Concatenation vs. Coalescent Models in Phylogenomics: Navigating Introgression and ILS for Accurate Species Tree Inference
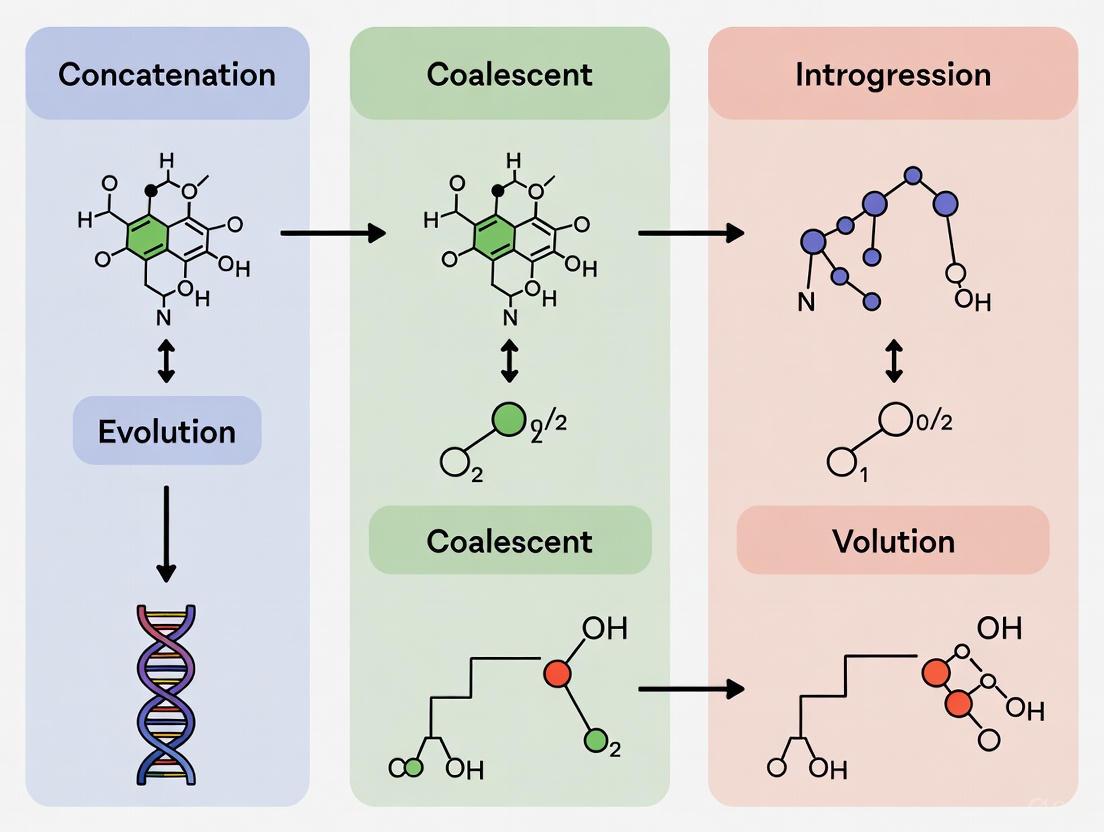
This article provides a comprehensive comparison of concatenation and multispecies coalescent (MSC) approaches for phylogenetic inference, with a special focus on datasets impacted by introgression and incomplete lineage sorting (ILS).
Concatenation vs. Coalescent Models in Phylogenomics: Navigating Introgression and ILS for Accurate Species Tree Inference
Abstract
This article provides a comprehensive comparison of concatenation and multispecies coalescent (MSC) approaches for phylogenetic inference, with a special focus on datasets impacted by introgression and incomplete lineage sorting (ILS). Aimed at researchers and bioinformaticians, we explore the foundational principles of both methods, detail their application workflows, and address key challenges like model violation and gene tree estimation error. Through empirical case studies and statistical validation frameworks, we demonstrate why the MSC model often outperforms concatenation in complex evolutionary scenarios. The review concludes with practical guidance for method selection and discusses the implications of these phylogenomic advancements for tracing the evolutionary origins of biomedically relevant traits and genes.
The Roots of Discordance: Understanding Gene Tree Conflict, ILS, and Introgression
The central challenge in modern phylogenomics lies in reconciling the differences between gene trees and species trees. A gene tree represents the evolutionary history of a single gene or locus, based on the genetic sequences of different individuals or species. In contrast, a species tree represents the actual evolutionary history of the species themselves—the true pattern of lineage splitting and descent over time [1]. The paradox emerges from the widespread observation that these trees are often incongruent, meaning they display different branching patterns. This incongruence presents a fundamental challenge for phylogeneticists who must choose analytical approaches that can accurately recover species relationships from conflicting genetic signals.
The debate between two primary methodological frameworks—concatenation versus multispecies coalescent (MSC) models—forms the core of contemporary discussions on addressing this paradox. Concatenation approaches combine data from all genes into a single "supermatrix" and infer one phylogenetic tree, implicitly assuming all genes share the same evolutionary history. Conversely, MSC approaches explicitly model gene tree variation resulting from biological processes like incomplete lineage sorting (ILS), providing a more sophisticated but computationally demanding framework for species tree inference [2]. This guide provides an objective comparison of these competing methodologies, evaluating their performance, underlying assumptions, and applicability to empirical phylogenomic data sets.
Biological Foundations of Incongruence
Primary Mechanisms Creating Gene Tree Discordance
Gene tree-species tree incongruence arises from several distinct biological processes that cause individual genes to have evolutionary histories that diverge from the overall species history.
Incomplete Lineage Sorting (ILS): ILS occurs when multiple gene lineages persist through successive speciation events. This happens when the genetic polymorphisms in an ancestral population are not fully sorted into distinct monophyletic lineages by the time subsequent speciation occurs [1] [3]. The probability of ILS increases when the time between speciation events is short relative to the population size, creating a situation where gene trees may reflect the random sorting of ancestral polymorphism rather than species divergence history. ILS is considered one of the most common biological sources of gene tree variation [2].
Hybridization and Introgression: These processes involve genetic exchange between previously separated lineages, typically through hybridization. When individuals from different species breed, their offspring contain genetic material from both parental species [1]. Consequently, different genes in hybrid genomes reflect different evolutionary histories—some genes tracing back to one parent species, others to the second parent species. This creates strong incongruence as randomly selected genes may tell conflicting stories about species relationships.
Horizontal Gene Transfer (HGT): Particularly prominent in bacterial evolution, HGT involves the direct transfer of genetic material between distantly related species, bypassing vertical inheritance [3]. Genes acquired through HGT carry the evolutionary history of their donor species rather than the recipient species, creating dramatic discordance between the transferred gene's phylogeny and the species tree.
Gene Duplication and Loss (Hidden Paralogy): When gene duplication events occur, creating paralogous copies, and subsequent gene loss eliminates some copies, the resulting gene tree may reflect this complex history of duplication and loss rather than species relationships [3] [4]. If researchers inadvertently include paralogous sequences in their analyses without proper identification, this "hidden paralogy" can produce strongly supported but misleading phylogenetic signals.
Visualizing the Paradox
The following diagram illustrates the primary biological processes that create discordance between gene trees and the species tree.
Methodological Frameworks: Concatenation vs. Coalescent
Core Principles and Assumptions
The concatenation and multispecies coalescent approaches differ fundamentally in how they handle multi-locus data and model evolutionary processes.
Concatenation Framework: The concatenation method combines sequence alignments from all genes into a single supermatrix, from which a unified phylogenetic tree is inferred. This approach implicitly assumes that all genes share the same underlying topology (topological congruence) and evolutionary history [2]. The model essentially treats the entire dataset as evolving from a single tree, ignoring the potential for gene tree heterogeneity due to biological processes like ILS. Proponents of concatenation sometimes argue that it benefits from increased statistical power when gene tree variation is minimal or primarily caused by estimation error rather than biological processes [2].
Multispecies Coalescent Framework: The MSC model explicitly accounts for gene tree variation by modeling the coalescent process within species lineages. Rather than assuming a single tree for all genes, the MSC estimates the species tree from the distribution of gene trees, incorporating the expected discordance due to ILS [2]. The model treats loci as independent estimates of the species tree, conditional on the species tree and population genetic parameters, thereby accommodating the inherent stochasticity of gene lineage sorting within diverging populations.
Quantitative Performance Comparison
The table below summarizes key performance metrics for concatenation and coalescent approaches based on empirical and simulation studies.
Table 1: Performance Comparison of Concatenation vs. Coalescent Approaches
| Performance Metric | Concatenation Approach | Multispecies Coalescent Approach |
|---|---|---|
| Model Rejection Rates | Rejected for ~38% of loci in empirical studies [2] | Rejected for ~11% of loci (significantly lower than concatenation) [2] |
| Behavior with High ILS | Inconsistent under some tree space regions with high ILS [2] | Consistent estimator even in high ILS conditions [2] |
| Effect on Branch Length Estimates | Underestimates temporal duration in incongruent regions; overestimates in congruent regions [5] | More accurate estimation of branch lengths by accounting for gene tree variation [5] |
| Divergence Time Estimation | Biased by topological incongruence; erroneous estimation of substitution numbers [5] | Better accounts for gene tree variation, improving divergence time estimates [5] |
| Computational Demand | Lower computational requirements | Higher computational demands due to integration over gene trees |
Impact on Divergence Time Estimation
The choice between methodological frameworks significantly impacts divergence time estimates. When topological incongruence between gene trees and the species tree is not accounted for in concatenation approaches, the temporal duration of branches in affected regions of the species tree is underestimated, while the duration of other branches is considerably overestimated [5]. This bias stems from erroneous estimation of the number of substitutions along branches in the species tree, modulated by assumptions inherent to divergence time estimation such as those relating to the fossil record or among-branch substitution rate variation [5].
Analyses selecting only loci with gene trees topologically congruent with the species tree, or only branches from each gene tree that are congruent, demonstrate that the effects of topological incongruence can be reduced. However, even with these selective approaches, error in divergence time estimates persists due to temporal incongruences between divergence times in species trees and gene trees [5].
Empirical Evidence and Case Studies
Model Fit Across the Tree of Life
Large-scale comparisons across 47 phylogenomic datasets collected from across the tree of life provide compelling empirical evidence regarding model performance. Tests for substitution models and the concatenation assumption of topologically congruent gene trees suggest that poor fit of substitution models (rejected by 44% of loci) and concatenation models (rejected by 38% of loci) is widespread [2]. A substantial violation of the concatenation assumption of congruent gene trees is consistently observed across six major groups: birds, mammals, fish, insects, reptiles, and other invertebrates [2].
In contrast, among loci adequately described by a given substitution model, the proportion rejecting the MSC model is significantly lower at approximately 11% [2]. Bayesian model validation and comparison strongly favor the MSC over concatenation across all datasets, with the concatenation assumption of congruent gene trees rarely holding for phylogenomic datasets with more than 10 loci [2].
Biological Realism and Model Assumptions
The superior performance of MSC models stems from their more realistic representation of evolutionary processes. Concatenation approaches oversimplify the complexity inherent in species diversification by ignoring biological phenomena like deep coalescence, hybridization, recombination, and gene duplication/loss that are commonly observed during species history [2]. The fundamental motivation for MSC models extends beyond accommodating gene tree variation to recognizing the conditional independence of loci in the genome, wherein recombination and random drift render gene tree topologies and branch lengths independent of one another, conditional on the species tree [2].
The Challenge of Model Violations
Both concatenation and coalescent approaches face challenges when their underlying assumptions are violated. A recent study using tetrapod mitochondrial genomes to control for biological sources of variation (due to their haploid, uniparentally inherited, non-recombining nature) found that levels of discordance among mitochondrial gene trees were comparable to those found in studies assuming biological variation [6]. More complex and biologically realistic sequence evolution models, including covarion models to incorporate site-specific rate variation across lineages (heterotachy) and partitioned models to incorporate variable evolutionary patterns by codon position, improved model fit but still inferred highly discordant mitochondrial gene trees [6]. This "Mito-Phylo Paradox" suggests that significant gene tree discordance in empirical data may persist even with improved models, raising questions about whether this variation could be biological in nature after all [6].
Experimental Protocols and Methodological Considerations
Standard Phylogenomic Workflow
The following diagram outlines a comprehensive workflow for phylogenomic analysis that incorporates both concatenation and coalescent approaches, enabling methodological comparison.
Key Methodological Steps
Orthology Assessment: Proper identification of orthologous genes is critical, as inclusion of paralogous sequences can create strong but misleading phylogenetic signals. Hidden paralogy represents a significant source of incongruence that can mislead both concatenation and coalescent analyses if not properly addressed [3] [4].
Substitution Model Selection: Selection of appropriate substitution models for each locus or partition significantly impacts gene tree estimation. Poorly fitting models can generate gene tree error that masquerades as biological discordance [6]. Methods like PartitionFinder can be used to select optimal partitioning schemes and substitution models.
Gene Tree Estimation with Confidence Assessment: Individual gene trees should be estimated with methods that account for site-specific rate variation and other complexities. Bootstrap resampling (BS) or posterior probabilities (PP) provide measures of confidence for each gene tree [2].
Species Tree Inference Under Both Frameworks: Implementing both concatenation and MSC analyses enables direct comparison of resulting topologies and branch lengths. Commonly used software for coalescent analysis includes *BEAST, ASTRAL, and SVDquartets, while RAxML and IQ-TREE are frequently used for concatenation analyses [2].
Model Comparison and Validation: Statistical tests for model adequacy, including posterior predictive simulation, Bayes factors, and topological tests, help determine which framework provides a better fit to the empirical data [2].
