. |
| Ensemble Genotyping | A method that combines multiple variant-calling algorithms to significantly reduce false positive variant calls without a major loss of sensitivity [35]. |
| Logistic Regression (LR) Filter | A model that uses variant quality metrics (e.g., genotype quality, genomic context) to calculate the probability a variant is a true positive, allowing for effective variant prioritization [35]. |
| PBSIM/PBSIM2 | A software package for simulating long-read sequencing data (both HiFi and ONT), useful for benchmarking alignment and variant calling methods [34]. |
From Data to Discovery: Optimizing Parameters and Troubleshooting Common Pitfalls
Frequently Asked Questions
1. What is the fundamental difference between overfitting and underfitting in the context of data analysis?
- Overfitting occurs when a model is too complex and learns not only the underlying signal but also the noise in the training data [36] [37]. An overfitted model performs well on training data but poorly on new, unseen data because it has essentially memorized the training set, including its random fluctuations [38].
- Underfitting occurs when a model is too simple to capture the underlying pattern in the data [36] [37]. An underfitted model performs poorly on both training and test data because it fails to learn the essential trends and complexities [38].
2. How does the bias-variance tradeoff relate to overfitting and underfitting?
The balance between bias and variance is crucial for model performance [36].
- High bias leads to underfitting, where the model makes overly simplistic assumptions [36] [37].
- High variance leads to overfitting, where the model is too sensitive to the specific training data [36] [37].
- The goal is to find an optimal balance where both bias and variance are minimized, resulting in good generalization to new data [36].
3. Why is parameter sensitivity analysis important for statistical methods like the D statistic in introgression research?
Statistical methods can be sensitive to parameters like genomic window size. The D statistic, for example, can produce spurious inferences of gene flow when applied to genomic regions with low nucleotide diversity, as the variance of the statistic is maximized in these windows [5]. Tuning analysis parameters is therefore essential to mitigate false positive signals.
Troubleshooting Guide: Identifying and Resolving Fit Issues
| Symptom | Diagnosis | Corrective Actions |
|---|---|---|
| Model performs well on training data but poorly on evaluation/unseen data. [38] [37] | Overfitting: Model is too complex and has learned noise. | • Reduce model complexity. [36]• Increase training data. [36]• Apply regularization techniques (e.g., Ridge, Lasso). [36]• Perform feature selection to use fewer, more relevant features. [38] |
| Model performs poorly on both training and evaluation data. [38] [37] | Underfitting: Model is too simple to capture data trends. | • Increase model complexity. [36]• Add new, domain-specific features and perform feature engineering. [36] [38]• Decrease the amount of regularization used. [38]• Increase training duration or the number of training epochs. [36] |
| Statistical method (e.g., D) gives inconsistent or unreliable inferences in local genomic regions. [5] | Parameter Sensitivity: Method is sensitive to regional variations like low nucleotide diversity. | • Use a more robust statistic designed for local analysis, such as the D+ statistic, which leverages both shared derived and ancestral alleles to improve precision [5].• Adjust window size parameters for genomic scans.• Validate findings with complementary methods or simulations. |
Experimental Protocol: Mitigating False Positives with the D+ Statistic
The following protocol details the application of the D+ statistic, a method designed to improve the detection of local introgression and reduce false positives that can arise from ancestral polymorphism [5].
1. Objective To precisely identify local regions of introgression in a genome by leveraging both shared derived (ABBA/BABA) and shared ancestral (BAAA/ABAA) allele patterns, thereby improving upon the precision of the standard D statistic [5].
2. Background The D statistic can give spurious inferences in small genomic windows with low diversity [5]. The D+ statistic incorporates an additional class of informative sites—those where ancestral alleles are shared between populations—increasing the number of informative sites per region and improving the ability to identify true local introgression [5].
3. Methodology
- Population Tree Configuration: Define the four populations in the analysis:
- P1 & P2: The two potential recipient populations for gene flow.
- P3: The putative donor population.
- P4: An outgroup population, used to determine the ancestral allele state [5].
- Site Pattern Identification: For each genomic window, count the occurrences of four key site patterns [5]:
- ABBA: Derived allele shared between P3 and P2.
- BABA: Derived allele shared between P3 and P1.
- BAAA: Ancestral allele shared between P3 and P2, derived allele only in P1.
- ABAA: Ancestral allele shared between P3 and P1, derived allele only in P2.
- D+ Calculation: Compute the D+ statistic for each window using the formula: D+ = Σ(ABBA - BABA) + (BAAA - ABAA) / Σ(ABBA + BABA) + (BAAA + ABAA) [5]
- Interpretation: A significant excess of ABBA and BAAA site patterns indicates introgression from P3 into P2. A significant excess of BABA and ABAA site patterns indicates introgression from P3 into P1.
Parameter Sensitivity in Model Fitting
The following diagram illustrates the logical workflow for diagnosing and correcting model fit issues by tuning key parameters, drawing an analogy to filtering noise from a signal.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function |
|---|---|
| D+ Statistic | A statistical measure that uses both shared derived and ancestral alleles to detect introgression in genomic windows with improved precision and a lower false positive rate compared to the D statistic [5]. |
| Regularization (L1/Lasso, L2/Ridge) | A technique used to reduce model complexity (variance) by adding a penalty to the loss function, which helps to prevent overfitting [36] [37]. |
| Cross-Validation | A resampling technique used to assess how a model will generalize to an independent dataset. It is vital for evaluating model performance and tuning parameters without overfitting the test set [37]. |
| STRUCTURE Software | A widely used population analysis tool that uses a Bayesian clustering algorithm to infer population structure and assign individuals to populations based on genetic data [32]. |
| Feature Selection Tools | Methods and software used to identify and select the most relevant features (e.g., genetic markers) for analysis, which helps to reduce model complexity and the risk of overfitting [38]. |
Frequently Asked Questions
| Question | Answer |
|---|---|
| What causes elevated false positive rates in familial search? | False positives occur when unrelated individuals are incorrectly identified as relatives. This risk is significantly higher when the allele frequencies used in calculations are misspecified and do not match the genetic ancestry of the query profile [39]. |
| How can genetic ancestry inference mitigate this issue? | Performing ancestry inference on a query DNA profile allows for the use of more appropriate, ancestry-specific allele frequencies in the likelihood calculations. This avoids the extreme misspecifications that lead to the highest false positive rates [39]. |
| What are the limitations of using a single population database? | Relying on allele frequencies from a single population, such as a European-centric database, for analyses of profiles from other ancestries (e.g., Sub-Saharan African, East Asian) can dramatically inflate false positive rates due to genetic variation between populations [39]. |
| Besides false positives, what other signals can confound introgression research? | Exceptional similarity between species can also be caused by incomplete lineage sorting (ILS) or regions of low mutation rate, which can be mistaken for genuine introgression signals. Specific methods are needed to distinguish these [4]. |
| What statistical measures are used to detect introgression? | Several summary statistics are used, including dmin (minimum sequence distance), dXY (average sequence distance), and Gmin (dmin/dXY). These are powerful for detecting recent gene flow [4]. |
Troubleshooting Guides
Issue 1: High False Positive Rates in Familial Search
Problem: Your familial search analysis is producing an unacceptably high rate of false positive matches, particularly when analyzing DNA profiles from underrepresented populations.
Solution: Implement an ancestry inference step prior to the familial search to select appropriate allele frequencies [39].
Experimental Protocol: Ancestry-Informed Familial Search
Genetic Ancestry Inference
- Tool: Use software like STRUCTURE for unsupervised genetic clustering [39].
- Input Data: The query DNA profile genotyped at your panel of forensic or ancestry-informative markers (e.g., 13 CODIS STR loci) [39].
- Parameters: Run with the admixture and correlated allele frequencies models. Set the number of clusters (K) based on your reference data. Use a sufficient burn-in period (e.g., 10,000 steps) followed by a high number of iterations for posterior calculation [39].
- Output: Obtain the estimated ancestry proportion vector (q̂) for the query profile, representing its membership in each inferred genetic cluster.
Generate Ancestry-Informed Allele Frequencies
- Input: The estimated cluster-specific allele frequencies (p̂) from STRUCTURE and the query's ancestry proportions (q̂).
- Calculation: Compute a weighted allele frequency distribution specific to the query individual using the formula:
p̂ℓ,a = q̂1p̂1,ℓ,a + q̂2p̂2,ℓ,a + q̂3p̂3,ℓ,a + q̂4p̂4,ℓ,a[39]. (Where ℓ is the locus and a is the allelic type).
Perform Familial Search with Corrected Frequencies
- Calculation: Use the individualized allele frequency distribution (
p̂ℓ,a) from the previous step in the Likelihood Ratio (LR) calculations for relatedness, instead of frequencies from a generic or mismatched population [39]. - Result: This method has been shown to reduce false positive rates to levels similar to those achieved when allele frequencies are perfectly matched to the population of origin [39].
- Calculation: Use the individualized allele frequency distribution (
Issue 2: Distinguishing Introgression from Ancestral Polymorphism
Problem: It is challenging to determine whether a signal of similarity between two species is due to true introgression (hybridization) or ancestral polymorphism (Incomplete Lineage Sorting).
Solution: Apply a suite of summary statistics robust to mutation rate variation and capable of detecting recent gene flow.
Experimental Protocol: Robust Introgression Detection
Data Preparation: Obtain phased haplotype data from the two sister species (P1 and P2) and an outgroup (O). The outgroup should not have experienced introgression with P1 or P2 [4].
Calculate Summary Statistics
- dXY: Calculate the average number of sequence differences between all pairs of sequences from P1 and P2. This measures overall divergence but is sensitive to variation in mutation rate [4].
- dmin: Identify the minimum sequence distance between any single haplotype in P1 and any single haplotype in P2. This is powerful for detecting rare, recent introgression [4].
- RNDmin: Compute a relative measure to account for mutation rate variation. This is the quotient of dmin and the average distance from P1 and P2 to the outgroup O.
RNDmin = dmin / doutwheredout = (dP1O + dP2O)/2[4]. - Gmin: Calculate the ratio of the minimum distance to the average distance.
Gmin = dmin / dXY[4].
Identify Candidate Introgressed Regions
- Method: Scan the genome in sliding windows, calculating the statistics from step 2 for each window.
- Identification: Genomic regions with exceptionally low values of dmin, RNDmin, or Gmin are strong candidates for recent introgression. These values indicate haplotypes that are much more similar than the genomic background [4].
Statistical Validation
- Null Model: Compare the observed values of your statistics against a null distribution generated by coalescent simulations without migration.
- Significance: Windows where the observed statistic falls in the lower tail of the null distribution (e.g., below a 5% threshold) provide significant evidence for introgression [4].
| Statistic | Formula | Description | Strengths | Weaknesses |
|---|---|---|---|---|
| dmin | min<sub>x∊X,y∈Y</sub>{d<sub>x,y</sub>} |
Minimum sequence distance between any two haplotypes in two species [4]. | High power to detect recent, strong migration; sensitive to rare introgressed lineages [4]. | Confounded by variation in mutation rate; requires phased haplotypes [4]. |
| dXY | Mean of all d<sub>x,y</sub> |
Average number of sequence differences between all pairs of sequences from two species [4]. | Simple to compute; does not require phased data [4]. | Not sensitive to low-frequency migrants; confounded by mutation rate variation [4]. |
| Gmin | d<sub>min</sub> / d<sub>XY</sub> |
Ratio of the minimum distance to the average distance [4]. | Robust to variation in mutation rate; relatively sensitive to recent migration [4]. | Requires phased haplotypes for dmin calculation [4]. |
| RNDmin | d<sub>min</sub> / d<sub>out</sub> |
Minimum distance normalized by divergence to an outgroup [4]. | Robust to mutation rate variation; reliable with inaccurate divergence time estimates [4]. | Requires a suitable outgroup [4]. |
Table 2: Essential Research Reagent Solutions
| Reagent / Material | Function in Analysis |
|---|---|
| Reference Population Datasets (e.g., HGDP) | Provides genotype data from globally diverse populations to serve as a baseline for accurate ancestry inference and allele frequency estimation [39]. |
| Ancestry Inference Software (e.g., STRUCTURE) | Performs unsupervised genetic clustering to estimate the most likely ancestral populations contributing to an individual's genotype [39]. |
| Phased Haplotype Data | Data requirement for several powerful introgression statistics (dmin, Gmin), allowing for the analysis of individual ancestral chromosomes [4]. |
| Outgroup Genome Sequence | A genome from a closely related species that diverged before the species pair of interest. Essential for calculating relative metrics like RNDmin to control for mutation rate variation [4]. |
Experimental Workflow Visualizations
Ancestry-Informed Familial Search
Introgression Detection Logic
Navigating Copy Number Alterations and Tumor Purity in Somatic Variant Calling
Frequently Asked Questions (FAQs)
How do tumor purity and ploidy impact the accurate calling of somatic copy number alterations (CNAs)?