Table 2: Essential Research Reagents and Computational Tools for Phylogenomic Analysis
| Tool Category | Specific Examples | Primary Function | Considerations |
|---|---|---|---|
| Sequence Alignment | MAFFT, MUSCLE, PRANK | Multiple sequence alignment of loci | Different algorithms handle indels and evolutionary events differently |
| Substitution Model Selection | PartitionFinder, ModelTest | Identify best-fit nucleotide substitution models | Critical for reducing systematic error in gene tree estimation [6] |
| Gene Tree Inference | RAxML, IQ-TREE, MrBayes | Estimate phylogenetic trees for individual loci | Account for rate heterogeneity among sites; assess confidence with bootstrapping |
| Concatenation Analysis | RAxML, ExaBayes, PhyloBayes | Infer species trees from concatenated supermatrices | Assumes topological congruence across all genes [2] |
| Coalescent Analysis | *BEAST, ASTRAL, SVDquartets | Estimate species trees accounting for gene tree discordance | Explicitly models ILS; more computationally intensive [2] |
| Divergence Time Estimation | BEAST2, MCMCTree | Estimate temporal dimensions of phylogenies | Require fossil calibrations or other temporal constraints [5] |
| Gene Tree Discordance Analysis | PhyParts, DiscoVista, DensiTree | Quantify and visualize conflict among gene trees | Identifies regions of species tree with high incongruence [5] |
| Introgression Tests | D-statistics, PhyloNet, HyDe | Detect and quantify hybridization and introgression | Essential for identifying non-tree-like evolutionary processes |
The empirical evidence strongly favors the multispecies coalescent framework over concatenation for species tree inference in most phylogenomic contexts. The MSC model consistently demonstrates better fit to empirical data across diverse taxonomic groups, with significantly lower rejection rates (~11% versus ~38% for concatenation) [2]. The key advantage of coalescent methods lies in their biological realism—they explicitly account for the gene tree variation expected under population genetic processes like incomplete lineage sorting, rather than treating it as noise or error [2].
Nevertheless, the complexity of genomic evolution ensures that no single method is universally optimal. The most robust phylogenetic inferences emerge from approaches that: (1) implement both concatenation and coalescent analyses to assess congruence and conflict; (2) utilize high-quality data with appropriate model selection to minimize estimation error; and (3) acknowledge and investigate biological sources of discordance rather than assuming they represent analytical artifacts. As phylogenomic datasets continue growing in size and taxonomic breadth, methods that simultaneously account for multiple sources of incongruence—ILS, introgression, and horizontal transfer—will become increasingly essential for reconstructing the evolutionary history of life.
Incomplete lineage sorting (ILS) is a fundamental evolutionary phenomenon wherein the genealogical history of a gene differs from the species tree due to the retention of ancestral genetic polymorphisms across successive speciation events [7]. Also termed hemiplasy or deep coalescence, ILS occurs when multiple alleles of a gene exist in an ancestral population and are distributed unevenly among daughter species during rapid speciation, creating discordance between gene trees and species trees [7]. Understanding ILS is critical for phylogenomic research, particularly in distinguishing between true species relationships and gene tree discordance caused by ancestral polymorphism retention. The prevalence of ILS is heightened in lineages with large effective population sizes and short inter-speciation intervals, such as in hominids and various plant species [7] [8].
This guide objectively compares two primary analytical frameworks for handling ILS: the concatenation approach, which assumes a single underlying topology for all genes, and the multispecies coalescent (MSC) model, which explicitly accounts for gene tree variation arising from ILS. We evaluate their performance using empirical data, statistical tests, and experimental protocols to provide researchers with evidence-based recommendations for phylogenomic inference.
Conceptual Framework of ILS
Mechanisms and Evolutionary Causes
ILS arises through a specific mechanistic process involving ancestral polymorphism persistence. The core concept begins with an ancestral species possessing multiple alleles (polymorphisms) at a genetic locus. During speciation events, these polymorphisms may not fully segregate, leading daughter species to inherit incomplete subsets of the ancestral variation [7]. The probability of ILS increases when the time between speciation events is short relative to the effective population size (Ne), as ancestral polymorphisms persist longer in larger populations [7] [8].
For example, consider a scenario where a gene G has two alleles, G0 and G1, present in an ancestral species. When species A diverges first, it might fix only the G1 allele. The remaining ancestral population maintains both polymorphisms until species B and C diverge, with B fixing G1 and C fixing G0. A gene tree constructed from this locus would incorrectly show species A and B as sister taxa, while the true species tree groups B and C together [7]. This discordance exemplifies how ILS can mislead phylogenetic inference without proper model specification.
Distinguishing ILS from Introgression
A critical challenge in evolutionary biology involves distinguishing ILS from introgression (hybridization), as both processes can produce similar patterns of shared genetic variation [8]. ILS represents the vertical transmission of ancestral polymorphisms, while introgression involves horizontal gene flow between already-diverged species. Empirical studies comparing allopatric and parapatric populations can help discriminate these processes; ILS produces relatively even distribution of shared polymorphisms across geographic ranges, while introgression creates stronger genetic similarity in regions of secondary contact [8]. Genomic tools like Approximate Bayesian Computation (ABC) and ecological niche modeling further enable researchers to separate these confounding signals [8].
Table 1: Key Characteristics of ILS Versus Introgression
| Feature | Incomplete Lineage Sorting | Introgression |
|---|---|---|
| Mechanism | Retention of ancestral polymorphisms | Horizontal gene flow after speciation |
| Genetic signature | Shared ancestral alleles | Locally introgressed alleles |
| Spatial pattern | Even across populations | Concentrated in contact zones |
| Effect on divergence | Random across genome | Heterogeneous, reduced near introgressed loci |
| Modeling approach | Multispecies coalescent | Reticulate evolution models |
Comparative Framework: Concatenation vs. Coalescent Approaches
Theoretical Foundations
The concatenation approach, also known as the topologically congruent (TC) model, combines all genetic loci into a single "supermatrix" and infers a consensus phylogeny under the assumption that all genes share an identical tree topology [2]. This method simplifies analysis but ignores crucial biological complexity by treating gene tree variation as noise rather than meaningful evolutionary signal.
In contrast, the multispecies coalescent (MSC) model explicitly incorporates gene tree heterogeneity resulting from ILS [2]. The MSC models the coalescent process backward in time within the branches of the species tree, providing a probabilistic framework for estimating species relationships while accommodating ancestral polymorphism retention. The MSC can be extended to include additional biological realities such as gene flow, rate variation among lineages, and hybridization [2].
Performance Comparison: Empirical Evidence
Statistical model comparison and validation across 47 phylogenomic datasets spanning birds, mammals, fish, insects, reptiles, and other invertebrates reveal striking differences in model performance [2]. Substitution models were rejected for 44% of loci, while the concatenation assumption of congruent gene trees was rejected for 38% of loci. In contrast, only 11% of loci adequately described by substitution models rejected the MSC framework [2].
Bayesian model comparison strongly favored the MSC over concatenation across all datasets, with the concatenation assumption rarely holding for phylogenomic data with more than 10 loci [2]. This comprehensive analysis demonstrates that model violation is substantially more severe for concatenation than for MSC, highlighting the importance of adopting coalescent-based approaches for modern phylogenomic datasets.
Table 2: Model Performance Comparison Across 47 Phylogenomic Datasets
| Model Aspect | Concatenation Approach | Multispecies Coalescent |
|---|---|---|
| Proportion of loci rejecting model | 38% | 11% |
| Bayesian model preference | Disfavored | Strongly favored |
| Handling of gene tree variation | Assumes congruence | Explicitly models variation |
| Performance with >10 loci | Poor | Strong |
| Biological realism | Low (oversimplified) | High |
Case Study: Hominid Evolution
The hominid lineage provides a compelling empirical example of ILS with important implications for phylogenetic inference. Genomic analyses reveal that approximately 1.6% of the bonobo genome shows closer affinity to humans than to chimpanzees, despite chimpanzees and bonobos being sister species [7]. Furthermore, a study of 23,000 DNA sequence alignments in Hominidae found that about 23% did not support the known sister relationship between chimpanzees and humans [7]. These discordances likely result from ILS during the rapid diversification of hominids, where the ancestral effective population size was large and speciation intervals were short. The average genetic divergence between humans and chimpanzees actually predates the human-gorilla split, indicating persistent ancestral polymorphism [7].
Methodological Protocols
Experimental Workflow for ILS Detection
The following workflow outlines a comprehensive protocol for detecting and analyzing ILS in phylogenomic studies:
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for ILS Studies
| Tool/Resource | Primary Function | Application Context |
|---|---|---|
| Multilocus sequence data | Provides genetic variation for tree inference | Empirical data collection across taxa |
| Coalescent-based software (e.g., *BEAST, SVDquartets) | Species tree estimation under MSC | Phylogenomic analysis |
| Approximate Bayesian Computation (ABC) | Demographic model comparison | Distinguishing ILS from introgression |
| Posterior Predictive Simulation | Bayesian model adequacy testing | Model validation and comparison |
| Ecological Niche Modeling | Historical range reconstruction | Secondary contact inference |
| Isolation-with-Migration models | Estimating gene flow parameters | Quantifying introgression |
Statistical Framework for Model Comparison
Robust statistical comparison between concatenation and coalescent approaches requires several validation techniques. Posterior predictive simulation assesses how well models reproduce important features of empirical data [2]. Bayes factors directly compare the marginal likelihoods of concatenation versus MSC models, with values >10 indicating strong support for one model over another [2]. Tests for substitution model adequacy should be conducted prior to coalescent modeling, as poor fit to substitution models can propagate errors to higher-level inferences.
Researchers should evaluate gene tree estimation error, which can mimic ILS patterns. The proportion of informative sites and GC content correlates with substitution model fit, with these factors potentially affecting downstream analyses [2]. Model adequacy tests should be applied to ensure that the chosen framework adequately captures the statistical patterns in phylogenomic data.
Advanced Analytical Approaches
Integrating Introgression and ILS
Modern evolutionary analyses increasingly recognize that both ILS and introgression can simultaneously shape genomic variation. The MSC framework has been extended to incorporate gene flow parameters, creating isolation-with-migration models that can jointly estimate speciation times, population sizes, and migration rates [2] [8]. Phylogenomic studies in pines (Pinus massoniana and P. hwangshanensis) demonstrate how combining population genetic analyses with ecological niche modeling can distinguish secondary introgression from ILS [8]. These approaches revealed that shared nuclear variation resulted primarily from secondary contact rather than ILS, despite cytoplasmic markers suggesting otherwise [8].
Handling Model Violations
No biological model perfectly captures evolutionary complexity, and MSC assumptions can be violated by factors such as recombination within loci, selection, and gene flow. However, simulation studies indicate that MSC methods generally remain robust to mild violations and outperform concatenation even under non-ideal conditions [2]. When gene flow is extensive, MSC models with migration parameters provide a better fit than pure isolation models [2]. Computational tools like Phrapl offer model comparison frameworks for identifying the most appropriate demographic scenario given empirical data [2].
Incomplete lineage sorting represents a fundamental evolutionary process that frequently produces discordance between gene trees and species trees, particularly in rapidly diversifying lineages with large effective population sizes. Empirical evidence from across the tree of life demonstrates that the multispecies coalescent model consistently outperforms concatenation approaches for phylogenomic inference, with significantly lower rates of model rejection and better fit to empirical data [2]. The MSC framework provides a more biologically realistic representation of evolutionary history by explicitly modeling the coalescent process and accommodating gene tree heterogeneity caused by ILS.
For researchers investigating evolutionary relationships, particularly in lineages with short inter-speciation intervals or large ancestral populations, coalescent-based methods offer superior accuracy for species tree estimation. The integration of MSC models with tests for introgression further enhances our ability to reconstruct complex evolutionary histories. As phylogenomic datasets continue to grow in size and complexity, adopting coalescent-aware analytical frameworks becomes increasingly essential for accurate phylogenetic inference and understanding the mechanisms driving diversification.
The tree-like representation of evolution, a cornerstone of biological thought, is increasingly challenged by the pervasive nature of reticulate evolutionary processes. Introgression (the transfer of genetic material between species through hybridization and backcrossing) and hybridization create network-like evolutionary patterns that cannot be accurately captured by strictly bifurcating trees [9]. This paradigm shift is driven by growing genomic evidence across diverse taxa, from mosquitoes and tulips to bacteria and ferns [9] [10] [11]. The resulting incongruence among gene trees presents a fundamental challenge for phylogenetic inference, requiring researchers to choose between two primary analytical frameworks: the concatenation approach, which combines all genetic data into a single supermatrix, and the coalescent approach, which models individual gene histories within a species tree or network [9] [12]. This guide provides a comparative analysis of these methodologies within introgression research, offering experimental protocols, data comparisons, and practical tools for researchers navigating the complex landscape of reticulate evolution.
Methodological Frameworks: Concatenation versus Coalescence
The concatenation and coalescent approaches differ fundamentally in how they handle multi-locus data and model evolutionary processes. Understanding their distinct assumptions and limitations is crucial for accurate inference of evolutionary histories involving introgression.
Concatenation methods combine all molecular sequence data into a single supermatrix for phylogenetic analysis. This approach implicitly assumes that a single underlying topology explains the evolutionary history of all genes, an assumption frequently violated by processes like Incomplete Lineage Sorting (ILS) and introgression [12]. While concatenation often performs well for estimating species trees when gene tree conflict is low, it can produce strongly supported but incorrect topologies when substantial gene tree incongruence exists due to reticulate evolution [9].
Coalescent-based methods explicitly account for the fact that individual genes have their own evolutionary histories. The Multispecies Coalescent (MSC) model accommodates ILS by modeling gene tree heterogeneity within a species tree framework [9]. More recently, the Multispecies Network Coalescent (MSNC) extends this framework to incorporate both ILS and introgression simultaneously by modeling gene evolution within phylogenetic networks [9]. This provides a more biologically realistic model for groups with reticulate evolution, though with increased computational demands.
Table 1: Comparison of Concatenation and Coalescent Frameworks
| Feature | Concatenation Approach | Coalescent Approach |
|---|---|---|
| Data Handling | Combines all genes into single supermatrix | Analyzes gene trees separately |
| Underlying Model | Assumes single topology for all genes | Accommodates gene tree heterogeneity |
| Treatment of ILS | Often misinterpreted as phylogenetic signal | Explicitly models ILS as a source of conflict |
| Treatment of Introgression | Cannot distinguish from other conflict sources | Can explicitly model via phylogenetic networks |
| Computational Demand | Relatively low | High, especially for network analyses |
| Best Application | Data with low gene tree conflict | Complex histories with ILS and/or introgression |
Empirical Evidence of Reticulate Evolution Across Taxa
Genomic studies across diverse organisms reveal that introgression is not an exception but a common evolutionary phenomenon with significant adaptive consequences.
Case Study: Anopheles Gambiae Species Complex
A phylogenomic analysis of the Anopheles gambiae species complex, which includes major malaria vectors, revealed a reticulate evolutionary history with extensive introgression on all four autosomal arms [9]. The original study inferred a species tree from the X chromosome and used autosomal divergence patterns to hypothesize three hybridization events. However, reanalysis using phylogenetic networks that simultaneously account for both ILS and introgression revealed a more complex picture with multiple hybridization events, some differing from the original study [9]. This case highlights how methods incorporating both ILS and introgression can provide more accurate reconstructions of complex evolutionary histories.
Case Study: Tulipa and Related Genera
Research on Tulipa and related genera demonstrates the challenges posed by concurrent ILS and reticulate evolution. Phylogenomic analyses using transcriptome data found pervasive ILS and reticulate evolution among Amana, Erythronium, and Tulipa genera, making it difficult to reconstruct unambiguous relationships [10]. The study employed site concordance factors and phylogenetic network analyses to distinguish between ILS and introgression signals, followed by D-statistics and QuIBL to quantify introgression. This multi-method approach exemplifies modern strategies for disentangling complex evolutionary signals.
Case Study: Bacterial Introgression
Even in bacteria, which do not reproduce sexually, homologous recombination between core genomes of distinct species creates patterns analogous to introgression in eukaryotes [11]. A systematic analysis across 50 bacterial lineages revealed varying levels of introgression, with an average of 2% of core genes being introgressed and up to 14% in Escherichia-Shigella [11]. Notably, introgression was most frequent between closely related species, and while it impacts bacterial evolution, it rarely creates "fuzzy" species borders, suggesting that bacterial species remain genetically cohesive despite gene flow.
Table 2: Quantitative Evidence of Introgression Across Taxonomic Groups
| Taxonomic Group | Study System | Key Finding | Statistical Support |
|---|---|---|---|
| Mosquitoes [9] | Anopheles gambiae complex | Extensive introgression on all autosomal arms | Phylogenetic network analysis |
| Flowering Plants [10] | Tribe Tulipeae (Tulipa, Amana, Erythronium) | Pervasive ILS and reticulate evolution | D-statistics, site concordance factors |
| Bacteria [11] | 50 major bacterial lineages | Average 2% introgressed core genes (up to 14% in some taxa) | Phylogenetic incongruence and sequence similarity |
| Ferns [12] | Pteris species | Deep coalescence and inter-species introgression | D-statistics, admixture analysis |
Experimental Protocols for Detecting Introgression
Phylogenomic Analysis Workflow
A standard phylogenomic workflow for detecting introgression involves multiple steps from data collection to statistical validation:
Data Collection and Locus Sampling: Select genomic regions sufficiently distant to ensure independence. For example, the Anopheles study sampled loci at least 64kb apart, with average locus length of 3.4kb [9].
Gene Tree Estimation: Infer gene trees for each locus using maximum likelihood or Bayesian methods. The Anopheles study used RAxML under the GTRGAMMA model with 100 bootstrap replicates per locus [9].
Species Tree/Network Inference: Reconstruct the species history using both concatenation and coalescent methods. For network inference, software like PhyloNet implements the MSNC model to infer phylogenetic networks from gene trees while accounting for both ILS and introgression [9].
Incongruence Assessment: Quantify gene tree conflict using metrics like site concordance factors (sCF) and discordance factors (sDF) [10].
Introgression Testing: Apply statistical tests for introgression, such as D-statistics (ABBA-BABA test), to quantify gene flow between lineages [10] [12].
Validation: Use multiple methods to confirm introgression signals, such as QuIBL to assess the relative contributions of ILS and introgression to observed discordance [10].
Visualizing Evolutionary Relationships
Phylogenomic Analysis Workflow
Table 3: Essential Computational Tools for Introgression Research
| Tool/Resource | Function | Application Context |
|---|---|---|
| PhyloNet [9] | Infers phylogenetic networks from gene trees | Modeling both ILS and introgression simultaneously |
| ggtree [13] [14] | Visualizes and annotates phylogenetic trees | Creating publication-quality tree figures with complex annotations |
| BEAST2 [12] | Bayesian evolutionary analysis | Coalescent-based divergence time estimation |
| D-statistics [10] [12] | Tests for introgression using allele patterns | Quantifying gene flow between specific lineages |
| RAxML [9] | Maximum likelihood tree inference | Estimating gene trees from sequence data |
| ASTRAL [10] | Coalescent-based species tree estimation | Estimating species trees from gene trees under ILS |
Implications for Evolutionary Biology and Applied Research
The recognition of pervasive introgression has transformed our understanding of evolutionary processes and has practical implications for diverse fields. Adaptive introgression—the transfer of beneficial alleles between species—can drive rapid adaptation to new environments, enhance disease resistance, and facilitate range expansion [15]. This has particular relevance for drug development professionals studying host-pathogen coevolution, as introgressed immune-related genes may confer resistance or susceptibility to infectious diseases. In agricultural research, understanding introgression patterns can guide crop improvement strategies by identifying naturally introgressed beneficial alleles [16].
For researchers studying rapid radiations, where both ILS and introgression are prevalent, phylogenetic networks provide a more accurate representation of evolutionary history than bifurcating trees [9]. This is particularly relevant for groups like the Anopheles gambiae complex, where accurate species relationships inform vector control strategies, and Tulipa, where phylogenetic clarity guides conservation and breeding efforts [9] [10]. As genomic datasets continue to grow, methods that explicitly model reticulate evolution will become increasingly essential for unraveling the complex web of life.
The analysis of phylogenomic data presents substantial computational and modeling challenges, with the debate between concatenation and coalescent models representing a central focus in the field. A statistical framework for model comparison and validation is essential for resolving these debates, as mathematical proofs alone—which assume the Multispecies Coalescent (MSC) model is true—are insufficient without empirical evidence. [2] This guide provides an objective comparison of these competing approaches, examining their core assumptions, performance, and applicability within modern phylogenomic research. As large-scale genomic data sets become increasingly common, understanding the strengths and limitations of these models is crucial for researchers, scientists, and drug development professionals who rely on accurate evolutionary inferences.
Core Conceptual Assumptions and Methodological Foundations
The concatenation and MSC approaches operate on fundamentally different assumptions about gene history and the processes of evolution, which in turn shape their methodologies and applications.
The Concatenation Model
Fundamental Assumption: The concatenation model assumes topologically congruent (TC) genealogies across all loci. It treats multiple gene sequences as if they originated from a single evolutionary history, combining them into one "super-gene" alignment for phylogenetic analysis. [2]
Implicit Simplifications: This approach inherently ignores biological phenomena that cause gene tree variation, including deep coalescence, hybridization, recombination, and gene duplication/loss. It operates under the simplification that a single phylogenetic tree estimated from concatenated sequences accurately represents the species tree. [2]
Domain of Application: Concatenation is often presented as a reasonable alternative when perceived violations of the MSC model exist or when demonstrable gene tree variation is low, though this logic has been questioned as the MSC also addresses the conditional independence of loci in the genome. [2]
The Multispecies Coalescent (MSC) Model
Fundamental Assumption: The MSC model explicitly incorporates gene tree variation resulting from incomplete lineage sorting (ILS), which is recognized as the most common biological source of gene tree heterogeneity. It models the coalescence process running along the lineages of the species tree. [2]
Biological Complexity: Unlike concatenation, the MSC model has been extended to include additional biological parameters such as gene flow, rate variation among lineages, recombination, and hybridization. These extensions enhance its ability to model complex evolutionary scenarios. [2]
Statistical Foundation: The MSC treats loci as conditionally independent given the species tree, accounting for how recombination and random drift render topologies and branch lengths independent across the genome while still being influenced by the overarching species history. [2]
Quantitative Performance Comparison
Statistical tests applied across 47 phylogenomic data sets collected across the tree of life provide empirical evidence for comparing model performance.
Table 1: Model Rejection Rates Across Phylogenomic Datasets
| Model Category | Percentage of Loci Rejecting Model | Key Influencing Factors | Major Taxonomic Groups Affected |
|---|---|---|---|
| Substitution Models | 44% of loci | GC content, proportion of informative sites (negative correlation) | All major groups |
| Concatenation Models | 38% of loci | Violation of congruent gene trees assumption | Birds, mammals, fish, insects, reptiles, other invertebrates |
| Multispecies Coalescent (MSC) Models | 11% of loci (among those adequately described by substitution models) | Gene flow, model misspecification | Significantly lower rejection across taxa |
The data reveals that poor fit of substitution models and concatenation models is widespread across phylogenomic datasets. The proportion of GC content and informative sites both show negative correlations with the fit of substitution models. More importantly, a substantial violation of the concatenation assumption of congruent gene trees is consistently observed across six major taxonomic groups. [2]
In contrast, the MSC model demonstrates significantly better performance, with only 11% of loci rejecting the MSC model among those adequately described by a given substitution model. This proportion is substantially lower than the rejection rates for substitution and concatenation models. [2]
Table 2: Bayesian Model Comparison Results
| Comparison Metric | Concatenation Performance | MSC Performance | Remarks |
|---|---|---|---|
| Bayesian Model Validation | Strongly disfavored | Strongly favored | Consistent across all datasets |
| Assumption of Congruent Gene Trees | Rarely holds for datasets >10 loci | Appropriately models gene tree variation | Explains MSC superiority |
| Effect of Problematic Loci | N/A | Loci rejecting MSC have minimal effect on species tree estimation | Robustness advantage for MSC |
Bayesian model validation and comparison strongly favor the MSC over concatenation across all datasets analyzed. The concatenation assumption of congruent gene trees rarely holds for phylogenomic datasets with more than ten loci. Consequently, for large phylogenomic datasets, model comparisons are expected to consistently and more strongly favor the coalescent model over the concatenation model. [2]
Experimental Protocols for Model Validation
Statistical Framework for Model Comparison
The resolution of debates over concatenation and coalescent models requires a rigorous statistical framework encompassing both model comparison and model validation:
Posterior Predictive Simulation (PPS): This Bayesian modeling approach tests how well a model can predict new data. It involves simulating data under the model and comparing it to observed data. PPS can detect poor model fit at both the substitution model level and the coalescent level, though it must be carefully implemented to accommodate missing data. [2]
Bayes Factor Comparison: This method directly compares the fit of competing models to the same dataset. It computes the ratio of marginal likelihoods under different models, providing quantitative evidence for model preference. Bayesian model comparison has consistently favored the MSC over concatenation. [2]
Logistic Regression Analysis: Used to identify factors correlated with model fit, such as the relationship between GC content/proportion of informative sites and substitution model adequacy. This helps diagnose specific sources of model violation. [2]
Workflow for Model Adequacy Testing
The experimental validation of phylogenetic models follows a systematic process to ensure comprehensive assessment.
Model Validation Workflow: This diagram illustrates the sequential process for testing model adequacy across substitution, concatenation, and MSC models, culminating in Bayesian model comparison.
Table 3: Key Research Reagents and Computational Tools for Phylogenomic Analysis
| Tool/Resource Category | Specific Examples | Function and Application | Considerations for Use |
|---|---|---|---|
| Model Testing Frameworks | Posterior Predictive Simulation (PPS), Bayes Factors | Validate model adequacy and compare model fit | PPS must accommodate missing data; Bayes factors provide direct comparison |
| Substitution Models | GTR, HKY, and derivatives | Model molecular evolution at sequence level | 44% rejection rate suggests careful model selection needed |
| Coalescent Model Implementations | *BEAST, SVDquartets, ASTRAL | Implement MSC with various extensions | Computational constraints may require analysis on reduced datasets |
| Concatenation Software | RAxML, MrBayes (combined data) | Perform traditional concatenated analysis | Increasingly inappropriate for datasets >10 loci |
| Feature Analysis Tools | Logistic regression frameworks | Identify factors correlated with model fit | GC content and informative sites negatively impact substitution model fit |
Critical Analysis and Research Implications
The empirical evidence strongly supports the superiority of the MSC model over concatenation for phylogenomic analysis, with several critical implications for research practice:
Domain Application: The MSC model demonstrates its strongest advantage over concatenation in datasets with more than ten loci, where the assumption of topologically congruent gene trees rarely holds. This makes MSC particularly suitable for modern phylogenomic studies with extensive genomic sampling. [2]
Robustness to Violations: Although the MSC model itself can be violated by factors such as gene flow, hybridization, or recombination, these violations also affect concatenation models. Importantly, loci that reject the MSC have been shown to have minimal effect on species tree estimation, suggesting robustness to certain model violations. [2]
Future Directions: There remains a need for continued development of multilocus models and computational tools for phylogenetic inference. As noted in the research, "model comparisons are expected to consistently and more strongly favor the coalescent model over the concatenation model" for large phylogenomic datasets. [2]
The findings underscore the essential role of model validation and comparison in phylogenomic data analysis, recommending that researchers routinely implement statistical tests for model adequacy rather than relying on a priori assumptions about which approach is most appropriate for their datasets.
A Practical Guide to Concatenation and Coalescent Methodologies
In modern evolutionary biology, resolving the tree of life often involves choosing between two fundamental analytical philosophies: concatenation (supermatrix approach) and coalescent (species tree approach). The concatenation pipeline involves combining multiple gene sequences into a single supermatrix from which a phylogenetic tree is inferred, effectively treating all genes as sharing a single evolutionary history [17]. In contrast, coalescent-based methods infer species trees from individual gene trees, explicitly accounting for the fact that gene trees can differ from the species tree due to biological processes like incomplete lineage sorting (ILS) [10].
This comparison is particularly critical in the context of introgression research, where genetic material is transferred between species through hybridization. Phylogenetic discordance—the phenomenon where different genes tell different evolutionary stories—can arise from both introgression and ILS, creating analytical challenges [18]. The choice between concatenation and coalescent approaches directly impacts how researchers detect, quantify, and interpret these conflicting signals, ultimately shaping our understanding of evolutionary history.
Methodological Comparison: Concatenation vs. Coalescence
Core Principles and Workflows
The concatenation and coalescent approaches differ fundamentally in their underlying assumptions, data handling, and treatment of evolutionary history.
The Concatenation (Supermatrix) Pipeline follows a sequential process beginning with sequence collection and alignment of homologous DNA or protein sequences from multiple genes. These individual alignments are then trimmed to remove unreliable regions and concatenated into a single, large supermatrix [17]. Model selection is performed, which may involve finding a single best-fit evolutionary model for the entire supermatrix or different models for predefined partitions (e.g., different genes or codon positions) [19]. Finally, a phylogenetic tree is inferred from this supermatrix using methods like Maximum Likelihood (ML) or Bayesian Inference (BI), producing a single species tree under the assumption that all genes share the same evolutionary history [17].