Tumor purity (the fraction of cancer cells in a sample) and ploidy (the number of chromosome sets) are critical because they directly change the observed magnitude of genomic signals. In a sample with 100% tumor cells and a diploid genome, a single-copy loss would theoretically result in a copy number of 1 and a halving of the read depth. However, in a real-world sample with 50% tumor purity, the same single-copy loss would be observed as a more subtle change, as the copy number from the normal cells dilutes the signal. The observed copy ratio (fold change) is a function of tumor purity (p), tumor copy number (C), and ploidy. The relationship can be modeled as follows [40] [41]:
Observed Copy Ratio = [ p * C + (1-p) * 2 ] / [ p * Ploidy + (1-p) * 2 ]
Failure to account for low purity can lead to missed CNAs (reduced sensitivity), while incorrect ploidy estimates can result in misclassifying gains as losses or vice versa.
A significant source of false positives in unmatched (tumor-only) analyses is the misclassification of private (non-database) germline variants as somatic mutations. This problem is exacerbated for individuals of non-European ancestry due to the limited diversity of current public polymorphism databases (e.g., dbSNP, gnomAD), which do not fully represent global genetic variation [42]. Population-specific characteristics like admixture or recent expansions can further inflate the germline false-positive rate.
Mitigation strategies include:
- Leveraging Allelic Frequency: Tools like LumosVar exploit the difference in allelic frequency between somatic and germline variants in impure tumors. In a mixed tumor-normal sample, somatic mutations in cancer cells have an allelic frequency less than 50%, while germline heterozygous SNPs are expected to be close to 50%. This pattern can be used to distinguish them [42].
- Integrated Callers: Use specialized algorithms designed for tumor-only data, such as PureCN or LumosVar. PureCN, for example, uses a Bayesian model that jointly analyzes copy number log-ratios and allelic fractions of SNPs to estimate purity, ploidy, and classify SNVs without a matched normal [41].
- Ancestry-Informed Filtering: Be aware that standard germline filters based on population databases are less effective for under-represented populations. For studies involving these groups, methods that do not rely solely on database filtering are particularly beneficial [42].
What computational methods are available for estimating tumor purity and absolute copy number from sequencing data?
Multiple computational methods have been developed to infer tumor purity and copy number. They can be broadly categorized by their input data and methodology. The table below summarizes several key tools.
Table 1: Computational Methods for Estimating Tumor Purity and Absolute Copy Number
| Method Name | Key Input Data | Core Methodology / Principle | Supports Tumor-Only (Unmatched) Analysis? |
|---|---|---|---|
| PureCN [41] | Targeted/WES/WGS; Copy number log-ratios & SNP allelic fractions | A likelihood model that jointly analyzes coverage and allelic fractions to fit purity and ploidy; uses a grid search and simulated annealing. | Yes |
| AITAC [43] | WES/WGS; Read depths (RD) | Creates a non-linear model correlating tumor purity with observed and expected RDs in regions with copy number losses; performs an exhaustive search for the optimal purity. | Information missing |
| ABSOLUTE [43] [44] | SNP array / NGS; Segmented copy number data | Incorporates segmented copy number and prior probabilities from cancer karyotypes to search for the best solution for purity and ploidy. | Information missing |
| ASCAT [44] | SNP array / NGS; Allele-specific data | Allele-Specific Copy number Analysis of Tumors; estimates purity and ploidy by analyzing allele-specific intensities and shifts in B-allele frequency. | Information missing |
| FACETS [45] | WGS/WES/Targeted | Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing; jointly segments total and allele-specific copy numbers. | Information missing |
| LumosVar [42] | WES/WGS; Allelic frequencies | A Bayesian tumor-only caller that leverages differences in allelic frequency between somatic and germline variants in impure tumors. | Yes (specifically designed for it) |
How consistent are tumor purity estimates across different estimation methods?
Tumor purity estimates can show considerable variation depending on the method and the type of molecular data (DNA vs. RNA) used. A systematic benchmarking study on a cohort of 333 prostate cancers found significant inter-method variation when comparing pathology reviews and multiple in silico estimates from DNA (copy number, methylation), mRNA, and microRNA profiles [44]. Furthermore, there is often limited concordance between DNA-derived and RNA-derived purity estimates, a phenomenon observed across 12 cancer types [44]. This highlights that purity is not a single absolute value but is context-dependent. It is recommended to parameterize genomic analyses with tumor purity estimated from the same molecular analyte (e.g., DNA for CNA analysis, RNA for expression analysis) being investigated [44].
Troubleshooting Guides
Issue: Inflated False Positive Somatic Variants in Tumor-Only Sequencing
Problem: Your analysis of unmatched tumor samples is producing an unusually high number of putative somatic variants, many of which are suspected to be private germline variants.
Solution:
- Step 1: Confirm Ancestry Bias. Check the self-reported or genetically inferred ancestry of your samples. Be aware that individuals of non-European ancestry are more susceptible to this issue due to gaps in reference databases [42].
- Step 2: Employ a Specialized Tool. Switch from a standard somatic caller to one designed for tumor-only data.
- Option A (LumosVar): This tool is explicitly noted for greatly reducing false positives from private germline variants by modeling the expected allelic frequency distributions for somatic and germline variants in impure tumors [42].
- Option B (PureCN): This tool uses a joint model of copy number and allelic fractions to estimate purity and classify SNVs. It can help re-classify variants of uncertain significance (VUS) as germline or somatic, even without a matched normal [41].
- Step 3: Validate Findings. If possible, use an orthogonal method (e.g., targeted sequencing of a matched normal sample) to confirm a subset of the called somatic variants, especially those in critical genes.
Issue: Inaccurate or Failed Tumor Purity and Ploidy Estimation
Problem: Your purity/ploidy estimation tool fails to produce a result, or the result seems biologically implausible (e.g., extremely low ploidy).
Solution:
- Step 1: Check Data Quality. For DNA-based methods, "quiet" genomes with a low number of somatic single nucleotide variants (SNVs) or few copy number alterations can cause methods to fail. This is common in cancers like prostate cancer [44]. Verify that your data has sufficient coverage and shows some evidence of genomic aberrations.
- Step 2: Try an Alternative Method. If one method fails, another might succeed. For example, if a read-depth-based method like AITAC fails, try an allele-frequency-based method like PureCN or FACETS [43] [41] [45].
- Step 3: Manual Review. Use a tool like PureCN, which provides a list of potential (purity, ploidy) solutions. Manually inspect the fit of different solutions by plotting the predicted copy number states against the observed log-ratios and B-allele frequencies to select the most biologically reasonable one [41].
- Step 4: Consult Pathology. Compare your in silico estimate with a pathologist's assessment if available. While they may differ due to tumor heterogeneity, a large discrepancy might indicate an issue with the computational estimate [44].
Experimental Protocols & Workflows
Detailed Methodology: PureCN Workflow for Tumor-Only Analysis
The following workflow is adapted from the PureCN software, which is optimized for targeted sequencing data but applicable to WES and WGS [41].
1. Data Pre-processing:
- Input: Tumor BAM file(s) and a pool of normal BAM files.
- Coverage Calculation: Calculate the average read depth for all targeted genomic intervals from the tumor BAM and all normal BAMs using tools like
GATK DepthOfCoverageor PureCN's built-in function. - GC Normalization: Correct the coverage data in the tumor and normal pools for GC-content bias.
2. Copy Number Normalization and Segmentation:
- Normal Pool PCA: Use Principal Component Analysis (PCA) on the GC-normalized coverage of the normal pool to select the most suitable normal sample(s) for normalizing the tumor sample. This corrects for library-specific biases.
- Calculate Log-Ratios: Compute log-ratios of coverage between the tumor and the selected normal(s).
- Segmentation: Segment the log-ratios using a segmentation algorithm (e.g., Circular Binary Segmentation). PureCN weights targets by their reliability (inverse of variance in the normal pool) to improve segmentation.
3. Integrating SNP Allelic Fractions:
- SNP Calling: Obtain germline SNP candidates from the tumor BAM using a standard SNV caller.
- Breakpoint Refinement: Use the allelic fractions of known germline SNPs (from dbSNP) to refine segmentation breakpoints. Breakpoints are confirmed if the allelic fractions in adjacent segments are significantly different.
- Copy Number Neutral LOH Detection: Recursively test segments for regions where the total copy number is neutral but have undergone loss of heterozygosity (LOH).
4. Purity and Ploidy Estimation:
- 2D Grid Search: Perform a grid search over a range of purity and ploidy values to find combinations that best fit the observed segment log-ratios. The model assumes log-ratios are normally distributed around the value predicted by the equation:
log2( (p * C + (1-p) * 2) / (p * Ploidy + (1-p) * 2) ), whereCis the integer copy number of the segment [41]. - Simulated Annealing: The top candidate solutions from the grid search are optimized using simulated annealing, where integer copy numbers are assigned to all segments, and the purity estimate is fine-tuned.
- Variant Classification: The optimal (purity, ploidy) solution is used to calculate the expected allelic fractions for germline and somatic variants. A Bayesian model then classifies SNVs into germline heterozygous, germline homozygous, somatic, or LOH categories.
Workflow: General Somatic Copy Number Alteration Analysis
The diagram below outlines a generalized workflow for somatic CNA analysis, incorporating best practices for handling tumor purity.
General CNA analysis workflow.
Table 2: Key Resources for CNA and Tumor Purity Analysis
| Resource Name | Type | Function / Purpose |
|---|---|---|
| Pool of Normal Samples [41] | Experimental Biological Reagent | A collection of normal tissue sequencing data (e.g., from blood or adjacent normal tissue) used to normalize tumor sequencing data, account for technical biases, and improve the accuracy of CNA detection and purity estimation. |
| Reference Genome (e.g., HG38) [45] | Reference Data | A standardized, annotated human genome sequence used as a baseline for aligning sequencing reads and identifying variations, including copy number changes. |
| Public Polymorphism Database (e.g., dbSNP) [42] [41] | Reference Database | A catalog of known germline polymorphisms used to filter out common germline variants during somatic variant calling. Note: Limited diversity in these databases can lead to ancestry-related false positives [42]. |
| PureCN (R/Bioconductor) [41] | Software Package | Estimates tumor purity, ploidy, copy number, and loss of heterozygosity (LOH), and classifies SNVs as somatic or germline. Optimized for targeted sequencing and supports unmatched tumor samples. |
| FACETS [45] | Software Algorithm | Estimates fraction and allele-specific copy numbers from tumor sequencing data. Used for joint segmentation of total and allele-specific copy numbers. |
| ASCAT [44] [45] | Software Algorithm | Allele-Specific Copy number Analysis of Tumors; widely used for estimating tumor purity and ploidy from SNP array or sequencing data by analyzing allele-specific signals. |
| The Cancer Genome Atlas (TCGA) [46] | Data Repository | A public resource containing genomic, epigenomic, transcriptomic, and clinical data for over 20,000 primary cancers across 33 cancer types, essential for benchmarking and discovery. |
Logical Relationship: Connecting Tumor Purity to Variant Classification
This diagram illustrates the core logical relationship between tumor purity, copy number, and the accurate classification of single nucleotide variants (SNVs), which is fundamental to mitigating false positives.