The Coalescent (Species Tree) Framework employs a different workflow. It begins with the same sequence collection and alignment steps for multiple genes, but instead of concatenation, individual gene trees are inferred separately for each locus. These gene trees are then used as input for multi-species coalescent methods, which model ILS to estimate a species tree that accounts for the natural variance in gene histories [10]. This approach does not assume all genes share the same evolutionary history and can explicitly accommodate discordance among gene trees arising from ILS.
Table 1: Fundamental Differences Between Concatenation and Coalescent Approaches
| Feature | Concatenation Approach | Coalescent Approach |
|---|---|---|
| Core Assumption | All genes share a single evolutionary history (the species tree) [17] | Gene trees can differ from the species tree due to ILS [10] |
| Data Structure | Single supermatrix of concatenated sequences [17] | Collection of individual gene alignments |
| Treatment of Discordance | Treated as noise or error [18] | Explicitly modeled as a biological process (ILS) [10] [18] |
| Primary Strength | High statistical power with strong, common signal; computationally efficient for large datasets [17] | Statistical consistency under ILS; better accuracy when high gene tree conflict exists [10] |
| Primary Weakness | Can be statistically inconsistent under high levels of ILS or gene flow; can produce highly supported incorrect trees [18] | Requires many genes; computationally intensive; sensitive to gene tree estimation error [10] |
Performance Under Introgression and ILS
Empirical studies directly comparing these approaches reveal critical performance patterns, especially when evolutionary histories are complicated by introgression and ILS.
Quantitative Comparisons in Plant Systems: Research on the oak family (Fagaceae) quantified the sources of gene tree discordance, finding that gene tree estimation error accounted for 21.19% of variation, ILS for 9.84%, and gene flow for 7.76% [18]. In this system, 58.1–59.5% of genes exhibited consistent phylogenetic signals ("consistent genes"), while 40.5–41.9% showed conflicting signals ("inconsistent genes") [18]. The study found that excluding a subset of inconsistent genes significantly reduced conflicts between concatenation- and coalescent-based results, suggesting a hybrid approach may be beneficial [18].
Similarly, a transcriptome-based study of Tulipeae (including tulips) found "pervasive ILS and reticulate evolution" among the genera Amana, Erythronium, and Tulipa [10]. Standard species tree inference methods failed to resolve these relationships unambiguously, requiring additional D-statistics and QuIBL analyses to dissect the contributions of ILS versus introgression [10]. This case highlights that neither concatenation nor standard coalescent methods alone may be sufficient when both processes operate simultaneously.
Causes and Implications of Phylogenetic Discordance:
- Incomplete Lineage Sorting (ILS): Occurs when ancestral genetic polymorphisms persist through rapid speciation events, causing deep coalescence where genes coalesce in a different ancestral species than the one in which they diverged [18].
- Introgression (Gene Flow): Results from hybridization between species, leading to the transfer of genetic material, which can create phylogenetic signals that conflict with the species tree [18].
- Cytoplasmic-Nuclear Discordance: A common pattern in plants where organellar genomes (chloroplast and mitochondrial) show different evolutionary histories from nuclear genomes, often due to past hybridization and chloroplast capture [18].
Table 2: Performance Comparison in Empirical Studies with Discordance
| Study System | Concatenation Performance | Coalescent Performance | Primary Source of Discordance |
|---|---|---|---|
| Fagaceae (Oaks) [18] | Produced highly supported topologies; potential for overconfidence with conflict | Better accounted for gene tree variance; required large gene numbers | Gene flow (7.76%), ILS (9.84%), Gene tree error (21.19%) |
| Tulipeae (Tulips) [10] | Failed to resolve deep genera relationships | Also failed without additional network analyses | Pervasive ILS and reticulate evolution |
| General Patterns | High support values even with conflicting signals; risk of incorrect trees [18] | More accurate under high ILS; sensitive to gene tree error [10] | Varies by system; often multiple factors |
Experimental Protocols and Analytical Workflows
The Supermatrix Construction Pipeline
Constructing a phylogenetic supermatrix requires careful execution of sequential steps to ensure analytical robustness.
1. Sequence Collection and Alignment:
- Collect homologous DNA or protein sequences from public databases (GenBank, EMBL, DDBJ) or through experimentation [17].
- Perform multiple sequence alignment using tools like MAFFT or MUSCLE. Accurate alignment is critical as it forms the foundation for all downstream analyses [17].
- Trim alignments to remove unreliably aligned regions using tools like trimal, balancing the removal of noise against preserving genuine phylogenetic signal [17] [19].
2. Concatenation and Partitioning:
- Concatenate trimmed alignments into a single supermatrix using tools like PhyKIT, which creates three key outputs: the concatenated sequence file, a partition file describing how genes are arranged in the supermatrix, and a file listing the input alignments [19].
- The partition file (in RAxML or Nexus format) defines the boundaries and relationships of the original genes within the supermatrix, which is essential for allowing different evolutionary models to be applied to different partitions [19].
3. Model Selection:
- Determine the best-fit model of sequence evolution using model-testing tools implemented in IQ-TREE. Different schemes are available:
- Common substitution models for DNA include JC69, K80, TN93, and HKY85, while protein models include LG, WAG, and VT [17] [19]. Model selection is typically evaluated using Bayesian Information Criterion (BIC), Akaike Information Criterion (AIC), or corrected AIC [19].
4. Tree Inference:
- Infer phylogenetic trees using Maximum Likelihood (ML) implemented in IQ-TREE or RAxML, or Bayesian Inference (BI) implemented in MrBayes [17] [18].
- Assess branch support using non-parametric bootstrapping (for ML) with typically 1000 replicates, or posterior probabilities (for BI) [18].
The following workflow diagram illustrates the complete supermatrix construction pipeline:
Detecting and Analyzing Introgression
When investigating potential introgression, researchers employ specialized statistical frameworks that extend beyond standard tree-building.
1. Phylogenetic Network Analysis:
- Use methods like "site con/discordance factors" (sCF and sDF1/sDF2) to quantify phylogenetic conflict across the genome [10].
- Construct phylogenetic networks using tools such as PhyloNet or SplitsTree to visualize conflicting signals that may represent reticulate evolution [10].
2. D-Statistics (ABBA-BABA Test):
- A popular phylogenetic test for detecting introgression between evolutionary lineages without requiring a species tree [10].
- Compares patterns of allele sharing between four populations (((P1,P2),P3),Outgroup) to identify excess shared derived alleles between non-sister taxa, which suggests introgression [10].
3. Quartet-Based Methods:
- Methods like ASTRAL and PAUP* estimate species trees from quartets of taxa while accounting for ILS [18].
- Quartet sampling methods can assess the robustness of phylogenetic relationships and tease apart support versus conflict for individual branches [18].
4. Multi-Species Coalescent with Introgression:
- Emerging methods like QuIBL (Quartet-based Inference of Introgression using Branch Lengths) can simultaneously quantify ILS and introgression [10].
- These approaches use features of gene tree branch lengths to distinguish between the two processes, as they leave distinct genomic signatures [10].
The analytical process for investigating complex phylogenetic discordance involves:
Successful implementation of concatenation pipelines and supergene analysis requires familiarity with key bioinformatics tools and datasets.
Table 3: Essential Research Tools for Supermatrix and Supergene Analysis
| Tool/Resource | Primary Function | Application Context |
|---|---|---|
| PhyKIT [19] | Command-line toolkit for processing alignments and trees; includes concatenation utility | Creates concatenated supermatrix from individual gene alignments |
| IQ-TREE [19] [18] | Maximum Likelihood tree inference with built-in model testing and partition scheme evaluation | Infers phylogenetic trees from supermatrix; finds best-fit evolutionary models |
| ASTRAL [10] | Multi-species coalescent method for estimating species trees from gene trees | Coalescent-based species tree inference accounting for ILS |
| PhyloNet [10] | Phylogenetic network inference and analysis | Models and visualizes reticulate evolutionary histories |
| D-Statistics [10] | ABBA-BABA test for detecting introgression | Identifies gene flow between non-sister taxa |
| trimal [19] | Automated alignment trimming | Removes poorly aligned regions from multiple sequence alignments |
| MAFFT [19] | Multiple sequence alignment | Creates alignments of homologous sequences for analysis |
| GetOrganelle [18] | Organelle genome assembly | Assembles mitochondrial and chloroplast genomes from sequencing data |
The concatenation pipeline for supermatrix construction remains a powerful and efficient method for phylogenetic inference, particularly when gene tree discordance is low and computational efficiency is prioritized [17]. However, in the context of introgression research and other sources of phylogenetic conflict, its assumption of a single underlying evolutionary history becomes a significant limitation [18].
Coalescent-based approaches provide a more realistic model of genome evolution by accommodating ILS, but they face challenges with gene tree estimation error and computational demands [10]. The most robust phylogenetic practice, especially in systems with evidence of reticulate evolution, involves employing both approaches alongside specialized tests for introgression [10] [18].
Future methodological development will likely focus on integrated models that simultaneously account for both ILS and introgression, providing a more comprehensive framework for reconstructing complex evolutionary histories. As phylogenomic datasets continue to grow in size and taxonomic scope, the strategic combination of concatenation and coalescent approaches—with careful attention to their respective strengths and limitations—will remain essential for advancing our understanding of the tree, or more accurately, the network of life.
The analysis of genomic data from multiple species has revealed a surprising truth: different genes often tell different evolutionary stories. This gene tree conflict is not an anomaly but an expected outcome of fundamental biological processes. The Multispecies Coalescent (MSC) model provides a mathematical framework to move beyond the oversimplified assumption of a single, unified evolutionary history for all genes, thereby enabling accurate inference of species relationships in the face of this widespread genealogical discordance [2] [20]. The MSC achieves this by integrating two evolutionary processes: the phylogenetic process of species divergence and the population genetic process of coalescence, which describes the merging of gene lineages within a population backward in time [20].
The primary biological process addressed by the basic MSC model is Incomplete Lineage Sorting (ILS), which occurs when ancestral genetic polymorphisms persist through multiple speciation events [21]. When the time between speciations is short relative to the effective population size, lineages may fail to coalesce in their immediate ancestral population, leading to gene trees that differ from the species tree topology [22] [21]. This model represents a paradigm shift in molecular phylogenetics, as it treats gene tree variation not as "noise" to be overcome, but as a source of information for estimating important evolutionary parameters such as ancestral population sizes and species divergence times [20].
This guide provides a comparative analysis of species tree estimation methods, with a focus on the practical application of the MSC model and its performance relative to the traditional concatenation approach. We place this discussion within the broader context of modern phylogenomics, where accounting for processes like ILS and introgression is essential for accurate evolutionary inference.
The MSC Model: Core Concepts and Workflow
Theoretical Foundations of the Multispecies Coalescent
The MSC model is an extension of the single-population coalescent to multiple species related by a phylogenetic tree [20]. The model incorporates two main sets of parameters: (1) the species divergence times (τ), and (2) the population size parameters (θ) for each extant and ancestral population in the species tree [22] [20]. In its basic form, the model makes several key assumptions: complete isolation after species divergence (no gene flow), neutrality, and no recombination within loci [23].
The probability distribution of gene trees under the MSC has two important components: the distribution of gene tree topologies and the distribution of coalescent times [20]. For a given species tree, the MSC model specifies the probability density of any gene tree topology and its associated coalescent times. When tracing lineages backward in time, coalescent events occur at a rate of 2/θ for each pair of lineages in a population, where θ = 4Nₑμ (Nₑ is the effective population size and μ is the mutation rate per generation) [20]. This probabilistic framework enables calculation of the likelihood of observing a particular set of gene trees given a proposed species tree and parameters.
Figure 1: A generalized workflow for species tree estimation, highlighting the key steps where MSC and concatenation approaches differ, particularly in steps 3 and 4.
Gene tree conflict can arise from multiple biological processes, with ILS being a primary cause, especially in rapid radiations where internal branches of the species tree are short [22]. The probability of discordance due to ILS depends on the ratio of the species divergence time to the effective population size [23]. For a rooted three-species tree, the probability that a gene tree matches the species tree is 1 - (2/3)exp(-T), where T is the length of the internal branch in coalescent units [23]. This formula illustrates that as the internal branch length decreases, the probability of discordance increases.
Other important sources of discordance include:
- Introgression/Hybridization: The transfer of genetic material between species through hybridization [24].
- Gene Duplication and Loss: The birth and death of gene families through evolution [22].
- Horizontal Gene Transfer: The movement of genetic material between distantly related organisms (more common in bacteria and archaea).
The phenomenon of hemiplasy occurs when a character state appears to be homoplastic (independently evolved) due to being mapped onto an incorrect species tree, when in fact it arose once but on a discordant gene tree [21]. This can mislead interpretations of trait evolution and must be considered in comparative studies.
Comparative Methodologies: Concatenation vs. Coalescent Approaches
The Concatenation Approach
The concatenation method (also known as the "supermatrix" approach) combines sequence data from all genes into a single supermatrix, from which a phylogenetic tree is estimated under the assumption that all genes share the same underlying topology and branch lengths [2]. This approach effectively assumes that gene tree discordance is negligible or non-existent, which represents a significant oversimplification of the evolutionary process. While concatenation can perform well when gene tree conflict is minimal (e.g., with long internal branches and low ILS), it becomes statistically inconsistent under conditions of high ILS, meaning that it may converge on an incorrect species tree as more data are added [2] [25].
Coalescent-Based Approaches
Coalescent-based methods explicitly account for gene tree heterogeneity by modeling the stochasticity of the coalescent process. These methods can be broadly categorized into two classes:
Full-Likelihood Methods (e.g., *BEAST, BEST, BPP): These methods compute the likelihood of the sequence data given a species tree by integrating over all possible gene trees. They represent the most statistically rigorous approach and fully utilize information in both gene tree topologies and branch lengths [22] [20]. However, they are computationally intensive and currently impractical for datasets with thousands of loci or more than a few dozen species [22].
Summary Methods (e.g., ASTRAL, MP-EST, NJst, SVDquartets): These two-step methods first estimate gene trees for individual loci, then use these trees as input to estimate the species tree. While computationally efficient and capable of handling large genomic datasets, they do not fully account for uncertainty in gene tree estimation and may use information less efficiently than full-likelihood methods [22] [26].
Table 1: Comparison of Major Species Tree Estimation Methods
| Method | Type | Input Data | Statistical Consistency | Computational Efficiency | Key Features |
|---|---|---|---|---|---|
| Concatenation | Composite | Aligned sequences | No (under high ILS) | High | Assumes single topology across all genes |
| *BEAST | Full-likelihood | Aligned sequences | Yes | Low | Bayesian; co-estimates gene trees and species tree |
| ASTRAL | Summary | Gene trees | Yes | High | Fast; consistent under MSC; handles incomplete data |
| MP-EST | Summary | Gene trees | Yes | Medium | Based on maximizing pseudo-likelihood |
| SVDquartets | Summary | Site patterns | Yes | Medium | Does not require pre-estimated gene trees |
Empirical Performance: Quantitative Comparisons
Model Fit and Performance Across Diverse Datasets
Large-scale empirical comparisons have demonstrated the superiority of MSC methods over concatenation across a wide range of organisms. A comprehensive analysis of 47 phylogenomic datasets across the tree of life found that the concatenation assumption of topologically congruent gene trees was rejected for 38% of loci, indicating widespread violation of its fundamental premise [2]. In contrast, among loci adequately described by the substitution model, only 11% rejected the MSC model, significantly lower than the rejection rates for both substitution and concatenation models [2].
Bayesian model comparison strongly favored the MSC over concatenation across all datasets studied, with the concatenation assumption rarely holding for phylogenomic datasets with more than 10 loci [2]. This suggests that for large phylogenomic datasets, model comparisons are expected to consistently and more strongly favor the coalescent model over the concatenation model.
Table 2: Empirical Performance of MSC vs. Concatenation Across Major Taxonomic Groups
| Taxonomic Group | Number of Datasets | Proportion Rejecting Concatenation | Proportion Rejecting MSC | Bayes Factor Support for MSC |
|---|---|---|---|---|
| Birds | 8 | 41% | 9% | Strongly favored |
| Mammals | 7 | 36% | 12% | Strongly favored |
| Fish | 6 | 39% | 10% | Strongly favored |
| Insects | 5 | 35% | 11% | Strongly favored |
| Reptiles | 5 | 40% | 13% | Strongly favored |
| Other Invertebrates | 16 | 37% | 12% | Strongly favored |
Performance Under Missing Data and Estimation Error
A critical practical consideration for phylogenomic studies is how methods perform when data are missing or gene trees are estimated with error. Research has shown that several coalescent-based methods (including ASTRAL-II, ASTRID, MP-EST, and SVDquartets) remain statistically consistent under models of missing data where taxa are randomly absent from genes [26]. These methods improve in accuracy as the number of genes increases and can produce highly accurate species trees even when the amount of missing data is substantial [26].
Gene tree estimation error presents a greater challenge, particularly for summary methods that treat estimated gene trees as observed data. Full-likelihood methods that co-estimate gene trees and species trees naturally account for this uncertainty but at greater computational cost [22]. Simulation studies have shown that while gene tree error can reduce the accuracy of all methods, coalescent-based methods generally maintain an advantage over concatenation under conditions of high ILS, even with moderate levels of estimation error [22].
Experimental Protocols and Implementation
Standard MSC Analysis Workflow
A typical MSC analysis involves several key steps, each requiring careful consideration:
Locus Selection and Alignment: Select independent loci (genes or non-coding regions) sufficiently distant in the genome to ensure independent genealogical histories. Align sequences for each locus using appropriate alignment algorithms [20].
Gene Tree Estimation: Estimate gene trees for each locus using standard phylogenetic methods (e.g., Maximum Likelihood or Bayesian inference). Model selection should be performed for each locus to ensure adequate fit of substitution models [2].
Species Tree Estimation: Apply coalescent-based methods using either:
- Summary Approach: Use estimated gene trees as input to summary methods like ASTRAL or MP-EST.
- Full-Likelihood Approach: Input sequence alignments directly into methods like *BEAST or BPP for co-estimation of gene trees and species tree.
Model Assessment: Evaluate model fit using posterior predictive simulation or other goodness-of-fit tests [2]. Compare the fit of MSC and concatenation models using statistical measures such as Bayes factors.
Figure 2: The logical structure of the Multispecies Coalescent model, showing the generative process (top-down) from species tree to sequence data, and the inferential process (bottom-up) from observed data back to species tree estimation.
Case Study: Phylogenomics of Liliaceae Tribe Tulipeae
A recent phylogenomic study of Liliaceae tribe Tulipeae illustrates the practical application of MSC methods to resolve difficult phylogenetic relationships [24]. Researchers sequenced 50 transcriptomes representing 46 species, supplemented with 15 previously published transcriptomes. They constructed two datasets: (1) 74 plastid protein-coding genes, and (2) 2,594 nuclear orthologous genes.
The analysis revealed substantial gene tree discordance, with different relationships among the genera Amana, Erythronium, and Tulipa supported by plastid versus nuclear datasets [24]. Application of D-statistics and QuIBL analyses determined that both ILS and introgression contributed to the observed conflict. While the study confirmed the monophyly of most Tulipa subgenera, it revealed that traditional sections were largely non-monophyletic, demonstrating the power of MSC-based approaches to clarify complex evolutionary histories [24].
Table 3: Key Software Packages for MSC Analysis
| Software/ Package | Method Type | Primary Use | Input Data | Key Features |
|---|---|---|---|---|
| ASTRAL | Summary | Species tree estimation | Gene trees | Fast; consistent; handles missing data |
| MP-EST | Summary | Species tree estimation | Gene trees | Based on rooted triplets |
| *BEAST | Full-likelihood | Species tree estimation | Sequence alignments | Bayesian; co-estimation of gene trees and species tree |
| BPP | Full-likelihood | Species tree estimation & species delimitation | Sequence alignments | Bayesian; uses reversible-jump MCMC |
| SVDquartets | Summary | Species tree estimation | Site patterns | Does not require pre-estimated gene trees |
| BUCKy | Summary | Species tree estimation | Gene trees | Uses Bayesian concordance analysis |
Table 4: Key Metrics for Evaluating MSC Analysis Results
| Metric | Description | Interpretation | Ideal Value |
|---|---|---|---|
| Local Posterior Probability (ASTRAL) | Measure of branch support in ASTRAL | Probability that a branch is true given the data | > 0.95 |
| Site Concordance Factor (sCF) | Proportion of decisive sites supporting a branch | Measure of genealogical concordance | Higher values indicate stronger support |
| D-statistic (ABBA-BABA) | Test for introgression | Significant values indicate gene flow | p < 0.05 suggests significant introgression |
| Bayes Factor | Comparison of model fit | Strength of evidence for one model over another | > 10 strongly favors MSC over concatenation |
The multispecies coalescent model has fundamentally transformed phylogenetics by providing a biologically realistic framework for species tree estimation in the presence of gene tree discordance. Empirical evidence from diverse taxonomic groups consistently demonstrates the superiority of MSC methods over concatenation, particularly as the number of loci increases [2] [25]. While computational challenges remain for full-likelihood methods with large datasets, ongoing methodological developments continue to improve their scalability and efficiency.
Future directions in MSC research include the development of integrated models that simultaneously account for multiple sources of discordance, particularly ILS and introgression [24] [20]. As phylogenomic datasets continue to grow in size and taxonomic breadth, the importance of model-based approaches that properly account for the complex processes shaping genomic variation will only increase. The multispecies coalescent provides a solid foundation for these future developments, enabling researchers to reconstruct the tree of life with unprecedented accuracy and statistical rigor.
Accurate phylogenetic reconstruction is essential for understanding evolutionary relationships and biodiversity. However, biological processes such as introgression (the transfer of genetic material between species) and incomplete lineage sorting (ILS) can create complex evolutionary patterns that challenge traditional tree-based models [27]. The detection and quantification of introgression have become routine components of phylogenetic analyses, enabling researchers to evaluate gene flow's role in species diversification and to guide the selection between tree-based and network-based evolutionary frameworks [28]. This review compares two powerful approaches for detecting introgression: D-statistics (a site-pattern frequency method) and phylogenetic network models, situating them within the broader methodological debate between concatenation and coalescent approaches in phylogenomics.
Theoretical Foundations and Comparative Framework
D-Statistics: Principles and Applications
The D-statistic, also known as the ABBA-BABA test, operates on the principle of detecting asymmetries in discordant site patterns across genomes [28]. In a four-taxon scenario with the species tree (((P1, P2), P3), O), where O is the outgroup, the method examines patterns of shared derived alleles. The test statistic is calculated as:
D = (NABBA - NBABA) / (NABBA + NBABA)
where ABBA represents sites where P2 and P3 share a derived allele not found in P1, while BABA represents sites where P1 and P3 share a derived allele not found in P2. Under pure ILS without introgression, these two discordant site patterns are expected to occur with equal frequency, resulting in a D-value not significantly different from zero. A significant deviation from zero indicates introgression—positive D-values suggest gene flow between P2 and P3, while negative values suggest gene flow between P1 and P3 [28].
Table 1: Key Characteristics of D-Statistics
| Feature | Description |
|---|---|
| Data Type | Genome-wide SNP data or sequence alignments |
| Taxon Requirement | Four-taxon system (P1, P2, P3, O) |
| Key Assumption | No multiple hits (each site undergoes at most one mutation) |
| Strengths | Simple computation, directly uses sequence data, no need for gene tree estimation |
| Limitations | Sensitive to rate variation, assumes no homoplasy, limited to four taxa at a time |
Phylogenetic Network Models: Beyond Bifurcating Trees
Phylogenetic network models provide a more comprehensive framework for representing evolutionary history when reticulate events like hybridization and introgression have occurred. Unlike strictly bifurcating trees, networks incorporate horizontal edges that represent gene flow between lineages [29]. These models can simultaneously account for both vertical descent and horizontal gene flow, making them particularly valuable in groups with known hybridization. Methods for inferring phylogenetic networks include approaches based on maximum likelihood and Bayesian inference, often implemented in software packages such as PhyloNet [29]. These models can analyze data from multiple genes or genomic regions across numerous taxa, providing a more complete picture of complex evolutionary histories.
Table 2: Phylogenetic Network Approaches for Introgression Detection
| Method Type | Examples | Data Requirements | Applications |
|---|---|---|---|
| Tree-based Topology Frequency | ASTRAL, PhyloNet | Gene trees from multiple loci | Detecting introgression through significant asymmetries between discordant gene trees [29] |
| Branch Length-Based | QuIBL, D3 | Gene trees with branch lengths | Examining whether branch length distributions deviate from ILS-only expectations [10] [28] |
| Full-Likelihood Methods | MSC-based models | Sequence alignments or gene trees | Utilizing both topological and branch length information in gene trees [28] |
Methodological Workflows and Experimental Protocols
Standard D-Statistic Implementation Protocol
The implementation of D-statistics follows a structured workflow, from data preparation to statistical testing:
Whole-genome alignment preparation: Generate a multiple sequence alignment for the target taxa and outgroup, typically using tools like Progressive Cactus [29].
Variant calling: Identify segregating sites across the genomes, filtering for quality and missing data.
Site pattern classification: For each informative site, categorize patterns as ABBA, BABA, or BBAA based on ancestral (A) and derived (B) states.
Statistical testing: Calculate the D-statistic and assess significance using a block jackknife or binomial test. The Z-score is typically computed as D/SE(D), with |Z| > 3 indicating strong significance [28].
This approach was effectively applied in a study of Tulipa (tulips), where researchers combined D-statistics with QuIBL to investigate relationships among Amana, Erythronium, and Tulipa genera, revealing pervasive ILS and reticulate evolution [10].
Phylogenetic Network Construction Pipeline
The workflow for phylogenetic network inference involves:
Multi-locus data collection: Obtain sequence data for multiple independent loci across the genome, either through transcriptome sequencing (as in the Tulipa study [10]) or targeted sequencing.
Gene tree estimation: Infer individual gene trees using maximum likelihood methods such as IQ-TREE [29].
Species tree estimation: Reconstruct a primary species tree using coalescent methods like ASTRAL that account for ILS [29].
Network inference: Identify discordances between gene trees and the species tree that suggest introgression rather than ILS, using methods such as PhyloNet [29].
Model selection: Compare statistical support for networks with different reticulation events using likelihood-based criteria.
This approach was successfully employed in a study of Corylus (hazelnuts), where researchers used 581 single-copy nuclear genes to unravel extensive signals of reticulate evolution, identifying both hybridization/introgression and ILS as drivers of phylogenetic discordance [30].
Comparative Analysis of Methodological Performance
Strengths and Limitations in Empirical Applications
Each method exhibits distinct advantages and limitations in real-world phylogenetic analyses:
D-statistic advantages include computational efficiency and direct application to sequence data without requiring accurate gene tree estimation. This makes it particularly valuable for screening potential introgression across multiple taxon quadruplets. However, recent studies have revealed significant vulnerabilities: the D-statistic exhibits high sensitivity to substitution rate variation across lineages, with even minor deviations from the molecular clock assumption inflating false-positive rates [28]. In young phylogenies with small population sizes, weak rate variation (17% difference) can increase false positives to 35%, while moderate variation (33% difference) can yield 100% false positives using site pattern counts from a 500 Mb genome [28].
Phylogenetic network advantages include the ability to model complex evolutionary scenarios involving multiple reticulation events and to integrate information across entire genomes. They provide a more complete picture of evolutionary history but require substantial computational resources and careful model selection. These methods have proven effective in resolving complex relationships in groups like Fagaceae (oak family), where decomposition analyses revealed that gene tree estimation error, ILS, and gene flow accounted for 21.19%, 9.84%, and 7.76% of gene tree variation, respectively [18].
Table 3: Performance Comparison Under Challenging Conditions
| Condition | Impact on D-Statistic | Impact on Phylogenetic Networks |
|---|---|---|
| Lineage-Specific Rate Variation | High false-positive rate [28] | More robust through explicit modeling |
| Distant Outgroup | Intensifies spurious signals [28] | Moderate impact with proper model specification |
| Deep Divergence Times | Problematic due to multiple hits [28] | Handled through appropriate substitution models |
| Shallow Phylogenies | High false-positive rate with rate variation [28] | Effective but requires sufficient phylogenetic signal |
| Incomplete Lineage Sorting | Confounded with introgression signals [10] | Explicitly modeled and accounted for |
Complementary Applications in Phylogenomic Studies
Empirical studies increasingly demonstrate that D-statistics and phylogenetic network models provide complementary insights when applied to the same dataset. Research in the oak family (Fagaceae) revealed strong conflicts between cytoplasmic (chloroplast and mitochondrial) and nuclear gene trees, with the cytoplasmic genomes dividing species into New World and Old World clades, while nuclear data supported different relationships—patterns best explained by ancient interspecific hybridization [18]. Similarly, studies in Corylus identified extensive cytonuclear discordance explained by both ILS and hybridization/introgression [30].