Variant classification logic.
Frequently Asked Questions
Q1: What is a spurious introgression signal, and why is it a problem? A spurious, or false positive, introgression signal occurs when statistical tests indicate gene flow between species that did not actually happen. These false signals can mislead researchers into proposing incorrect evolutionary histories, wasting resources on validating non-existent biological processes and obscuring the true phylogenetic relationships between taxa [10].
Q2: How does the choice of outgroup lead to such false signals? The outgroup is used to root the evolutionary tree and determine the ancestral state of genetic variants. A more distantly related outgroup has had more time for multiple mutations to occur at the same site in its genome. When these multiple hits are not accounted for, they can create patterns in the data that mimic the signal of hybridization between the ingroup species. Research has confirmed that employing a more distant outgroup intensifies these spurious signals [10].
Q3: Besides outgroup distance, what other factors can cause false positives? Lineage-specific rate variation is a major factor. Even minor differences in substitution rates between sister lineages can create asymmetry in site patterns, which is misinterpreted as introgression by methods like the D-statistic. This is particularly impactful in shallow phylogenies where a molecular clock is often assumed to hold [10].
Q4: Are some statistical methods more vulnerable than others? Yes, site pattern-based methods like the D-statistic and HyDe are highly sensitive to violations of the "no multiple hits" assumption and are therefore pervasively vulnerable to these artifacts, especially when coupled with rate variation or a distant outgroup [10].
Q5: How can I mitigate the risk of false positives in my analysis? You can:
- Choose the closest practical outgroup to minimize the number of multiple hits.
- Test for lineage-specific rate variation before applying introgression tests.
- Use methods that explicitly account for rate heterogeneity or employ full-likelihood models that use both topological and branch length information from gene trees [10].
- Be cautious in interpreting significant results from summary methods, especially in shallow phylogenies.
Troubleshooting Guide: Resolving Spurious Signals
Problem: Consistently Significant D-Statistics with No Biological Evidence
Potential Cause: The chosen outgroup is too evolutionarily distant from the ingroup, leading to an accumulation of undetected multiple substitutions that create ABBA/BABA asymmetry [10].
Solution:
- Re-run with a Closer Outgroup: If possible, select a different, more closely related species as the outgroup and re-calculate the D-statistic. A significant signal that disappears with a closer outgroup strongly suggests an artifact.
- Use a Saturation Test: Check for multiple hits by analyzing the transition/transversion ratio as a function of divergence distance. A leveling off of this ratio indicates saturation.
- Employ a Robust Method: Consider switching to methods less sensitive to these issues, such as full-likelihood tests that incorporate branch length information [10].
Problem: High False Positive Rate in Young, Rapidly Diverging Clades
Potential Cause: Underlying variation in substitution rates between the sister lineages you are testing (lineage-specific rate variation). Even a 17% difference in rates in a young phylogeny can inflate false-positive rates up to 35% with a 500 Mb genome [10].
Solution:
- Perform a Relative Rate Test: Quantify and confirm whether significant rate variation exists between your lineages of interest.
- Apply a Correction Model: Use phylogenetic methods that incorporate models of rate variation across lineages.
- Simulate Your Data: Under a model of no introgression but with the estimated rate variation. If your empirical D-statistics fall within the distribution of D-statistics from these simulations, the "introgression" signal is likely false [10].
Quantitative Data on False Positive Drivers
The following data, synthesized from simulation studies, illustrates how outgroup distance and rate variation interact to produce false positives.
Table 1: Impact of Rate Variation and Phylogenetic Age on False Positives (500 Mb genome) [10]
| Phylogenetic Age (Generations) | Rate Variation Between Sisters | False Positive Rate | Key Condition |
|---|---|---|---|
| 300,000 (Young) | Weak (17% difference) | Up to 35% | Small population size |
| 300,000 (Young) | Moderate (33% difference) | Up to 100% | Small population size |
| >1,000,000 (Deep) | Present | Significant | Confirmed by simulation studies |
Table 2: Effect of Outgroup Distance on Signal Strength [10]
| Outgroup Distance | Impact on Spurious D-Statistic Signal |
|---|---|
| Close | Signal is minimized or absent |
| Distant | Signal is significantly intensified |
Experimental Protocol: Validating Introgression Signals
This protocol provides a framework to test whether a significant D-statistic result is a true biological signal or an artifact of evolutionary distance and rate heterogeneity.
Objective: To distinguish true introgression from false signals caused by multiple hits and rate variation.
Materials & Computational Tools:
- Genomic sequence data for ingroup taxa and multiple potential outgroups.
- Software for population genetic analysis (e.g.,
popgentools for D-statistic). - Software for phylogenetic analysis and molecular clock testing (e.g.,
HYPHY,PAML,BEAST2). - Simulation software (e.g.,
mswithseq-gen, orSLiM).
Methodology:
- Test for Rate Variation: Use a relative rate test or a likelihood ratio test in a phylogenetic framework to determine if a molecular clock can be rejected for your taxa.
- Assess Outgroup Saturation: Plot the number of substitutions per site against divergence time for the outgroup. A non-linear relationship indicates saturation.
- The Sensitivity Analysis: Re-calculate the D-statistic using a series of outgroups at increasing phylogenetic distances. A signal that strengthens systematically with outgroup distance is suspect.
- The Coalescent Simulation: a. Model Parameters: Use a coalescent simulator without migration. Input parameters like effective population size and divergence times should be estimated from your data. b. Incorporate Rate Variation: Apply different branch-specific substitution rates to the simulated trees, reflecting the rate variation you detected. c. Generate Sequence Data: Evolve sequences along the simulated trees. d. Analysis: Calculate the D-statistic on the simulated datasets (where no introgression is possible) to generate a null distribution of D-values.
- Interpretation: Compare your empirical D-value to the null distribution from the simulations. If your value falls within the 95% range of the simulated false positives, the signal cannot be trusted.
This workflow can be visualized in the following diagram:
Table 3: Key Analytical Tools for Introgression Analysis
| Tool / Resource | Type | Primary Function | Relevance to Mitigating False Positives |
|---|---|---|---|
| D-Statistic (ABBA-BABA) | Statistical Test | Detects asymmetry in site patterns to infer gene flow. | The baseline method whose results require validation using the tools below. [10] |
| Relative Rate Test | Statistical Test | Tests for significant substitution rate differences between two lineages using an outgroup. | Diagnoses lineage-specific rate variation, a key source of false positives. [10] |
| HyDe | Statistical Test | Detects hybrid speciation from site pattern frequencies. | Similarly vulnerable to multiple hits and rate variation; results need caution. [10] |
| Coalescent Simulators (e.g., ms, SLiM) | Software | Simulates genetic data under evolutionary models without gene flow. | Generates a null distribution to test if an empirical signal exceeds what is expected by chance alone. [10] |
| Full-Likelihood Methods | Statistical Model | Uses comprehensive information from gene tree topologies and branch lengths. | More robust to violations like rate heterogeneity compared to summary statistics. [10] |
FAQ: Core Concepts for NGS in Introgression Research
What is the critical difference between sequencing depth and coverage? These are two distinct but related metrics crucial for assessing data quality [47].
- Sequencing Depth: Also called read depth, this refers to the number of times a specific nucleotide is read during sequencing [47]. For example, 30x depth means a base was sequenced 30 times on average. Higher depth increases confidence in variant calls [47].
- Coverage: This is the percentage of the target genome or region that has been sequenced at least once [47]. For example, 95% coverage means nearly the entire target region has been read [47].
A successful project balances both: high enough depth for accurate variant calling and comprehensive coverage to prevent information gaps [47].
Why is high sequencing depth particularly important for detecting introgression? High sequencing depth improves the detection of low-frequency variants, which is essential for identifying subtle introgression signals and distinguishing true introgression from sequencing errors [48]. In the context of statistics like Patterson's D, spurious inferences of local introgression can occur in genomic regions with low nucleotide diversity; higher depth helps mitigate this by providing more data points per region [49].
How can specific NGS methods be optimized for studies of ancestral polymorphism? Targeted gene panels, which focus on known disease-associated genes, allow for greater depth of coverage, thereby increasing analytical sensitivity and specificity [50]. This focused approach is beneficial for introgression studies as it allows for deeper sequencing of specific loci of interest, improving the precision of local statistics like D+ [49]. Furthermore, for regions with low coverage, follow-up Sanger sequencing can be used to fill gaps and improve clinical sensitivity [50].
Troubleshooting Common NGS Issues
Problem: Low Sequencing Depth or Inadequate Coverage Low depth can lead to false negatives and an inability to detect true variants, especially low-frequency ones [48].
- Verification Step: Check the depth and coverage metrics in your sequencing output. Most analysis pipelines provide a mean depth and coverage percentage.
- Solution: Increase the total number of sequencing reads per sample. This may require adjusting the library concentration loaded onto the sequencer or re-evaluating the number of samples pooled together in a single run (multiplexing) [50].
- Preventative Action: During the study design phase, use coverage calculators to determine the total sequencing output required to achieve your desired mean depth across all targeted regions, especially for heterogeneous samples or rare variant detection [47].
Problem: Spurious Introgression Signals in Genomic Windows The D statistic can produce unreliable inferences when applied to small genomic regions or areas of low nucleotide diversity [49].
- Verification Step: Check the variance of the D statistic; it is often maximized in windows of low diversity [49].
- Solution: Use the D+ statistic, which leverages both shared derived and shared ancestral alleles. This increases the number of informative sites per region and has been shown to have better precision and a lower false positive rate for detecting local introgression compared to the D statistic [49].
- Preventative Action: When designing an analysis to find local targets of introgression, plan to use statistics like D+ that are specifically designed for this purpose and are more robust in small windows [49].
Problem: Ion S5/S5 XL System - Chip Check Failure The instrument fails to recognize or properly interface with the sequencing chip.
- Verification Step: Open the chip clamp, remove the chip, and inspect for physical damage or signs of moisture outside the flow cell [51].
- Solution:
- If the chip appears damaged, replace it with a new one.
- Ensure the chip is properly seated in its socket.
- Close the clamp firmly and rerun the Chip Check.
- If the failure persists, the issue may be with the chip socket itself, and technical support should be contacted [51].
Problem: Ion PGM System - "W1 Empty" Error The system reports that the W1 wash solution bottle is empty when it is not.
- Verification Step: Confirm that there is at least 200 mL of solution in the W1 bottle and that all bottles and sippers are securely attached [51].
- Solution:
- Run the "line clear" procedure to clear any potential blockages in the fluidics line.
- If the error persists, detach the reagent bottles and shut down the instrument.
- Check the chip port for any standing fluid and dry it carefully if needed.
- Replace the chip with a new or different one as a precaution.
- Replace the W1 solution with a fresh batch, restart the instrument, and re-initialize [51].
Essential Methodologies and Protocols
The D+ Statistic for Local Introgression Detection
The D+ statistic is an advanced method for detecting introgressed regions in a genome. It improves upon Patterson's D by incorporating information from shared ancestral alleles, not just shared derived alleles, thereby increasing the number of informative sites in a genomic window [49].
Workflow and Logical Relationship
The following diagram illustrates the conceptual workflow and logical relationships involved in using the D+ statistic to mitigate false positives from ancestral polymorphism.
Formula and Calculation The D+ statistic is calculated as follows [49]:
Where:
- ABBA: Sites where P3 and P2 share a derived allele, while P1 has the ancestral allele.
- BABA: Sites where P3 and P1 share a derived allele, while P2 has the ancestral allele.
- BAAA: Sites where P3 and P2 share the ancestral allele, while P1 has a derived allele.