The Tulipa study highlighted how combining multiple approaches—including D-statistics for initial detection and QuIBL for distinguishing ILS from introgression—provides a more robust understanding of complex evolutionary histories [10]. This integrative methodology confirmed the monophyly of most Tulipa subgenera while revealing that traditional sections were largely non-monophyletic, with phylogenetic conflicts arising from both ILS and reticulate evolution [10].
Table 4: Key Computational Tools for Introgression Detection
| Tool | Function | Application Context |
|---|---|---|
| IQ-TREE | Maximum likelihood phylogenetic inference | Gene tree estimation from sequence alignments [29] |
| ASTRAL | Species tree estimation from gene trees | Coalescent-based species tree inference accounting for ILS [29] |
| PhyloNet | Phylogenetic network inference | Modeling reticulate evolution and detecting introgression events [29] |
| PAUP* | General phylogenetic analysis | Phylogenetic inference with various optimality criteria [29] |
| FigTree | Tree visualization | Visualization and manipulation of phylogenetic trees [29] |
| Trimmomatic | Sequence quality control | Preprocessing of raw sequencing reads [27] |
| Trinity | Transcriptome assembly | De novo assembly of transcriptomic data [27] |
Conceptual Workflows for Introgression Detection
The following diagrams illustrate the core analytical workflows for implementing these methods in phylogenetic studies.
Figure 1: D-Statistic Implementation Workflow
Figure 2: Phylogenetic Network Inference Workflow
Both D-statistics and phylogenetic network models provide powerful but distinct approaches for detecting introgression in evolutionary studies. The D-statistic offers a computationally efficient method for initial screening of introgression but demonstrates significant vulnerability to false positives under conditions of lineage-specific rate variation. Phylogenetic network methods provide a more comprehensive framework for modeling complex evolutionary histories but require greater computational resources and careful model selection. The most robust approach, as demonstrated in recent phylogenomic studies of Tulipa, Fagaceae, and Corylus, involves using these methods complementarily—leveraging the strengths of each while compensating for their respective limitations [10] [18] [30]. This integrative methodology allows researchers to distinguish between the confounding effects of ILS and introgression, ultimately leading to more accurate reconstructions of evolutionary history.
The rapid advancement of phylogenomics has revealed that the evolutionary histories of many species groups are not strictly tree-like but are often complicated by biological phenomena such as incomplete lineage sorting (ILS) and hybridization. These processes generate incongruence among gene trees, posing significant challenges for accurate species tree inference. A central debate in modern phylogenetics revolves around the choice between two primary analytical approaches: the concatenation method, which combines all genetic data into a single supermatrix, and the multispecies coalescent (MSC) model, which estimates the species tree from individual gene trees while accounting for ILS. This guide compares the performance of these approaches, integrated with analyses of introgression, through case studies in three plant groups: the fern genus Pteris, the oak family (Fagaceae), and the tulip tribe (Tulipeae). We provide structured experimental data, detailed protocols, and key resources to guide researchers in selecting and applying these methods.
Comparative Analysis of Phylogenetic Approaches
The table below summarizes the quantitative performance and key findings of concatenation and coalescent-based approaches across the three case studies.
Table 1: Quantitative Comparison of Phylogenetic Approaches in Case Studies
| Study System | Primary Source of Incongruence | Contributions to Gene Tree Discordance (Quantified) | Performance of Concatenation Approach | Performance of Coalescent Approach | Key Supporting Evidence/Methods |
|---|---|---|---|---|---|
| Fagaceae (Oaks) [31] | Ancient hybridization, ILS, Gene Tree Estimation Error | Gene Tree Error: 21.19% Incomplete Lineage Sorting: 9.84% Gene Flow: 7.76% "Consistent" Genes: ~58.8% "Inconsistent" Genes: ~41.2% | Produced strongly supported but potentially misleading topologies due to violation of congruent gene tree assumption [31] [2]. | More robust to gene tree heterogeneity; significantly reduced incongruence after filtering inconsistent genes [31]. | Cytoplasmic vs. nuclear genome discordance; Decomposition analysis; D-statistics. |
| Tulipeae (Tulips) [24] | Pervasive ILS and Reticulate Evolution | Specific quantitative contributions of ILS vs. introgression were not clearly partitioned. | Conflicted with plastid and coalescent-based nuclear topologies, particularly in relationships among Amana, Erythronium, and Tulipa [24]. | Recovered a different, weakly supported topology for major genera; confirmed most subgeneric monophyly within Tulipa [24]. | Phylogenomic networks; Polytomy tests; D-statistics; QuIBL analysis. |
| Pteris (Ferns) [32] | Long-Distance Dispersal, Allopatric/Parapatric Speciation | Not quantitatively analyzed in the study. | Produced a global phylogeny using rbcL and matK plastid genes, but left genus monophyly and some deep relationships uncertain [32]. | Not applied in the cited study; modern phylogenomic approaches could address lingering uncertainties [32]. | Global taxon sampling; Biogeographic analysis using BioGeoBEARS; Morphological character evolution. |
Experimental Protocols and Workflows
The following workflows detail the core methodologies used in the featured case studies to dissect complex phylogenies.
Genomic Data Assembly and Phylogenetic Inference (Fagaceae)
This protocol outlines the comprehensive approach for generating and analyzing mitochondrial, chloroplast, and nuclear data to investigate phylogenetic discordance [31].
Transcriptome-Based Phylogenomics and Reticulate Evolution Analysis (Tulipeae)
This protocol describes a transcriptome-based methodology to resolve complex relationships and test for ILS and introgression [24].
Multi-Gene Plastid Phylogeny and Biogeographic Reconstruction (Pteris)
This protocol outlines a traditional but extensive multi-gene approach to establish a foundational phylogeny and investigate historical biogeography [32].
This section catalogs key bioinformatics tools, software, and analytical methods referenced in the case studies, providing a resource for designing phylogenomic studies.
Table 2: Essential Research Reagents and Resources for Phylogenomic Analysis
| Category | Item/Reagent | Primary Function/Purpose |
|---|---|---|
| Sequencing & Assembly | Illumina Short-Read Sequencing | Generating high-throughput genomic or transcriptomic data [31] [24]. |
| GetOrganelle | De novo assembly of organellar genomes (plastid, mitochondrial) [31]. | |
| Unicycler | Hybrid assembly of genomes, improving continuity of mitochondrial scaffolds [31]. | |
| Alignment & Mapping | BWA | Mapping sequencing reads to a reference genome for SNP calling [31]. |
| SAMtools | Processing, sorting, and indexing alignment files [31]. | |
| Variant Calling & Filtering | GATK HaplotypeCaller | Identifying single nucleotide polymorphisms (SNPs) from mapped reads [31]. |
| Depth & Quality Filters | Removing low-quality or unreliable sites (e.g., depth <10 or >300) [31]. | |
| BLASTN | Identifying and filtering out nuclear or chloroplast-derived sequences from mitochondrial data [31]. | |
| Phylogenetic Inference | IQ-TREE | Maximum likelihood tree inference with model selection and branch support [31]. |
| MrBayes | Bayesian inference of phylogenetic trees [31]. | |
| ASTRAL | Coalescent-based species tree estimation from gene trees [24]. | |
| Incongruence & Reticulation Analysis | Site Concordance/Discordance Factors (sCF/sDF) | Quantifying phylogenetic conflict and support at individual sites [24]. |
| D-statistics (ABBA-BABA) | Testing for gene flow (introgression) between taxa [24]. | |
| QuIBL | Quantifying the relative contributions of ILS and introgression to gene tree discordance [24]. | |
| Phylogenetic Networks | Visualizing and testing evolutionary hypotheses that include hybridization events [24]. | |
| Visualization | PhyloScape / ggtree | Interactive, customizable, and publication-ready visualization of phylogenetic trees [33] [13]. |
| Data Types | Plastid Protein-Coding Genes (PCGs) | A standard set of genes for constructing plastome-based phylogenies [24]. |
| Nuclear Orthologous Genes (OGs) | Hundreds to thousands of low-copy nuclear genes from transcriptomes or genomes for coalescent analysis [31] [24]. |
Overcoming Pitfalls: Model Violations, Error Sources, and Best Practices
Identifying and Mitigating Gene Tree Estimation Error (GTEE)
Gene tree estimation error (GTEE) represents a fundamental challenge in phylogenomics, potentially leading to incorrect inferences about species relationships and evolutionary history. As researchers increasingly rely on genomic-scale data to resolve difficult phylogenetic problems, the impact of GTEE becomes more pronounced, particularly in debates surrounding concatenation versus coalescent approaches. GTEE arises from multiple sources including insufficient phylogenetic signal, model misspecification, alignment artifacts, and biological complexities like recombination [2]. These errors are particularly problematic for coalescent methods that operate on estimated gene trees rather than directly on sequence data [34].
The multispecies coalescent (MSC) model provides a theoretical framework for accommodating gene tree heterogeneity due to incomplete lineage sorting (ILS), but its performance depends critically on accurate gene tree estimation [2]. Meanwhile, concatenation methods, while potentially more robust to gene tree error in some circumstances, make biologically unrealistic assumptions about identical gene histories across the entire genome [2] [34]. This comparison guide examines current methodologies for identifying and mitigating GTEE, providing researchers with evidence-based recommendations for navigating these complex analytical trade-offs.
Impact of Gene Tree Error on Phylogenetic Inference
Gene tree estimation errors propagate through phylogenetic analyses, affecting downstream species tree inference and potentially leading to strongly supported but incorrect evolutionary relationships. The severity of these impacts varies across methods and biological contexts.
Effects on Coalescent versus Concatenation Approaches
Coalescent methods exhibit differential sensitivity to GTEE. Shortcut coalescent methods (e.g., MP-EST, STAR) that use pre-estimated gene trees as input are particularly vulnerable, as their statistical consistency relies on the assumption that gene tree incongruence stems primarily from ILS rather than estimation error [34]. When applied to ancient divergences with limited phylogenetic signal, these methods can produce misleading results if gene trees are inaccurate [34]. In contrast, concatenation methods may be more robust to individual gene tree errors due to their pooling of signal across loci, but they risk inconsistency when MSC assumptions are violated [2].
Empirical studies reveal that the proportion of loci rejecting the MSC model (11%) is significantly lower than those rejecting substitution models (44%) or concatenation assumptions (38%), suggesting that poor fit of substitution models contributes substantially to GTEE [2]. Logistic regression analyses have identified that proportions of GC content and informative sites negatively correlate with substitution model fit, highlighting specific sequence features that predispose to estimation error [2].
Impact of Biological Complexities
Biological processes beyond ILS further complicate gene tree estimation and exacerbate GTEE:
Introgression: Widespread gene flow between lineages creates complex phylogenetic signals that contradict species boundaries. Studies of Neotropical true fruit flies (Anastrepha) reveal "signals of incomplete lineage sorting, vestiges of ancestral introgression between more distant lineages and ongoing gene flow between closely related lineages" [35]. These processes simultaneously affect phylogenetic signal and challenge accurate gene tree estimation.
Ancient Introgression: Cross-lineage gene flow can affect even deep evolutionary relationships. Research on Xanthoceras (Sapindaceae) uncovered "ancient introgression, incorporating approximately 16% of its genetic material from ancestral subfam. Sapindoideae lineages," causing persistent cyto-nuclear discordance [36].
Table 1: Impact of Gene Tree Error Across Phylogenetic Methods
| Method Type | Sensitivity to GTEE | Primary Error Consequences | Optimal Application Context |
|---|---|---|---|
| Shortcut Coalescent (MP-EST, STAR) | High | Inconsistent species trees with mis-rooted gene trees [34] | Recent radiations with strong phylogenetic signal [34] |
| Summary Methods (ASTRAL) | Moderate | More robust to mis-rooting than other coalescent methods [34] | Various timescales with moderate ILS [34] |
| Weighted Quartet Methods (wTREE-QMC) | Lower | Improved accuracy despite missing data and homology errors [37] | Large datasets with taxon incompleteness [37] |
| Concatenation | Variable | Inconsistency under high ILS; more robust to gene tree error [2] [34] | Low ILS scenarios or when gene tree error dominates [2] |
Methodological Approaches for Mitigating GTEE
Weighted Quartet Methods and Gene Tree Filtering
Recent algorithmic advances address GTEE through weighting schemes that account for gene tree uncertainty. Weighted TREE-QMC incorporates branch lengths and support values to weight quartets, demonstrating "improved robustness to gene tree incompleteness, estimation errors, and systematic homology errors" compared to unweighted approaches [37]. This method maintains accuracy even with extreme missing data, making it suitable for phylogenomic datasets with heterogeneous taxon sampling.
Empirical tests show that weighting quartets by gene tree branch lengths "can improve robustness to systematic homology errors and can be as effective as removing the impacted taxa from individual gene trees or removing the impacted gene trees entirely" [37]. This approach provides a valuable alternative to filtering strategies that reduce dataset size and potentially discard useful phylogenetic signal.
Model Validation and Comparison
Statistical framework for model comparison offers powerful approach for identifying GTEE and selecting appropriate inference methods:
- Posterior Predictive Simulation: Bayesian approach evaluating model fit by comparing observed data to data simulated under the model [2]
- Bayes Factor Comparison: Quantitative framework for comparing fit of MSC versus concatenation models [2]
- Heterogeneous Tree Assumptions: Robust regression techniques that mitigate effects of tree misspecification in comparative analyses [38]
Studies applying these validation methods consistently favor MSC over concatenation across diverse datasets, with concatenation assumptions "rejected by 38% of loci" and rarely holding "for phylogenomic data sets with more than 10 loci" [2].