- ABAA: Sites where P3 and P1 share the ancestral allele, while P2 has a derived allele.
By using both shared derived (ABBA, BABA) and shared ancestral (BAAA, ABAA) patterns, D+ increases the number of informative sites, improving power and precision for identifying local introgression [49].
NGS Wet-Lab Protocol for High-Quality Data
This protocol outlines the key steps for generating NGS data suitable for sensitive introgression analysis.
Library Preparation and Target Enrichment
- DNA Fragmentation and Library Generation: Fragment genomic DNA to a specific size range (e.g., 100-500 bp) and ligate platform-specific adapter sequences to both ends [50].
- Barcoding (Indexing): Add unique molecular barcodes to each sample's library via PCR. This allows multiple libraries to be pooled (multiplexed) and sequenced in a single run [50].
- Target Enrichment: Unless performing whole-genome sequencing, enrich for regions of interest. For introgression studies focusing on specific loci, use:
- Multiplex PCR-based methods: Suitable for a relatively small number of targeted genes.
- Hybridization-based capture: Used for larger target sets, such as exomes or custom panels [50].
- Sequencing: Load the pooled and enriched libraries onto a next-generation sequencer (e.g., Illumina, Ion Torrent) following the manufacturer's instructions.
Research Reagent Solutions
The following table details key materials and reagents used in targeted NGS workflows for introgression studies.
| Item | Function/Benefit |
|---|---|
| Targeted NGS Panels | Focuses sequencing on specific genomic regions (e.g., disease-associated genes or candidate introgression loci), allowing for greater depth of coverage and improved sensitivity for variant detection [50]. |
| Hybridization Capture Kits | Used for exome sequencing or large custom panels to enrich for target regions prior to sequencing, improving the efficiency of data generation [50]. |
| Unique Molecular Identifiers (UMIs) | Short random nucleotide sequences added to each DNA fragment before PCR amplification. UMIs help correct for PCR duplicates and sequencing errors, increasing the accuracy of variant calling, which is critical for low-frequency variants [48]. |
| High-Fidelity DNA Polymerase | An essential enzyme for library amplification and target enrichment that has a low error rate, minimizing the introduction of false-positive mutations during PCR steps [50]. |
Table 1: Recommended Sequencing Depth for Various Applications
This table consolidates guidelines for sequencing depth based on different study objectives, which is critical for planning NGS experiments in introgression research.
| Application / Study Goal | Recommended Minimum Depth | Key Rationale |
|---|---|---|
| Variant Calling (General) | High depth is required [47] | Increases confidence that a called variant is real and not a sequencing error [47]. |
| Rare Variant Detection | Higher depth is crucial [47] | Essential for finding variants present at low frequencies in a sample [47]. |
| Targeted Gene Panels | Allows for greater depth [50] | Increased analytical sensitivity and improved ability to detect mosaicism or heterogeneity [50]. |
| Measurable Residual Disease (MRD) | Very high depth is required [48] | Necessary for capturing ultra low-frequency variants that indicate residual disease [48]. |
Table 2: NGS Troubleshooting Guide for Common Scenarios
A quick-reference table for diagnosing and addressing frequent NGS issues.
| Problem Scenario | Possible Cause | Recommended Action |
|---|---|---|
| Low VAF Sensitivity | Insufficient sequencing depth [48] | Increase total sequencing reads; use deeper coverage [48]. |
| High False Positive Variants | Low sequencing specificity; high error rate [48] | Employ UMIs; increase depth of coverage to reduce error impact [48]. |
| Chip Check Failure (Ion S5) | Clamp not closed; chip not seated; damaged chip [51] | Reseat or replace chip; ensure clamp is closed; contact support if unresolved [51]. |
| "W1 Empty" Error (Ion PGM) | Fluidics line blockage; loose sippers [51] | Run line clear procedure; check and secure all bottles and sippers [51]. |
Ensuring Accuracy: Validation Protocols and Comparative Analysis of Method Performance
Frequently Asked Questions (FAQs)
1. Why does my analysis show high false-positive rates for local introgression when using Patterson's D statistic on small genomic windows?
The D statistic is designed for genome-wide introgression detection and becomes unreliable in small genomic regions or windows with low nucleotide diversity. In these areas, the statistic's variance increases significantly, leading to spurious inferences of gene flow. It is recommended to use the D+ statistic, which incorporates both shared derived and shared ancestral alleles, providing more precision for local analyses by increasing the number of informative sites per region [5].
2. My Identity-by-Descent (IBD) analysis in Plasmodium falciparum yields many false negatives. What is the likely cause and how can I mitigate this?
High false negative rates in IBD analysis, particularly for shorter segments, are often caused by low SNP density per centimorgan (cM), a direct consequence of the exceptionally high recombination rate in P. falciparum genomes. To mitigate this:
- Optimize IBD caller parameters, especially those controlling for minimum SNP density and segment length [52] [53].
- Use IBD callers specifically designed or optimized for high-recombining genomes. Benchmarking studies recommend hmmIBD for such contexts, as it provides more accurate segment detection and less biased estimates of effective population size ((N_e)) [52] [53].
3. How does the choice of evolutionary scenario affect the performance of selection detection tools in Evolve and Resequence (E&R) studies?
The performance of selection detection tools varies dramatically across different evolutionary scenarios. Some tools excel in detecting selective sweeps but perform poorly under models of polygenic adaptation (e.g., stabilizing selection) or truncating selection. For instance:
- LRT-1 and CLEAR are among the top-performing methods across multiple scenarios [54].
- Tools that utilize multiple population replicates generally outperform those that use only a single dataset [54]. You should choose a tool whose underlying model matches your experimental selection regime. Refer to the benchmarking table below for specific performance metrics [54].
4. What are the critical factors to consider when simulating genomes for benchmarking studies on species with high recombination rates?
For accurate benchmarking in high-recombining species like Plasmodium, your simulation framework must incorporate:
- Species-specific recombination and mutation rates: The recombination rate in P. falciparum is about 70 times higher than in humans per physical unit, while mutation rates are similar. This results in a much lower SNP density per genetic unit [52] [53].
- Appropriate demographic history: Implement a demographic model that reflects the species' population history, such as a decreasing effective population size ((N_e)) for P. falciparum [52] [53].
- A unified accuracy metric: Use consistent metrics like False Negative (FN) and False Positive (FP) rates, calculated against a ground truth from the simulation's ancestral recombination graph (ARG) [52].
Troubleshooting Guides
Issue: High False-Positive Rates in Local Introgression Inference
Problem: You are detecting apparently significant local introgression signals that may be spurious, especially when analyzing small genomic windows.
Solution: Implement the D+ Statistic Workflow The D+ statistic reduces false positives by leveraging both shared derived (ABBA, BABA) and shared ancestral (BAAA, ABAA) allele patterns, providing more reliable local inference [5].
Experimental Protocol: Detecting Local Introgression with D+
- Define Populations: Identify your four populations according to the required phylogeny: two sister populations (P1, P2), a putative donor population (P3), and an outgroup (P4).
- Generate Site Pattern Counts: For each genomic window, tabulate the counts of ABBA, BABA, BAAA, and ABAA sites. A site must be biallelic, and the ancestral allele is determined by the outgroup P4.
- ABBA: Derived allele in P3 and P2, ancestral in P1.
- BABA: Derived allele in P3 and P1, ancestral in P2.
- BAAA: Ancestral allele in P3 and P2, derived in P1.
- ABAA: Ancestral allele in P3 and P1, derived in P2 [5].
- Calculate the D+ Statistic: Use the following formula to compute D+ for each window:
D+ = [Σ(ABBA - BABA) + Σ(BAAA - ABAA)] / [Σ(ABBA + BABA) + Σ(BAAA + ABAA)][5]. - Interpret Results: A significant excess of ABBA and BAAA sites indicates introgression between P3 and P2. A significant excess of BABA and ABAA sites indicates introgression between P3 and P1.
The following diagram illustrates the logical workflow and the site patterns used by the D+ statistic:
Issue: Inaccurate Identity-by-Descent (IBD) Segments in High-Recombining Genomes
Problem: IBD detection tools yield segments with high false-positive or false-negative rates, compromising downstream inferences of relatedness, population size ((N_e)), and selection.
Solution: A Unified Benchmarking and Optimization Framework Follow a structured workflow to evaluate and optimize IBD detection methods using simulated data that mirrors the high recombination and demographic history of your study species [52] [53].
Experimental Protocol: Benchmarking IBD Callers
- Simulate Genomes: Use a simulator (e.g.,
msprimeorSLiM) to generate genomic data with species-specific parameters: high recombination rate, realistic mutation rate, and demographic history (e.g., decreasing (N_e)). - Extract Ground Truth IBD: From the simulated data, use a tool like
tskibdto extract the true IBD segments from the Ancestral Recombination Graph (ARG), which serves as your benchmark [52] [53]. - Run IBD Callers: Execute the IBD detection tools you wish to benchmark (e.g., hmmIBD, isoRelate, hap-IBD, RefinedIBD) on the simulated data.
- Calculate Accuracy Metrics:
- Optimize Parameters: Systematically adjust key parameters in the IBD callers—especially those related to minimum SNP density, minimum segment length, and genotype error tolerance—and re-run the benchmarking to find the set that minimizes FN and FP rates for your specific data.
- Validate with Empirical Data: Apply the optimized tools to a subset of your real empirical data (e.g., from a database like MalariaGEN Pf 7) to confirm that the findings from the simulation hold [52] [53].
The following diagram illustrates the comprehensive benchmarking workflow:
Performance Data Tables
Table 1: Benchmarking Selection Detection Tools in Evolve & Resequence Studies
This table summarizes the performance of various software tools for detecting selected SNPs under three different evolutionary scenarios, based on a partial Area Under the Curve (pAUC) metric at a low false-positive rate threshold [54].
| Software Tool | Supports Replicates? | Requires Time Series? | Selective Sweeps | Truncating Selection | Stabilizing Selection |
|---|---|---|---|---|---|
| LRT-1 | Yes | No | Best Performance | High Performance | High Performance |
| CLEAR | Yes | Yes | High Performance | Best Performance | High Performance |
| CMH Test | Yes | No | High Performance | High Performance | Medium Performance |
| χ2 Test | No | No | High Performance | Medium Performance | Medium Performance |
| LLS | Yes | Yes | Medium Performance | Low Performance | Low Performance |
Table 2: Impact of Recombination Rate and SNP Density on IBD Detection Accuracy
This table illustrates the inverse relationship between recombination rate and SNP density, and its subsequent effect on the accuracy of IBD segment detection, specifically for the hmmIBD tool [52] [53].
| Recombination Rate (per bp/gen) | Example Genome | SNP Density (SNPs/cM) | False Negative Rate | False Positive Rate |
|---|---|---|---|---|
| 1.00E-08 | Human-like | ~1,660 | Low | Low |
| 1.00E-07 | --- | ~166 | Moderate | Moderate |
| 1.00E-06 | P. falciparum-like | ~25 | High | High |
Research Reagent Solutions
Essential Software and Databases for Genomic Benchmarking
| Item Name | Type | Function/Brief Explanation |
|---|---|---|
| hmmIBD | Software Tool | An IBD detection method recommended for high-recombining genomes like Plasmodium falciparum. It provides more accurate estimates of effective population size ((N_e)) compared to other tools in this context [52] [53]. |
| D+ Statistic | Statistical Method | A tool for detecting local introgression that leverages both shared derived and ancestral alleles, offering improved precision and reduced false positives compared to the Patterson's D statistic in small genomic windows [5]. |
| MalariaGEN Pf 7 | Empirical Database | A publicly available whole-genome sequencing database for Plasmodium falciparum. Used for validating findings from simulation studies and applying optimized methods to real-world data [52] [53]. |
| tskibd | Software Tool | A utility used with simulated data to extract the "ground truth" IBD segments from the Ancestral Recombination Graph (ARG), which is essential for calculating accuracy metrics like false positive and false negative rates [52] [53]. |
| E&R Benchmark Data Sets | Simulated Data | Standardized, simulated genomic data sets generated under different selection regimes (e.g., selective sweeps, truncating selection) used to consistently evaluate and compare the performance of selection detection tools [54]. |
Troubleshooting Guides
Guide 1: Troubleshooting False Positive Signals in Introgression Analysis
Problem: Your analysis detects local introgression signals that are potentially false positives, especially in genomic regions with low nucleotide diversity.
Explanation: The widely used D statistic (ABBA-BABA test) can produce spurious inferences of gene flow in small genomic windows or areas with low diversity, as its variance increases under these conditions [5]. This is a significant concern when working with ancestral polymorphisms.
Solution: Implement the D+ statistic, which incorporates both shared derived and shared ancestral alleles to improve precision.
- Step 1: Verify the Issue. Check the nucleotide diversity in your genomic windows. The D statistic is known to be less reliable in low-diversity regions [5].
Step 2: Calculate the D+ Statistic. Use the following formula to leverage additional information from ancestral alleles [5]:
D+ = Σ [ (ABBA - BABA) + (BAAA - ABAA) ] / Σ [ (ABBA + BABA) + (BAAA + ABAA) ]- ABBA: Derived allele shared between P3 (donor) and P2 (recipient).
- BABA: Derived allele shared between P3 and P1.
- BAAA: Ancestral allele shared between P3 and P2, while P1 has the derived allele.
- ABAA: Ancestral allele shared between P3 and P1, while P2 has the derived allele.
- Step 3: Interpret Results. The D+ statistic is more precise than the D statistic due to a lower false positive rate in small genomic regions, making it more reliable for identifying local targets of introgression [5].
The following workflow outlines the diagnostic and resolution process for this issue:
Guide 2: Addressing the Multiple Testing Problem in Genomic Studies
Problem: You have performed hundreds or thousands of statistical tests across the genome (e.g., for introgression or association), and you need to prevent an avalanche of false positive findings.
Explanation: When multiple hypothesis tests are performed, the probability of incorrectly rejecting at least one null hypothesis (Type I error/false positive) increases dramatically. With a standard significance level (α) of 5%, performing just 20 independent tests leads to a ~64% chance of at least one false positive [55]. This overall error rate across all tests is known as the Family-Wise Error Rate (FWER).
Solution: Apply a multiple testing correction to control the FWER or the False Discovery Rate (FDR).
- Step 1: Choose a Correction Method. Select a method based on the number of hypotheses and the need for statistical power. Common methods are listed below.
- Step 2: Apply the Correction. Adjust your raw p-values using your chosen method.
- Step 3: Use Adjusted p-values for Inference. Compare your adjusted p-values to your significance level (e.g., 0.05). Only results with adjusted p-values below the threshold should be considered statistically significant.
The table below summarizes the primary correction methods:
| Method | Core Function | Best Use Case | Key Advantage |
|---|---|---|---|
| Bonferroni | Controls FWER by multiplying raw p-values by the number of tests (m). | Confirmatory studies with a small number of tests. | Very simple to implement and understand [56]. |
| Holm's Step-Down | Controls FWER by sequentially adjusting p-values in a step-wise manner. | General use when control of any false positive is critical. | More statistical power than Bonferroni while controlling the same error rate [56]. |
| Benjamini-Hochberg | Controls the False Discovery Rate (FDR), the proportion of false positives among significant results. | Exploratory studies with a large number of tests (e.g., genomics) [56]. | Provides a better balance between discovering true effects and limiting false positives than FWER methods in large-scale studies [56]. |
The decision process for selecting and applying a multiple testing correction is as follows:
Frequently Asked Questions (FAQs)
Q1: What is a p-value, and how is it interpreted in the context of introgression? A p-value is a plausibility parameter. It represents the probability of observing your data (or something more extreme) if the null hypothesis is correct [55]. For introgression, a null hypothesis might be "There has been no gene flow between P3 and P2." A small p-value (typically ≤ 0.05) indicates that the observed excess of shared alleles (e.g., ABBA sites) is unlikely under the null hypothesis of no gene flow, providing evidence for introgression.
Q2: Why is correcting for multiple hypotheses so important in genomic studies like introgression research? Genomic studies involve testing many thousands of hypotheses simultaneously (e.g., per gene or per window). If each test has a 5% chance of a false positive, the sheer number of tests guarantees a large number of false positives without correction [55]. Controlling for multiple comparisons ensures that the handful of significant results you find are likely to be real biological signals and not statistical flukes.
Q3: The Bonferroni correction is considered very conservative. What does this mean for my analysis? "Conservative" means that the Bonferroni correction is very strict in preventing false positives. The trade-off is that it can dramatically reduce your statistical power, which is the probability of detecting a true effect [56]. This means you might miss genuine but weakly significant introgression signals. For this reason, other methods like Holm or Benjamini-Hochberg are often preferred.
Q4: What is the difference between controlling the Family-Wise Error Rate (FWER) and the False Discovery Rate (FDR)? FWER controls the probability of making at least one false discovery among all hypotheses. Methods like Bonferroni and Holm control FWER. In contrast, FDR controls the proportion of false discoveries among all hypotheses that are declared significant. The Benjamini-Hochberg method controls FDR, which is often more appropriate for large-scale exploratory studies where tolerating some false positives is acceptable to find more true positives [56].
Q5: Beyond multiple testing corrections, how can I improve the reliability of my introgression analysis?
- Consult Experts: Engage with bioinformaticians and statisticians, especially when designing your study [57].
- Leverage Existing Tools: Use well-documented pipelines and workflows for consistency and reproducibility [57] [58].
- Document Everything: Maintain detailed records of software versions, parameters, and code in an electronic lab notebook or version control system like Git [57].
- Validate Biologically: Use simulations or orthogonal experimental methods to confirm key findings.
The Scientist's Toolkit: Key Research Reagents & Solutions
| Item | Function in Analysis |
|---|---|
| D Statistic (ABBA-BABA) | Detects genome-wide introgression by measuring an excess of shared derived alleles between populations [5]. |
| D+ Statistic | Improves local introgression detection by leveraging both shared derived and ancestral alleles, reducing false positives [5]. |
| Bonferroni Correction | A simple multiple testing correction method that strongly controls the Family-Wise Error Rate [56]. |
| Holm's Step-Down Correction | A sequential multiple testing correction method that provides more power than Bonferroni while controlling the same error rate [56]. |
| Benjamini-Hochberg Correction | A multiple testing correction method that controls the False Discovery Rate, ideal for large-scale exploratory studies [56]. |
| Workflow Management System (e.g., Nextflow, Snakemake) | Streamlines pipeline execution, manages software dependencies, and ensures reproducibility and scalability of analyses [57] [58]. |
| Version Control (e.g., Git) | Tracks changes in analysis scripts and parameters, which is crucial for reproducibility and collaboration [57]. |
Frequently Asked Questions (FAQs)
Q1: What is the primary advantage of using a method like Gmin over the more traditional FST for detecting introgression?
Gmin is a haplotype-based measure that calculates the ratio of the minimum between-population nucleotide difference to the average between-population difference in a genomic window [59]. Its key advantage over FST in a secondary contact model is greater sensitivity and specificity for detecting recent introgression [59]. FST requires shared alleles to have equal frequencies in both populations to approach zero, which is not always the case after recent or limited gene flow. In contrast, Gmin is directly sensitive to the presence of recently exchanged haplotypes, is computationally efficient for whole-genome scans, and its sensitivity is robust to variations in population mutation and recombination rates [59].
Q2: My D-statistic (ABBA-BABA) test suggests introgression, but I need additional verification. What complementary approach can I use?
Tree-based phylogenetic methods serve as an excellent complement to SNP-based tests like the D-statistic [60]. The D-statistic assumes identical substitution rates and an absence of homoplasy, conditions that can be problematic when studying divergent species. You can extract numerous sequence alignment blocks from a whole-genome alignment, infer phylogenies (gene trees) for each block using maximum likelihood tools like IQ-TREE, and then analyze the distribution of these topologies. An over-representation of certain tree topologies can confirm introgression and is robust to conditions that may mislead the D-statistic [60].
Q3: How can I distinguish a true introgression signal from a false positive caused by ancestral polymorphism?
Cross-platform verification is crucial. A signal supported by multiple methods based on different principles is more robust. For instance, a genomic region flagged by both Gmin (a haplotype-based method) and the D-statistic (a site-pattern frequency method) presents a stronger case. Furthermore, tree-based methods can assess whether an excess of allele sharing is due to introgression (which creates a specific tree asymmetry) or ancestral polymorphism (which has a different, more random signature) [60]. Using an outgroup species is critical for polarizing alleles and making this distinction in many of these tests.
Q4: What are the key steps in a tree-based workflow to detect introgression from a whole-genome alignment?
A standard workflow involves: 1) Extraction: Pulling alignment blocks of a suitable length (e.g., 1,000 bp) from a whole-genome alignment (e.g., a MAF file) [60]. 2) Filtering: Removing alignments with high proportions of missing data or strong signals of within-alignment recombination to ensure phylogenetic reliability [60]. 3) Gene Tree Inference: Generating a phylogeny for each filtered alignment block using maximum likelihood software like IQ-TREE [60]. 4) Species Tree/Network Inference: Using programs like ASTRAL (for a species tree) or PhyloNet (for a network) to reconcile the gene trees and infer species relationships [60]. 5) Analysis: Quantifying asymmetry in gene tree topologies to infer introgression events.
Troubleshooting Guides
Issue: Inconsistent Introgression Signals Across Different Methods
Problem: Your analysis shows strong evidence for introgression with one tool (e.g., Gmin), but the signal is weak or absent with another (e.g., FST).
| Potential Cause | Explanation | Solution |
|---|---|---|
| Recency of Introgression | Gmin is highly sensitive to very recent gene flow, which may not yet have equilibrated allele frequencies. FST is less sensitive in this scenario [59]. | This is an expected outcome. Trust the method designed for recent gene flow and report the results from both. |
| Incomplete Lineage Sorting (ILS) | Shared ancestral polymorphism (ILS) can create patterns that mimic introgression in some tests, like the D-statistic. | Employ tree-based methods [60] or use a fuller suite of population genetic models to disentangle the contributions of ILS and introgression. |
| Method-Specific Assumptions | Each method has underlying assumptions about the demographic model, mutation rates, and absence of homoplasy that, if violated, can lead to false inferences. | Always state the assumptions and limitations of each method used. Use model-checking tools where available and prioritize signals consistently identified across multiple, disparate methods. |
Issue: High False Positive Rate in Introgression Scans
Problem: You are identifying many genomic regions as introgressed, but you suspect some may be false positives due to factors like ancestral polymorphism.
| Potential Cause | Explanation | Solution |
|---|---|---|
| Insufficient Filtering | Alignment blocks with high missing data or internal recombination can produce unreliable genealogies and spurious signals [60]. | Strictly filter genomic windows or alignment blocks for completeness, and use tools to detect and remove recombining segments. |
| Lack of an Outgroup | Without an outgroup to polarize alleles (determine which is ancestral and which is derived), it is impossible to distinguish introgressed alleles from those shared by common descent. | Always include one or more outgroup species in your analysis design for methods like the D-statistic and for rooting gene trees. |
| Incorrect Demographic Model | The null model against which introgression is tested does not reflect the true population history, leading to incorrect inference of gene flow. | Use model selection approaches to infer the underlying demography first. Methods like ∂a∂i or fastsimcoal2 can estimate divergence times and population sizes. |
Experimental Protocols & Data Presentation
The table below summarizes the core principles, strengths, and limitations of several major introgression detection tools.
| Method | Core Principle | Data Requirement | Key Strength | Primary Limitation |
|---|---|---|---|---|
| Gmin [59] | Ratio of min. between-population haplotype distance to the average distance. | Sequence data (haplotypes) from two populations. | High sensitivity for recent introgression; computationally inexpensive; robust to mutation/recombination rate [59]. | Less effective for ancient gene flow. |
| D-Statistic (ABBA-BABA) [60] | Compares frequencies of two site patterns ("ABBA" vs. "BABA") to detect asymmetry in allele sharing. | Genotype data for two sister populations and an outgroup. | Powerful for detecting minor gene flow; widely used and understood. | Assumes no homoplasy; can be confounded by certain patterns of ancestral polymorphism [60]. |
| Tree-Based Topology Analysis [60] | Infers phylogenies for many genomic regions and looks for asymmetry in tree topologies. | Multiple sequence alignments, often from a whole-genome alignment. | Robust to conditions that mislead D-statistic; provides a visual and intuitive framework [60]. | Computationally intensive; requires careful alignment filtering. |
| f-branch (fd) | An extension of the D-statistic that estimates the fraction of introgression from a specific source. | Similar to D-statistic, but often requires a candidate source population. | Provides a quantitative estimate of introgression proportion. | More complex and requires careful specification of test populations. |
Detailed Methodology: GminCalculation
Gmin is calculated in sliding windows across a whole-genome alignment. The procedure is as follows [59]:
- Data Preparation: Obtain nucleotide sequences for multiple individuals from two populations (Population 1 and Population 2), with sample sizes n₁ and n₂.