The following diagram illustrates the relationship between data quality issues, their impacts on gene tree estimation, and corresponding mitigation strategies:
Figure 1: Gene Tree Error Causes and Mitigation Strategies
Experimental Protocols for GTEE Assessment
Researchers can implement the following experimental protocol to quantify and mitigate GTEE in phylogenomic datasets:
Protocol 1: Gene Tree Quality Assessment and Filtering
- Gene Tree Estimation: Estimate individual gene trees using appropriate substitution models and methods (e.g., RAxML, IQ-TREE)
- Support Evaluation: Calculate branch support values (e.g., bootstrap, posterior probabilities) for each gene tree
- Model Fit Testing: Apply posterior predictive simulation to test fit of substitution models for each locus [2]
- Data Filtering: Filter genes based on:
- Substitution model adequacy (p > 0.05 in posterior predictive checks)
- Presence of sufficient informative sites
- Minimal missing data
- Branch support thresholds
- Comparative Analysis: Compare species trees estimated from filtered versus unfiltered datasets
Protocol 2: Coalescent Model Validation
- Multispecies Coalescent Analysis: Infer species tree under MSC model using all gene trees
- Posterior Predictive Simulation: Simulate gene trees under the estimated MSC parameters [2]
- Test Statistics Calculation: Compare test statistics (e.g., tree distance metrics) between observed and simulated gene trees
- Model Adequacy Assessment: Identify loci with significant deviations from MSC expectations
- Sensitivity Analysis: Compare species tree estimates with and without problematic loci
Table 2: Experimental Approaches for GTEE Mitigation
| Approach | Key Methodology | Data Requirements | Implementation Tools |
|---|---|---|---|
| Gene Tree Weighting | Weight quartets by branch lengths and support values [37] | Gene trees with branch supports | weighted TREE-QMC [37] |
| Bayesian Model Validation | Posterior predictive simulation under MSC model [2] | Multi-locus sequence alignments | BPP [2] |
| Robust Regression | Sandwich estimators to reduce sensitivity to tree misspecification [38] | Trait data and phylogenetic trees | Robust phylogenetic regression [38] |
| Introgression Testing | HyDe, PhyloNet analyses for detecting gene flow [35] [36] | Genome-scale sequence data | HyDe, PhyloNet [36] |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for GTEE Research
| Tool/Resource | Function | Application Context | Key Features |
|---|---|---|---|
| ASTRAL | Species tree estimation from gene trees | Coalescent-based phylogenetics [34] | Statistical consistency under MSC [34] |
| weighted TREE-QMC | Weighted quartet-based species tree inference | Datasets with missing data and gene tree error [37] | Branch length and support-based weighting [37] |
| PhyloNet | Reticulate evolution analysis | Detecting introgression and hybridization [36] | Network inference beyond tree-like evolution [36] |
| HyDe | Hybridization detection | Testing for ancient introgression [36] | Site pattern-based introgression tests [36] |
| PhyParts | Gene tree conflict analysis | Quantifying phylogenetic discordance [36] | Comparing gene trees to species tree [36] |
Gene tree estimation error remains a significant challenge in phylogenomics, but methodological advances provide powerful strategies for mitigation. Weighted quartet methods offer improved robustness to incomplete data and gene tree errors, while comprehensive model testing frameworks enable researchers to select appropriate analytical approaches based on empirical evidence rather than a priori assumptions [2] [37].
The integration of GTEE assessment into standard phylogenomic workflows is essential, particularly as datasets continue growing in size and complexity. By implementing the experimental protocols and tools outlined in this guide, researchers can substantially improve the accuracy of species tree estimation and produce more reliable evolutionary inferences. Future methodological development should focus on integrated models that simultaneously account for multiple sources of error and biological complexity, further bridging the gap between theoretical models and empirical data characteristics.
The reconstruction of evolutionary histories is a cornerstone of modern biology, with profound implications for understanding biodiversity, trait evolution, and disease origins. For decades, the concatenation approach—which combines all genetic data into a single supermatrix for analysis—dominated phylogenomic inference. However, the emergence of multispecies coalescent (MSC) methods promised greater accuracy by explicitly modeling fundamental biological processes like incomplete lineage sorting (ILS). Despite their theoretical sophistication, both approaches can produce conflicting results when applied to empirical data, creating critical uncertainty about their reliability under real-world conditions.
This guide objectively compares the performance of concatenation and MSC methods when confronted with the complex realities of empirical datasets. We synthesize current research to quantify how these models behave when biological and analytical challenges—including gene flow, ILS, and gene tree estimation error—violate their underlying assumptions. By providing structured experimental data and methodological protocols, we equip researchers with the framework needed to evaluate these competing approaches for their specific phylogenetic challenges.
Biological Realities Challenging Phylogenetic Models
Phylogenomic analyses routinely reveal extensive conflict among gene trees, which can stem from both biological processes and analytical artifacts:
Incomplete Lineage Sorting (ILS): The failure of gene lineages to coalesce in successive speciation events, particularly problematic during rapid radiations where short internodes provide limited time for allele sorting. ILS preserves ancestral polymorphisms that create genuine biological discordance between gene trees and species trees [18].
Gene Flow and Introgression: Hybridization and subsequent backcrossing can transfer genetic material between species, creating conflicting phylogenetic signals across the genome. This reticulate evolution produces patterns distinct from the bifurcating relationships assumed by most tree-building methods [10].
Gene Tree Estimation Error (GTEE): Analytical artifacts arising from methodological limitations, including insufficient phylogenetic signal, model misspecification, long-branch attraction, and alignment errors. GTEE introduces non-biological discordance that can mislead phylogenetic inference [18] [34].
Quantitative Contributions to Gene Tree Variation
Recent research has begun quantifying the relative contributions of these discordance sources. A 2025 study on Fagaceae decomposition analysis measured their impacts on nuclear gene tree variation, providing crucial empirical benchmarks [18].
Table 1: Quantitative Contributions to Gene Tree Discordance in Fagaceae
| Discordance Source | Contribution (%) | Biological Nature | Methodological Challenge |
|---|---|---|---|
| Gene Tree Estimation Error (GTEE) | 21.19% | Analytical artifact | Model misspecification, limited signal |
| Incomplete Lineage Sorting (ILS) | 9.84% | Biological process | Coalescent process modeling |
| Gene Flow/Introgression | 7.76% | Biological process | Reticulate evolution modeling |
| Consistent Phylogenetic Signal | 58.1–59.5% | - | - |
| Conflicting Phylogenetic Signals | 40.5–41.9% | - | - |
These data reveal that analytical artifacts (GTEE) can contribute more than twice as much to gene tree variation as biological processes like ILS in certain empirical datasets. This finding has profound implications for method selection, suggesting that approaches robust to gene tree error may outperform theoretically sophisticated but error-sensitive methods in practice.
Empirical Performance Comparison
Methodological Frameworks and Their Assumptions
Concatenation Methods: Combine all sequence alignments into a single "supermatrix" analyzed under the assumption of a shared evolutionary history across all loci. Implemented in maximum likelihood (IQ-TREE, RAxML) and Bayesian (MrBayes) frameworks, these methods effectively amplify phylogenetic signal through data combination but violate the reality of heterogeneous gene histories [18] [34].
Multispecies Coalescent (MSC) Methods: Explicitly model ILS by estimating gene trees individually before summarizing them into a species tree. "Shortcut" methods (ASTRAL, MP-EST, STAR) use pre-estimated gene trees, while full-likelihood methods co-estimate gene and species trees. MSC methods assume gene tree incongruence stems primarily from ILS rather than other biological processes or estimation error [34] [10].
Case Studies Quantifying Method Performance
Ancient Angiosperm Divergences
A critical test case involves rooting the angiosperm tree, where concatenation and coalescent methods have produced conflicting results regarding whether Amborella alone or (Amborella, Nymphaeales) form the sister lineage to all other flowering plants. A reappraisal study demonstrated that discrepant results were primarily caused by certain coalescent methods (MP-EST, STAR) not being robust to highly divergent and often mis-rooted gene trees [34] [39].
This research revealed that low phylogenetic signal and methodological artifacts in gene-tree reconstruction proved more problematic for these shortcut coalescent methods than concatenation's violation of the hierarchical unity assumption. The study identified that a third coalescent method, ASTRAL, demonstrated greater robustness to mis-rooted gene trees than MP-EST or STAR, highlighting significant performance variation within the MSC framework [34].
Rapid Radiations in Fagaceae and Tulipeae
Studies of rapidly radiating plant groups provide additional performance insights. Fagaceae research found that cytoplasmic (cpDNA and mtDNA) and nuclear genomes produced strongly conflicting topologies, with organellar genomes dividing taxa into New World and Old World clades while nuclear data supported different relationships—discordance attributed to ancient interspecific hybridization [18].
Table 2: Phylogenetic Method Performance Across Empirical Studies
| Study System | Concatenation Performance | Coalescent Performance | Primary Discordance Source |
|---|---|---|---|
| Angiosperm Rooting [34] [39] | Supported Amborella-alone hypothesis | MP-EST/STAR supported incorrect (Amborella, Nymphaeales) clade; ASTRAL more robust | Gene tree estimation error and mis-rooting |
| Fagaceae [18] | Produced highly supported but conflicting topologies across genomes | Revealed biological discordance from ILS and gene flow | Ancient hybridization (7.76%), ILS (9.84%), GTEE (21.19%) |
| Tulipeae [10] | Unable to resolve relationships among Amana, Erythronium, and Tulipa | Similarly unable to resolve deep relationships despite extensive data | Pervasive ILS and reticulate evolution obscuring signal |
| Pteris Ferns [12] | Effective for deeper taxonomic relationships | Revealed deep coalescence and ILS within genus | Incomplete lineage sorting and ancient hybridization |
In Tulipeae, neither concatenation nor coalescent approaches could reliably resolve relationships among Amana, Erythronium, and Tulipa genera despite extensive transcriptome sequencing, with researchers attributing this limitation to "especially pervasive ILS and reticulate evolution" that obscured phylogenetic signal [10].
Signal Quality Assessment Framework
A promising approach for improving phylogenetic accuracy involves differentiating genes based on their phylogenetic signal quality. Fagaceae research classified genes into "consistent" (58.1–59.5%) and "inconsistent" (40.5–41.9%) categories based on their likelihood and quartet-based phylogenetic signals [18].
Critically, the study found that excluding inconsistent genes significantly reduced conflicts between concatenation- and coalescent-based approaches, suggesting that data curation based on signal consistency may be more important than method selection for some challenging datasets. This filtering approach demonstrates that methodological performance depends heavily on underlying data quality, not just theoretical considerations.
Experimental Protocols for Method Evaluation
Gene Tree Discordance Decomposition Analysis
The decomposition protocol used in Fagaceae research provides a template for quantifying discordance sources [18]:
Dataset Preparation: Assemble multi-locus datasets with representative taxon sampling across the study group, including nuclear and organellar genomes where possible.
Phylogenetic Reconstruction: Generate individual gene trees using both maximum likelihood (IQ-TREE) and Bayesian (MrBayes) approaches, with appropriate model selection and support measures.
Incongruence Detection: Calculate gene tree conflicts using topological distance metrics and identify significantly supported discordances.
Source Attribution:
- Apply Patterson's D-statistics (ABBA-BABA tests) to detect gene flow
- Use quartet-based methods to quantify ILS contributions
- Estimate GTEE through comparison of bootstrap support, model adequacy, and congruence across inference methods
Proportion Quantification: Decompose the relative contributions of each discordance source using statistical frameworks that partition variance among biological and analytical factors.
Reticulate Evolution Analysis
For groups with suspected hybridization, the Tulipeae protocol offers a comprehensive approach [10]:
Multi-Method Tree Reconstruction: Generate species trees using both concatenation (maximum likelihood) and coalescent (ASTRAL) methods from the same nuclear dataset.
Site Concordance Analysis: Calculate "site con/discordance factors" (sCF and sDF1/sDF2) to identify phylogenetic nodes with conflicting signal.
Network Analysis: Apply phylogenetic network methods (PhyloNet, SplitsTree) to nodes displaying high or imbalanced sDF1/sDF2 values.
Polytomy Testing: Compare fit of bifurcating versus multifurcating models at contentious nodes to distinguish hard polytomies from method artifacts.
Introgression Tests: Implement D-statistics and QuIBL to quantify introgression signals and localize them on the phylogeny.
Visualizing Phylogenetic Analysis Workflows
Method Selection Decision Framework
Gene Tree Discordance Analysis Workflow
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Essential Research Tools for Phylogenomic Method Evaluation
| Tool/Resource | Function | Application Context |
|---|---|---|
| IQ-TREE [18] | Maximum likelihood phylogeny inference with model selection | Gene tree and concatenated phylogeny estimation |
| ASTRAL [34] [10] | Coalescent-based species tree inference from gene trees | Species tree estimation accounting for ILS |
| BEAST2 [12] | Bayesian evolutionary analysis sampling trees | Co-estimation of gene trees and species trees |
| D-Statistics [10] [12] | Test for gene flow and introgression | Detecting hybridization signals in genomic data |
| PhyloNet [10] | Phylogenetic network inference | Modeling reticulate evolutionary histories |
| BUSCO [40] | Benchmarking universal single-copy orthologs | Gene set assessment for phylogenomic analyses |
| GetOrganelle [18] | Organelle genome assembly | Assembling mitochondrial and chloroplast genomes |
| OrthoFinder [40] | Orthogroup inference and gene tree analysis | Identifying orthologous groups across species |
The empirical evidence demonstrates that both concatenation and multispecies coalescent methods can produce misleading results when their underlying assumptions are violated by biological complexities and analytical challenges. Rather than declaring a universal winner, our analysis reveals that method performance is context-dependent, influenced by factors including:
- The relative contributions of ILS versus gene flow in the study system
- The prevalence of gene tree estimation error due to limited signal or model misspecification
- The evolutionary timeframe, with coalescent methods potentially more vulnerable to error in deep phylogenies
- Data quality and the proportion of genes with consistent phylogenetic signals
For researchers navigating these methodological challenges, we recommend a pluralistic approach that employs both concatenation and coalescent methods, assesses gene tree signal quality, explicitly tests for biological sources of discordance, and interprets resulting phylogenies with appropriate caution regarding potential methodological artifacts. By understanding the conditions under which each model is most likely to be wrong, researchers can make more informed decisions about which approach—or combination of approaches—will yield the most reliable evolutionary insights for their specific biological system.
The widespread observation of gene tree discordance, where evolutionary histories differ across regions of the genome, has become a central focus in phylogenomics [18]. This discordance stems from multiple biological and analytical sources, primarily incomplete lineage sorting (ILS), introgression (gene flow), and gene tree estimation error (GTEE) [41] [42]. Disentangling their relative contributions is crucial for accurate phylogenetic inference and has profound implications for understanding speciation, selection, and evolutionary history.