- Calculate Pairwise Differences: For a given genomic window, compute the Hamming distance (p-distance), dXY, for every possible pair of sequences (X, Y) where X is from Population 1 and Y is from Population 2. There will be n₁ × n₂ such comparisons.
- Compute Average Difference: Calculate the average of all these pairwise between-population differences, denoted as d̄XY.
- Find Minimum Difference: Identify the single smallest pairwise difference, min(dXY), among all the n₁ × n₂ comparisons.
- Form the Ratio: Gmin is defined as the ratio of the minimum to the average distance: Gmin = min(dXY) / d̄XY
The value of Gmin ranges from 0 to 1. A value closer to 1 indicates that at least one haplotype is very similar between the two populations, which is a signature of recent introgression [59].
Detailed Methodology: Tree-Based Introgression Detection
This protocol outlines the steps for detecting introgression using phylogenies, as implemented in a workshop activity using cichlid fish data [60].
- Extract Alignment Blocks: Use a script (e.g., a custom Python script) to extract multiple sequence alignment blocks of a fixed length (e.g., 1,000 bp) from a whole-genome alignment file (e.g., in MAF format) [60].
- Filter Alignment Blocks: Filter the extracted alignments to ensure high data quality. Remove alignments with:
- A high proportion of missing data (incomplete sequences).
- Strong signals of internal recombination, which can violate the assumption of a single genealogy for the region [60].
- Infer Gene Trees: For each filtered alignment block, infer a phylogenetic tree (a "gene tree") using maximum likelihood software such as IQ-TREE [60].
- Infer Species Tree/Network: Use the collection of gene trees to infer the overall species relationship.
- Analyze Topology Frequencies: Examine the entire set of gene trees. An overabundance of gene trees that match a specific topology inconsistent with the species tree can provide direct evidence for introgression between particular taxa [60].
Method Comparison & Workflows
Research Reagent Solutions
The table below lists essential software and data types used in modern introgression detection studies.
| Item Name | Type | Primary Function in Introgression Detection |
|---|---|---|
| Whole-Genome Alignment | Data | Provides the coordinated, genome-wide sequence data necessary for methods like Gmin scans and for extracting alignment blocks for tree-based methods [60]. |
| MS/MSMOVE [59] | Software | Coalescent simulation software used to generate genomic data under user-specified demographic models (with or without gene flow). Essential for testing methods and interpreting real data. |
| IQ-TREE [60] | Software | A tool for efficient and effective maximum likelihood phylogenetic analysis. Used to infer gene trees from hundreds or thousands of genomic alignment blocks. |
| ASTRAL [60] | Software | Estimates a species tree from a set of input gene trees. It is statistically consistent under the multi-species coalescent model, even when gene trees are inaccurate. |
| PhyloNet [60] | Software | Infers phylogenetic networks from gene trees or sequence data, allowing for the explicit visualization and testing of introgression/hybridization events. |
Workflow for Cross-Platform Introgression Analysis
The following diagram illustrates a robust, integrated workflow for verifying introgression signals by combining multiple methods.
Method Selection Logic
This diagram provides a decision tree for selecting the most appropriate introgression detection method based on your specific research question and data.
Frequently Asked Questions (FAQs)
Q1: What is the primary challenge in distinguishing adaptive introgression from ancestral polymorphism?
A: The core challenge is that both processes can create similar genomic signatures, such as regions of reduced divergence between species. Ancestral polymorphism represents genetic variants retained from a shared common ancestor, while adaptive introgression results from the selective incorporation of beneficial alleles from another species. Without careful validation, neutral ancestral polymorphisms can be misinterpreted as adaptive introgression events, leading to false positives [61] [62].
Q2: What are the key methodological categories for detecting adaptive introgression?
A: Methods fall into three main categories [11]:
- Summary Statistics: Evolved methods using statistics like
Q95to identify genomic regions with unusually high ancestry proportions from a donor population. These are often computationally efficient and can perform well across diverse scenarios [11] [62]. - Probabilistic Modeling: Methods like
Ancestry_HMM-Suse Hidden Markov Models (HMMs) to incorporate evolutionary processes explicitly. They infer local ancestry and can quantify the strength of selection acting on introgressed loci [63]. - Supervised Learning: An emerging approach where methods like
genomatnn(a CNN) andFILET(using Extra-Trees) are trained to recognize patterns of adaptive introgression in genomic data. These can capture complex, non-linear patterns but may require retraining for non-model systems [11] [64] [23].
Q3: My analysis has identified a candidate region with high archaic ancestry. What is the minimal set of validations required?
A: A robust validation framework should include:
- Confirm Archaic Origin: Use methods like
Ancestry_HMM-Sorgenomatnnto confirm the haplotype's origin is from a donor (e.g., Neanderthal) and not ancestral variation [63] [64]. - Test for Selection: Apply multiple tests for positive selection (e.g., site frequency spectrum-based tests, extended haplotype homozygosity) on the candidate region in the recipient population [65].
- Functional Pathway Analysis: Check if the introgressed gene participates in a known functional pathway with other candidate genes. The co-introgression of functionally linked genes is a strong indicator of adaptation [65].
- Phenotypic Association: If possible, link the introgressed haplotype to a specific adaptive phenotype (e.g., via gene expression studies or association mapping) [61] [65].
Q4: How do I choose the best method for my specific study system?
A: Method selection depends on your data and system [62]:
- For well-studied models with reference genomes: Probabilistic HMM methods are powerful.
- For non-model systems or those with unknown demography: The
Q95statistic is a robust and straightforward starting point. - When you have large, phased genomic datasets: Supervised learning methods like CNNs can offer high accuracy.
- Crucially, benchmark performance using simulations that mirror your study system's known evolutionary history (e.g., divergence times, population sizes) to understand the false positive rate [62].
Troubleshooting Guides
Problem: High False Positive Rates in Adaptive Introgression Scan
| Symptom | Potential Cause | Solution |
|---|---|---|
| Many candidate loci are in regions of low recombination. | Incomplete lineage sorting (ILS) is misidentified as introgression. | Use a method that jointly models ILS and introgression. Compare against a carefully chosen outgroup to polarize alleles [62]. |
| Signals are weak and distributed across many loci. | Polygenic adaptation with weak selection on individual loci. | Employ methods designed for polygenic signals or gene set enrichment analyses instead of looking for hard sweeps [65]. |
| A method trained on human data performs poorly on your species. | Demographic differences confound the model. | Retrain machine learning models on data simulated with your species' specific demography, or switch to a more general summary statistic approach like Q95 [62]. |
| Strong introgression signal with no linked adaptive trait. | Neutral introgression or hitchhiking. | Analyze haplotype lengths to date the introgression event. Look for independent evidence of selection and rule out linked adaptive loci [61] [63]. |
Problem: Validating the Functional Impact of an Introgressed Haplotype
| Symptom | Potential Cause | Solution |
|---|---|---|
| An introgressed allele shows no frequency difference in cases vs. controls. | The allele may not be the causal variant or may have a context-dependent effect. | Perform finer-scale mapping to find the causal variant. Investigate gene-gene (epistatic) or gene-environment interactions [61]. |
| The gene's function is unknown or not obviously linked to reproduction. | The introgressed allele may have a regulatory role or a novel function. | Perform functional assays (e.g., CRISPR edits in cell lines) to characterize the allele's effect on gene expression or protein function [65]. |
| The introgressed haplotype is broken up by recombination. | The adaptive signal is difficult to track. | Focus on the core, non-recombined haplotype block and use phylogenetic methods to reconstruct the ancestral introgressed haplotype [63]. |
Experimental Protocols & Data
Protocol 1: Detecting Adaptive Introgression using Ancestry_HMM-S
This protocol uses a hidden Markov model to infer local ancestry and detect selection [63].
- Input Data Preparation: Collect whole-genome sequencing data from:
- Focal Population: The admixed population of interest.
- Reference Population 1: A population representing the donor ancestry.
- Reference Population 2: A population representing the recipient ancestry.
- Model Training: Run
Ancestry_HMM-Son the data to infer the baseline neutral admixture model and parameters. - Scan for Selection: Execute the model to scan the genome for regions where the donor ancestry proportion significantly exceeds the genome-wide baseline.
- Quantify Selection: For significant outlier regions, use the model's output to estimate the selection coefficient (s) acting on the introgressed haplotype.
- Validation: Compare the identified regions with independent tests for positive selection and known functional annotations.
Protocol 2: Validating Introgression with Convolutional Neural Networks (genomatnn)
This protocol uses a CNN to distinguish adaptive introgression from other evolutionary scenarios [64].
- Data Matrix Construction:
- Select a genomic window (e.g., 100 kbp).
- For the donor, recipient, and an outgroup population, create a genotype matrix. Sort haplotypes by similarity to the donor.
- Concatenate matrices into a single input for the CNN.
- Classification: Feed the input matrix into a pre-trained
genomatnnCNN. - Output Interpretation: The network outputs a probability score for adaptive introgression. High-probability regions are candidates.
- Saliency Mapping: Use the model's saliency map feature to identify which parts of the input data (e.g., specific haplotypes) most influenced the prediction.
Table 1: Comparison of Adaptive Introgression Detection Methods
| Method Name | Category | Key Principle | Data Requirements | Key Output |
|---|---|---|---|---|
Q95 |
Summary Statistic [62] | 95th percentile of local ancestry proportion | Genotype data, local ancestry estimates | Genomic regions with exceptionally high donor ancestry |
Ancestry_HMM-S |
Probabilistic Modeling [63] | HMM that incorporates selection into local ancestry inference | Unphased genotypes from admixed and reference populations | Loci under selection and estimated selection coefficient (s) |
genomatnn |
Supervised Learning (CNN) [64] | CNN trained on simulated data to classify genomic windows | Phased or unphased genotypes from donor, recipient, and outgroup | Probability of adaptive introgression for a genomic region |
FILET |
Supervised Learning (Extra-Trees) [23] | Machine learning classifier combining multiple summary statistics | Genomic data from two populations | Classified introgressed loci and direction of gene flow |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Tools for Adaptive Introgression Studies
| Item | Function/Description | Example Use Case |
|---|---|---|
| High-Coverage WGS Data | Provides the fundamental data for calling variants and haplotypes with high accuracy. | Essential for all methods to reliably detect introgressed segments and distinguish them from sequencing errors [65]. |
| Reference Panels (e.g., Neanderthal, Denisovan) | Serves as a baseline for identifying archaic-like haplotypes in modern genomes. | Used in Ancestry_HMM-S and genomatnn to assign local ancestry [63] [64]. |
| Forward Simulator (e.g., SLiM) | Simulates genomic data under complex evolutionary models including selection and admixture. | Generating training data for machine learning methods or creating null distributions for summary statistics [64] [62]. |
| Functional Annotation Databases | Provides information on gene function, regulatory elements, and biological pathways. | Annotating candidate introgressed regions to hypothesize their potential adaptive role (e.g., in immunity or reproduction) [65]. |
Methodological Visualization
Adaptive Introgression Detection Workflow
Ancestry_HMM-S Logical Architecture
Troubleshooting Guide: Common Challenges in Introgression Analysis
Q1: How can I distinguish true bidirectional adaptive introgression from false signals caused by ancestral polymorphism?
A1: Ancestral polymorphisms can create phylogenetic patterns that mimic introgression. To confirm true bidirectional adaptive introgression, employ this multi-faceted approach:
- Population Genetic Analysis: Calculate Patterson's D statistics to test for significant gene flow between species pairs. True introgression will show significant deviations from the null model of no gene flow.
- Coalescent Simulations: Use models like MSMC to simulate expected patterns under ancestral polymorphism and compare with observed data.
- Population Branch Statistic (PBS): Identify loci with extreme differentiation that indicate selection, which often accompanies adaptive introgression.
- Environmental Association Analysis: Correlate putatively introgressed loci with environmental variables to establish adaptive significance.
Q2: What quality control metrics are most critical for high-throughput sequencing data in population genomics studies?
A2: Based on the spruce case study methodology, implement these QC thresholds [66] [67]:
- Genotype Call Rates: Exclude samples with ≥10% missing data and SNPs with ≥10% missingness.
- Minor Allele Frequency: Filter out variants with MAF <1% to reduce sequencing error artifacts.
- Hardy-Weinberg Equilibrium: Remove markers with HWE p-values <0.001.
- Linkage Disequilibrium Pruning: Apply parameters (--indep-pairwise 200 25 0.2) to minimize non-independence of markers.
Q3: How can researchers validate that detected introgressed regions are truly adaptive rather than neutral?
A3: The spruce study employed these validation strategies [66]:
- Functional Annotation: Identify genes within introgressed regions and assess their biological functions.
- Phenotypic Correlation: Link introgressed regions to known adaptive traits (e.g., stress resilience, flowering time).
- Environmental Association: Test for correlations between allele frequencies and environmental gradients.
- Population Differentiation: Calculate FST for introgressed regions versus genome-wide background.
Experimental Protocols & Data Standards
Table 1: Key Quality Control Metrics for Introgression Studies
| QC Parameter | Threshold | Software Tool | Purpose in Introgression Analysis |
|---|---|---|---|
| Sample Call Rate | ≥90% | PLINK 1.9 | Ensures sufficient data quality per individual |
| SNP Call Rate | ≥90% | PLINK 1.9 | Maintains marker reliability |
| Minor Allele Frequency | ≥1% | PLINK 1.9 | Reduces false positives from rare variants |
| HWE p-value | ≥0.001 | PLINK 1.9 | Filters markers with genotyping errors |
| LD Pruning | R² < 0.2 | PLINK 1.9 | Ensures marker independence for structure analysis |
| ANI Threshold | 94-96% | PyANI | Defines species boundaries for bacterial studies [31] |
| Analysis Type | Key Finding | Statistical Support | Biological Significance |
|---|---|---|---|
| Genetic Differentiation | Distinct differentiation despite gene flow | FST analysis | Maintains species boundaries amid gene flow |
| Adaptive Introgression | Bidirectional between allopatric species | D-statistics | Challenges unidirectional gene flow assumptions |
| Candidate Genes | Dozens linked to stress resilience & flowering | PBS, FST outliers | Explains historical environmental adaptation |
| Gene Flow Level | Substantial between species | Migration rates (Nm) | Supports porous species boundaries in spruces |
Detailed Methodologies from Featured Studies
Protocol 1: Population Transcriptome Analysis for Introgression Detection (Adapted from Plant Diversity Study [66])
Sample Collection and Sequencing:
- Collect tissue samples from multiple populations across species' ranges
- Extract RNA and prepare sequencing libraries
- Sequence using Illumina platform (typically 150bp paired-end)
- Target: 20-30 individuals per species from sympatric and allopatric zones
Bioinformatic Processing:
- Quality control: FastQC and Trimmomatic for adapter removal
- Mapping: STAR or HISAT2 to reference genome
- Variant calling: GATK best practices pipeline
- Filtering: Apply QC metrics from Table 1
Introgression Analysis:
- Population structure: ADMIXTURE with cross-validation
- Phylogenetics: Maximum likelihood trees per gene and concatenated
- Introgression tests: Patterson's D, f4-statistics
- Adaptive loci: XP-CLR and PBS for selection scans
Protocol 2: Core Germplasm Population Construction (Adapted from Frontiers in Genetics Study [67])
SLAF-seq Library Construction:
- Digest genomic DNA with restriction enzymes (RsaI + HaeIII for spruce)
- Ligate sequencing adapters to digested fragments
- Perform selective amplification based on fragment size
- Sequence on Illumina platform (typically 100-125bp reads)
SNP Discovery and Analysis:
- Identify SLAF tags and group homologous tags
- Call SNPs across all samples with consistency filtering
- Develop 1,964,178 high-consistency SNP markers [67]
- Population analysis: phylogenetic trees, structure analysis
Core Germplasm Selection:
- Use Core Hunter II software with multiple genetic distance measures
- Select 20% of samples representing full diversity
- Validate core collection maintains allelic diversity
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Introgression Studies
| Reagent/Resource | Function | Example from Spruce Studies |
|---|---|---|
| SLAF-seq Library Kit | Simplified genome sequencing | 1,964,178 SNP markers in Qinghai spruce [67] |
| Human Origins Array | Genotyping for ancestry analysis | 597,569 SNPs for East Asian ancestry inference [68] |
| AISNP Panels | Ancestry-informative marker sets | 50-2,000 SNP panels for machine learning [68] |
| Core Hunter II | Core germplasm selection | Selected 33 core germplasms (20%) from 165 accessions [67] |
| PLINK 1.9 | Quality control and basic association | Applied genotype QC filters [68] |
| ADMIXTURE | Population structure inference | Estimated ancestry components with cross-validation [68] |
Analytical Workflows Visualization
Introgression Analysis Workflow
Introgression Signal Discrimination
Methods Comparison at a Glance
The following table summarizes the core characteristics, strengths, and limitations of the three hybridization detection methods.
| Method | Core Principle | Optimal Use Case | Key Strengths | Documented Limitations |
|---|---|---|---|---|
| IntroMap [69] [70] | Signal processing of read alignment data to detect drops in sequence homology. | Identifying precise introgressed regions in a hybrid individual relative to a reference genome. | Does not require variant calling or de novo assembly; provides visualizable, positional data on introgression [69] [70]. | Accuracy depends on parameter tuning (filter window, fit parameter, threshold); can over- or under-estimate region size if misconfigured [70]. |
| D-Statistic (ABBA-BABA) [71] [4] [20] | Compares frequencies of two discordant site patterns (ABBA, BABA) to test for genome-wide gene flow. | Testing for the presence and direction of introgression in a four-taxon system (P1, P2, P3, Outgroup). | Powerful for detecting the signal of hybridization; simple and widely used statistic [71] [4]. | High False Discovery Rate (FDR), especially with high incomplete lineage sorting (ILS); does not identify specific introgressed loci [71] [4]. |
| HyDe [71] [20] | Uses phylogenetic invariants (site pattern frequencies) under a coalescent model with hybridization. | Detecting hybridization and estimating the precise admixture proportion ((\gamma)) in a population. | Robust and accurate estimates of (\gamma); powerful for detecting hybridization; can handle multiple individuals per population [71] [20]. | Statistical power decreases under conditions of very high incomplete lineage sorting (ILS) [71]. |
Experimental Protocols & Workflows
IntroMap Protocol
IntroMap uses sequence alignment data to identify regions of reduced homology, indicating potential introgression, without requiring variant calling [69] [70].
Key Steps:
- Alignment: Align the Next-Generation Sequencing (NGS) reads from your hybrid sample to a reference genome (which should share high homology with one parental line) using an aligner like
bowtie2. The output is a BAM file [69] [70]. - Data Parsing: IntroMap parses the BAM file, specifically the
MDtags, which encode match/mismatch information. Each aligned read is converted into a binary vector where1represents a match and0a mismatch/indel [70]. - Score Calculation: The binary vectors are used to construct a matrix, and the mean score for each base position in the reference is computed, creating a sequence homology vector [70].
- Signal Processing: The raw homology signal is smoothed using a convolution-based low-pass filter (to reduce high-frequency noise from single SNPs) and a locally weighted scatterplot smoothing (LOWESS) regression. This produces a fitted curve representing overall homology [70].
- Calling Introgressed Regions: A threshold is applied to the smoothed signal. Genomic regions where the homology score drops below this threshold are identified and output as candidate introgressed regions, along with visualization plots [70].
D-Statistic (ABBA-BABA Test) Protocol
This method tests for a genome-wide excess of shared derived alleles between taxa, which is a signature of introgression [71] [4] [20].
Key Steps:
- Taxon Configuration: Define your four populations: two sister populations (P2 and P3), an outgroup (O), and a reference (P1). The test investigates whether P2 shares more derived alleles with P3 (no gene flow) or P1 (signaling gene flow between P1 and P2) [4] [20].
- Site Pattern Counting: Genome-wide, count the occurrences of two site patterns for biallelic SNPs (ancestral allele
A, derived alleleB):- ABBA: Sites where P1 and P3 have the ancestral allele (
A), but P2 and the outgroup have the derived allele (B). - BABA: Sites where P1 and P2 have the ancestral allele (
A), but P3 and the outgroup have the derived allele (B) [20].
- ABBA: Sites where P1 and P3 have the ancestral allele (
- Calculate D-Statistic: Compute Patterson's D using the formula:
D = (ABBA - BABA) / (ABBA + BABA)[20]. - Significance Testing: A significant deviation of D from zero (assessed via a block jackknife procedure across the genome) indicates gene flow. A significant positive D suggests gene flow between P1 and P2 [4] [20].
HyDe Protocol
HyDe uses phylogenetic invariants under a coalescent model with hybridization to detect hybrids and estimate their admixture proportions [20].
Key Steps:
- Data Preparation: Prepare phased genomic data (SNPs or sequences) for an outgroup (O), two parental populations (P1 and P2), and a putative hybrid population (Hyb). HyDe can utilize data from multiple individuals per population [20].
- Site Pattern Calculation: For all possible combinations of individuals (forming quartets of O, P1, Hyb, P2), HyDe calculates the observed frequencies of site patterns (e.g.,
OOAC,OOCAfor nucleotide data) [20]. - Model Testing and Estimation: HyDe tests whether the observed site pattern frequencies deviate significantly from the expectation under a null model of no admixture. It simultaneously estimates the admixture proportion ((\gamma)), which represents the genetic contribution from P2 to the hybrid [20]. The test statistic is asymptotically normally distributed, providing a p-value [20].
- Output and Analysis: The software outputs estimates of (\gamma), p-values, and can perform bootstrap resampling to assess support for individual hybrids or the overall population-level signal [20].
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function/Description | Relevance in Experiments |
|---|---|---|
| Reference Genome | A high-quality, assembled genomic sequence. | Essential for IntroMap's alignment step and for providing genomic coordinates for introgressed regions [69] [70]. |
| Outgroup Sequence | Genomic data from a species closely related to, but diverged before, the studied taxa. | Critical for polarizing alleles (ancestral vs. derived) in the D-statistic and for providing an evolutionary scale in methods like RNDmin [4] [20]. |
| Phased Genomic Data | Data where the alleles on homologous chromosomes have been assigned to their respective haplotypes. | Required for statistics that use minimum distances between haplotypes (e.g., dmin, RNDmin) and is beneficial for HyDe's site pattern analysis [4] [20]. |
| Parental Population Samples | Genomic data from pure, non-admixed populations that are the putative parents of the hybrid. | A fundamental requirement for all three methods (IntroMap, D-statistic, HyDe) to define the source pools for introgression [71] [69] [20]. |
Frequently Asked Questions (FAQs)
Q1: My analysis with the D-statistic shows a significant signal of introgression, but I cannot pinpoint the specific introgressed regions. What should I do? This is a known limitation of the D-statistic, which is designed for genome-wide tests, not locus-specific detection [4]. To identify specific regions, you should employ a secondary method. IntroMap is ideal if you have a hybrid individual and a reference genome, as it provides positional data [69] [70]. Alternatively, sliding-window approaches using statistics like RNDmin or Gmin can help localize introgression signals by being sensitive to rare, recent migrants and robust to mutation rate variation [4].
Q2: How can I determine if a significant signal is a true positive and not a false positive caused by ancestral population structure or incomplete lineage sorting? This is a critical challenge. The D-statistic is particularly known for a high False Discovery Rate under high ILS [71]. To mitigate this:
- Use Multiple Methods: A signal confirmed by different methods (e.g., a significant D-statistic plus a significant HyDe test with a well-estimated (\gamma)) is more robust [71].
- Leverage Method Strengths: HyDe is noted for its robust and accurate estimation of the admixture proportion ((\gamma)), which is less prone to error in the presence of ILS compared to the D-statistic's FDR [71].
- Examine Sequence Divergence: Methods like RNDmin and Gmin are explicitly designed to be robust to variation in mutation rates, which can be a confounder, by normalizing with distances to an outgroup or average within-species divergence [4].
Q3: My hybrid population is not a uniform admixture but contains only a few introgressed individuals. Which method is most appropriate? HyDe has a specific feature for this scenario. It can conduct hypothesis tests on individuals within the putative hybrid population and use bootstrap resampling to detect heterogeneity in the admixture signal [20]. This allows you to identify which specific individuals show evidence of introgression, making it superior to the D-statistic in cases of non-uniform gene flow. IntroMap also analyzes a single hybrid genome and would need to be run on each individual separately [69] [70].
Q4: I have low-coverage sequencing data and am concerned about the accuracy of variant calling. Which tool is most suitable? IntroMap is uniquely advantageous here. Its algorithm relies solely on the alignment of reads to a reference genome and does not require a variant calling step. It uses the raw match/mismatch information from the BAM file's MD tags, making it less susceptible to errors associated with low-coverage variant calling [69] [70].
Conclusion
Mitigating false positive introgression signals requires a multi-faceted approach that acknowledges the profound impacts of ancestral polymorphism and selection. The key takeaways are the critical need to move beyond methods that assume strict neutrality and a molecular clock, the power of integrating multiple lines of evidence from different bioinformatic pipelines, and the necessity of rigorous, ancestry-aware statistical validation. For future research, the development of new methods robust to selection and rate heterogeneity is paramount. In biomedical research, these refined detection strategies are essential for accurately interpreting the functional role of introgressed alleles in human disease, drug response, and cancer, ensuring that evolutionary insights translate into reliable clinical applications.
References
- 1. Importance of incomplete lineage sorting and introgression in the origin of shared genetic variation between two closely related pines with overlapping distributions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5315522/]
- 2. Incomplete lineage sorting - Wikipedia [https://en.wikipedia.org/wiki/Incomplete_lineage_sorting]
- 3. Distinguishing between Incomplete Lineage Sorting and Genomic Introgressions: Complete Fixation of Allospecific Mitochondrial DNA in a Sexually Reproducing Fish (Cobitis; Teleostei), despite Clonal Reproduction of Hybrids - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4074047/]
- 4. Powerful methods for detecting introgressed regions from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4899106/]
- 5. Leveraging shared ancestral variation to detect local ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10798638/]
- 6. Variation in DNA Substitution Rates among Lineages ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0009649]
- 7. Identifying and Mitigating False Positive Alerts [https://panther.com/blog/identifying-and-mitigating-false-positive-alerts]
- 8. 🚦How to reduce False Positive Alerts in Threat Detection ... [https://medium.com/@tahirbalarabe2/how-to-reduce-false-positive-alerts-in-threat-detection-sharpening-security-detections-d8382b93915a]
- 9. 9 ways to eliminate false positive SIEM alerts [https://www.connectwise.com/blog/9-ways-to-eliminate-siem-false-positives]
- 10. Detecting Introgression in Shallow Phylogenies [https://pmc.ncbi.nlm.nih.gov/articles/PMC12485364/]
- 11. Review Decoding genomic landscapes of introgression [https://www.sciencedirect.com/science/article/pii/S0168952525001660]
- 12. Revisiting phylogenetic signal; strong or negligible impacts of ... [https://bmcecolevol.biomedcentral.com/articles/10.1186/s12862-017-0898-y]
- 13. Pervasive sequence patents cover the entire human genome [https://pmc.ncbi.nlm.nih.gov/articles/PMC3706854/]
- 14. Pervasive misannotation of microexons that are ... [https://www.nature.com/articles/s41467-022-28449-8]
- 15. Support FAQ's - Tecan Genomics [https://lifesciences.tecan.com/genomics-support-faq]
- 16. : a IntroMap method for the detection of... signal analysis based [https://pubmed.ncbi.nlm.nih.gov/29202713/]
- 17. danshea/IntroMap: Introgression screening software ... [https://github.com/danshea/IntroMap]
- 18. Towards Reliable Detection of Introgression in the Presence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11639170/]
- 19. Clevenger | IntroMap : A Pipeline and Set of... | Peanut Science [https://peanutscience.com/article/id/1257/]
- 20. A Python Package for Genome-Scale Hybridization Detection [https://pmc.ncbi.nlm.nih.gov/articles/PMC6454532/]
- 21. HyDe: Hybridization Detection Using Phylogenetic Invariants ... [https://hybridization-detection.readthedocs.io/]
- 22. pblischak/HyDe: Hybridization detection using ... [https://github.com/pblischak/HyDe]
- 23. Supervised machine learning reveals introgressed loci in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5933812/]
- 24. Variant calling from RNA-Seq data reveals allele-specific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12119874/]
- 25. Detecting likely germline variants during tumor-based ... [https://www.jci.org/articles/view/190264]
- 26. Estimation of allele frequency and association mapping using ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-12-231]
- 27. Standards and Guidelines for the Interpretation and Reporting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5707196/]
- 28. A Simplified Approach to Somatic Analysis in NGS [https://www.euformatics.com/blog-post/a-simplified-approach-to-somatic-analysis-in-ngs]
- 29. Germline pathogenic variation impacts somatic alterations ... [https://www.nature.com/articles/s41467-025-65190-4]
- 30. AN EMPIRICAL BAYES APPROACH - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3065192/]
- 31. Introgression impacts the evolution of bacteria, but species ... [https://www.nature.com/articles/s41467-025-64947-1]
- 32. An overview of STRUCTURE: applications, parameter settings ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3665925/]
- 33. Application of Bayesian genomic prediction methods to ... [https://gsejournal.biomedcentral.com/articles/10.1186/s12711-022-00724-8]
- 34. RAfilter: an algorithm for detecting and filtering false ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10107899/]
- 35. Reducing false positive incidental findings with ensemble ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4112476/]
- 36. ML | Underfitting and Overfitting [https://www.geeksforgeeks.org/machine-learning/underfitting-and-overfitting-in-machine-learning/]
- 37. Understanding Signal, Noise, and Curve Fitting [https://trendspider.com/learning-center/understanding-signal-noise-and-curve-fitting/]
- 38. Model Fit: Underfitting vs. Overfitting [https://docs.aws.amazon.com/machine-learning/latest/dg/model-fit-underfitting-vs-overfitting.html]
- 39. Human-Genetic Ancestry Inference and False Positives in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7407470/]
- 40. Tumor Purity and Fold Change [https://support-docs.illumina.com/SW/DRAGEN_v310/Content/SW/DRAGEN/SomaticWESCNV_TumorPurity.htm]
- 41. PureCN: copy and SNV classification using targeted... number calling [https://scfbm.biomedcentral.com/articles/10.1186/s13029-016-0060-z]
- 42. A method to reduce ancestry related germline false positives in tumor ... [https://pubmed.ncbi.nlm.nih.gov/29052513/]
- 43. Accurate Inference of Tumor Purity and Absolute Copy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7205152/]
- 44. Systematic Assessment of Tumor Purity and Its Clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7529507/]
- 45. Copy Number Variation (CNV) Calling [https://www.cancer.gov/about-nci/organization/cbiit/training/library/cnv]
- 46. Signatures of copy number alterations in human cancer [https://www.nature.com/articles/s41586-022-04738-6]
- 47. Sequencing Depth vs. Coverage: Key Metrics in NGS ... [https://3billion.io/blog/sequencing-depth-vs-coverage]
- 48. Sequencing depth vs. VAF sensitivity - NGS [https://www.ogt.com/ca/resources/ngs-resources-and-support/ngs-resources/sequencing-depth-vs-vaf-sensitivity/]
- 49. Leveraging shared ancestral variation to detect local introgression - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10798638/]
- 50. ACMG clinical laboratory standards for next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4098820/]
- 51. NGS Sequencing Support-Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/next-generation-sequencing-support-center/ngs-support/ngs-support-troubleshooting.html]
- 52. Benchmarking and Optimization of Methods for the ... [https://elifesciences.org/reviewed-preprints/101924v1]
- 53. Benchmarking and Optimization of Methods for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11092787/]
- 54. Benchmarking software tools for detecting and quantifying ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1770-8]
- 55. Judging a Plethora of p-Values: How to Contend With ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2822959/]
- 56. Adjustment of p-values for multiple hypotheses [https://tidsskriftet.no/en/2021/09/medicine-and-numbers/adjustment-p-values-multiple-hypotheses]
- 57. 5 Tips for Tackling Your Data Analysis Without Experience in ... [https://bioinformatics.ccr.cancer.gov/btep/5-tips-for-tackling-your-data-analysis-without-experience-in-bioinformatics/]
- 58. Bioinformatics Pipeline Troubleshooting [https://www.meegle.com/en_us/topics/bioinformatics-pipeline/bioinformatics-pipeline-troubleshooting]
- 59. A New Method to Scan Genomes for Introgression in a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4396994/]
- 60. Introgression detection - Evolution and Genomics [https://evomics.org/learning/population-and-speciation-genomics/2025-population-and-speciation-genomics/introgression-detection/]
- 61. Adaptive Introgression as an Evolutionary Force: A Meta ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12181397/]
- 62. Benchmarking the detection of adaptive introgression from ... [https://evolbiol.peercommunityin.org/articles/rec?articleId=835]
- 63. Inferring Adaptive Introgression Using Hidden Markov ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8097282/]
- 64. Detecting adaptive introgression in human evolution using ... [https://elifesciences.org/articles/64669]
- 65. Archaic introgression contributed to shape the adaptive ... [https://elifesciences.org/articles/89815]
- 66. A complex interplay of genetic introgression and local ... [https://www.sciencedirect.com/science/article/pii/S2468265925000848]
- 67. Genetic evolution of parental populations and construction ... [https://www.frontiersin.org/articles/10.3389/fgene.2025.1712106/full]
- 68. Integrated genetic and geographic ancestry prediction via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570655/]
- 69. IntroMap: a signal analysis based method for the detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5716257/]
- 70. IntroMap: a signal analysis based method for the detection of ... [https://bmcgenomdata.biomedcentral.com/articles/10.1186/s12863-017-0568-5]
- 71. Comparative Performance of Popular Methods for Hybrid ... [https://pubmed.ncbi.nlm.nih.gov/33404632/]