This guide quantitatively compares the performance of two major phylogenetic approaches—concatenation and coalescent-based methods—in the presence of these confounding factors. We provide a structured analysis of experimental data and methodologies, offering a practical framework for researchers navigating phylogenomic conflict.
Empirical studies have begun to quantify the proportional contributions of ILS, introgression, and GTEE to overall gene tree variation. The table below summarizes key findings from recent phylogenomic investigations.
Table 1: Quantitative Contributions of Different Processes to Gene Tree Discordance
| Study System | Incomplete Lineage Sorting (ILS) | Introgression/Gene Flow | Gene Tree Estimation Error (GTEE) | Other/Consistent Signal | Key Citation |
|---|---|---|---|---|---|
| Fagaceae (Oak family) | 9.84% | 7.76% | 21.19% | 58.1–59.5% (consistent genes) | [18] [31] |
| Rattlesnakes | Dominant process in rapid radiations, alongside introgression. Quantified via network analyses. | Widespread, with frequent hybridization events. | Acknowledged as a key source of conflict. | — | [42] |
| Amaranthaceae | Major driver of deep-level discordance. | Ancient hybridization tested but not the sole dominant factor. | Addressed via model selection and data filtering. | Hard polytomy suggested for some nodes. | [41] |
| Malvaceae | Primary source of localized deep-level discordance. | Secondary role, with some detected introgression events. | — | — | [43] |
| Pancrustaceans | Significant contributor, confounding deep phylogeny. | Considered as a potential source. | long-branch attraction (LBA) a major analytical confounder. | — | [44] |
Experimental Protocols for Disentangling Discordance
Genome-Wide Data Assembly and Phylogenetic Inference
A standard workflow for phylogenomic analysis involves sequencing, assembly, and multiple layers of phylogenetic inference to test for robustness and conflict.
Diagram: Phylogenomic Analysis Workflow
Detailed Methodological Steps:
- Taxon Sampling and Sequencing: Researchers collect tissue samples representing the diversity of the group. Studies typically use whole-genome sequencing, transcriptomics (e.g., [41]), or targeted sequence capture (e.g., of hundreds of nuclear loci; [43]) to generate genomic data.
- Orthology Assessment: A critical step to avoid confounding signals from paralogous genes. This can be achieved using a phylogenetically-informed orthology approach [44] or by leveraging probe sets designed for conserved low-copy nuclear genes [43].
- Gene Tree Estimation: Individual maximum likelihood (ML) or Bayesian Inference (BI) gene trees are inferred for each locus. Software like IQ-TREE (for ML) and MrBayes (for BI) are commonly used [18] [31].
- Species Tree Inference:
- Concatenation Approach: All aligned loci are combined into a single "supermatrix," from which a species tree is inferred under ML or BI. This method assumes a single underlying evolutionary history for all genes [18].
- Coalescent Approach: Species trees are estimated from individual gene trees using methods that account for ILS, such as ASTRAL. This approach does not assume a single history and is more robust to ILS [18] [43].
Table 2: Experimental Methods for Ispecific Sources of Phylogenetic Discordance
| Target Process | Key Method/Software | Experimental Protocol Summary | Interpretation of Results |
|---|---|---|---|
| Gene Tree Estimation Error (GTEE) | "Consistent" vs. "Inconsistent" gene filtering based on likelihood/quartet signals [18]. | 1. Infer a reference species tree (e.g., via coalescent method).2. Classify genes as "consistent" if their signal supports the species tree and "inconsistent" if they conflict.3. Compare phylogenetic inferences with and without inconsistent genes. | A significant reduction in conflict between concatenation and coalescent results after filtering suggests GTEE was a major source of discordance [18]. |
| Incomplete Lineage Sorting (ILS) | Coalescent-based species tree inference (e.g., ASTRAL); Site Pattern Tests (e.g., D-statistics) [41] [42]. | 1. Estimate species trees with methods that model ILS.2. Use Patterson's D statistic to test for an excess of shared derived alleles between non-sister taxa, which can indicate introgression against a background of ILS [45] [42]. | High levels of gene tree heterogeneity, even in the absence of significant D-statistic signals, can point to ILS as the dominant process, especially in rapid radiations [46] [42]. |
| Introgression / Gene Flow | D-statistic (ABBA-BABA test) [45], Phylogenetic Networks (e.g., PhyloNet) [41] [42]. | 1. Apply the D-statistic to a 4-taxon system (P1, P2, P3, Outgroup) to detect genome-wide introgression.2. Use network inference software to identify specific hybridization events and estimate their direction and strength. | A significant D-statistic value indicates an excess of allele sharing between P3 and either P1 or P2, suggestive of introgression. Network analyses can visualize these reticulate relationships directly [41] [42]. |
| Combined ILS & Introgression | Multi-Species Coalescent Network (MSCN) models (e.g., SNaQ) [42]. | 1. Input a set of gene trees into an MSCN method.2. The model estimates a species network with reticulate branches, accounting for both ILS and hybridization. | MSCN approaches provide a more biologically realistic framework for groups with both rapid diversification and introgression, quantifying the relative impact of each [42]. |
Diagram: Decision Framework for Analyzing Discordance
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Reagents and Computational Tools for Phylogenomic Discordance Research
| Category | Item / Software | Primary Function in Analysis |
|---|---|---|
| Sequencing & Assembly | Illumina Short-Read Sequencing | Standard workhorse for generating genome-wide SNP data or transcriptomes [18] [46]. |
| GetOrganelle | Assembles organellar genomes (cpDNA, mtDNA) from NGS data [18]. | |
| BWA / GATK | Read mapping and variant calling for SNP-based phylogenetic datasets [18] [31]. | |
| Phylogenetic Inference | IQ-TREE | Maximum likelihood inference of gene trees and concatenated phylogenies; implements complex models and bootstrapping [18] [44]. |
| MrBayes | Bayesian inference of phylogenetic trees, useful for assessing node credibility [18] [31]. | |
| ASTRAL | Coalescent-based species tree estimation from gene trees, robust to ILS [43]. | |
| Discordance Analysis | D-Suite | Suite for calculating D-statistics and related metrics to detect introgression [45] [42]. |
| PhyloNet / SNaQ | Infers phylogenetic networks from gene trees, modeling both ILS and hybridization [41] [42]. | |
| Data Processing | HybPiper / PHYLUCE | Processes targeted sequence capture data, extracts loci, and assesses orthology [43]. |
| AMAS | Alignment manipulation and summary statistics. |
Concatenation vs. Coalescent Approaches in the Presence of Introgression
The performance of concatenation and coalescent methods is highly dependent on the source of discordance.
Under Incomplete Lineage Sorting (ILS): Coalescent-based methods are superior. They explicitly model the coalescent process and are statistically consistent in the face of ILS. In contrast, concatenation can be positively misleading, strongly supporting an incorrect species tree topology, especially in "anomaly zones" created by rapid radiations [42]. The Fagaceae study demonstrated that removing genes with inconsistent signals reduced conflict between the two approaches, highlighting the confounding effect of GTEE and ILS on concatenation [18].
Under Introgression: Both methods face challenges, but network-based extensions of the coalescent offer a solution. Standard coalescent models and concatenation both assume a strictly diverging tree-like species history. When widespread introgression occurs, as in rattlesnakes, this assumption is violated, and both approaches may yield unstable or incorrect phylogenies [42]. Multi-species coalescent network (MSCN) models represent the most advanced framework, as they can account for both ILS and introgression simultaneously, providing a more realistic evolutionary picture [42].
In practice, the most robust strategy is not to choose one over the other, but to apply a combination of approaches (concatenation, coalescent, networks, and tests for introgression) to thoroughly explore the data and identify the predominant evolutionary processes [41].
The analysis of large genomic datasets is fundamental to modern evolutionary biology and genetics. Researchers face critical decisions regarding the selection of genomic loci, the choice of phylogenetic models, and the computational strategies for handling data. These decisions are particularly pivotal in the context of the long-standing debate between concatenation and coalescent approaches, especially when the evolutionary history includes complex processes like introgression. This guide provides an objective comparison of these methodologies, supported by current experimental data and detailed protocols, to equip researchers with the knowledge to optimize their genomic analyses.
Locus Selection Strategies for Robust Phylogenomics
The choice of genomic loci is a primary determinant of success in phylogenetic inference and genome quality assessment. Strategic locus selection can mitigate model inadequacies and improve topological accuracy.
Universal Single-Copy Orthologs
Universal single-copy orthologs, such as Benchmarking Universal Single-Copy Orthologs (BUSCOs), are a mainstay in phylogenomics and assembly completeness benchmarking due to their high conservation and identifiability across deep evolutionary divergences [40].
- Evolutionary Influence: Analyses of 11,098 eukaryotic genomes reveal that evolutionary history significantly impacts BUSCO gene content. Some taxonomic groups show statistically significant deviations in BUSCO completeness, while others display elevated levels of duplicated orthologs, often tracing back to ancestral whole-genome duplication events [40].
- Gene Loss and Misidentification: Standard BUSCO searches are susceptible to undetected ancestral gene loss, leading to a 2.25% to 13.33% mean lineage-wise gene misidentification rate. Employing a Curated set of BUSCO orthologs (CUSCOs) can reduce false positives by up to 6.99% compared to standard searches [40].
Evolutionary Rate and Taxonomic Congruence
The evolutionary rate of selected sites directly impacts the accuracy of inferred phylogenies. A comprehensive study involving 3,566 phylogenetic trees across five major lineages (Eudicots, Ascomycota, Basidiomycota, Arthropoda, and Vertebrata) yielded critical insights [40].
Table 1: Impact of Site Evolutionary Rate on Phylogenetic Accuracy
| Site Category | Taxonomic Congruence | Terminal Bifurcation Variability | Recommended Use |
|---|---|---|---|
| Higher-rate sites | Up to 23.84% more congruent | At least 46.15% less variable | Optimal for taxonomic congruence |
| Lower-rate sites | Less congruent | More variable | Less reliable for deep phylogenies |
| Concatenated alignments | High congruence | Low variability | Preferred over coalescent trees for BUSCOs |
The study concluded that for BUSCO-derived phylogenies, higher-rate sites from concatenated alignments produce the most congruent and least variable phylogenies [40].
Model Choice: Coalescent vs. Concatenation in the Era of Introgression
The selection between coalescent and concatenation models is a central decision in phylogenomics. A statistical framework for model comparison and validation is essential for resolving debates about their application.
Widespread Inadequacy of Substitution and Concatenation Models
A large-scale analysis of 47 phylogenomic datasets across the tree of life revealed widespread inadequacy of simple models [2]:
- Substitution models were rejected by 44% of loci.
- Concatenation models, which assume topologically congruent gene trees, were rejected by 38% of loci.
- In contrast, the Multispecies Coalescent (MSC) model was rejected by only 11% of loci that were adequately described by a substitution model [2].
This demonstrates that the violation of the concatenation assumption is common in datasets with more than ten loci.
Model Comparison Strongly Favors the Coalescent
Bayesian model validation and comparison consistently favor the MSC model over concatenation. The assumption of congruent gene trees rarely holds for large phylogenomic datasets, making the coalescent model a consistently better fit for the data [2]. The concatenation model is best described as a special case of the MSC model where all gene trees are topologically identical [2].
Incorporating Introgression: The MSci Model and its Challenges
To account for gene flow, the Multispecies Coalescent with Introgression (MSci) model has been developed. However, it faces specific challenges, particularly with the Bidirectional Introgression (BDI) model.
- Unidentifiability Issue: The BDI model has a fundamental "mirror" unidentifiability problem. For any parameter set Θ, a mirror parameter set Θ' exists that has an identical probability of generating the observed gene tree data, G, such that f(G|Θ) = f(G|Θ') [47]. This is known as within-model unidentifiability.
- Impact on Inference: An MSci model with k BDI events can have 2^k unidentifiable modes in the posterior. This problem is pronounced when only one sequence is sampled per species, though it can be mitigated with multiple samples [47].
Table 2: Comparison of Concatenation and Coalescent Approaches
| Feature | Concatenation Approach | Coalescent Approach (MSC) | MSci with Introgression |
|---|---|---|---|
| Core Assumption | Topologically congruent gene trees | Gene tree variation from ILS | Gene tree variation from ILS and introgression |
| Model Rejection Rate | 38% of loci [2] | 11% of loci (post-substitution model filter) [2] | Subject to unidentifiability [47] |
| Computational Demand | Lower | Higher | Highest |
| Handles Incomplete Lineage Sorting (ILS) | No | Yes | Yes |
| Handles Introgression | No | No | Yes |
| Best Application | Small datasets (<10 loci) with low ILS | Large datasets with significant ILS | Datasets with known or suspected gene flow |
Handling Large Genomic Datasets: Methods and Workflows
The scale of modern genomic data requires robust bioinformatics tools and explicit workflows for detecting complex evolutionary signals.
Experimental Protocols for Detecting Selection and Introgression
Genome-Wide Scans for Selection Signatures
- Objective: To identify genomic regions that have undergone past selection by detecting deviations from neutral evolution patterns.
- Methodology:
- Data Genotyping: Utilize high-density SNP arrays (e.g., Illumina HD BeadChip) on hundreds of individuals from multiple populations [48].
- Quality Control: Filter SNPs for Hardy-Weinberg Equilibrium, minor allele frequency, and individual missing call rates. Remove related individuals based on genomic kinship [48].
- Population Structure Analysis: Use Principal Component Analysis (PCA) and ancestry coefficient estimation (e.g., with
sNMF) to understand population relationships [48]. - Signature Identification: Employ multiple statistics (e.g.,
treemixfor population splits/mixtures) to identify regions with extreme SNP and haplotype frequency differences between populations, indicating potential selection [48]. - Validation: For candidate regions, such as coat color genes, confirm allelic heterogeneity by re-sequencing the locus in populations under selection [48].
PhyloNet-HMM for Detecting Introgression
- Objective: To accurately identify introgressed genomic regions while accounting for confounding factors like Incomplete Lineage Sorting (ILS) [49].
- Methodology:
- Data Preparation: Generate whole-genome multiple sequence alignments from the studied species and an outgroup.
- Model Construction: Combine a phylogenetic network that represents the species history including potential introgression events with a Hidden Markov Model (HMM) that accounts for dependencies between adjacent sites in the genome [49].
- Genome Scanning: The HMM scans the alignment, identifying regions where the local genealogy better fits the introgressed history versus the vertical descent history [49].
- Application: This method successfully detected a known adaptive introgression event involving the Vkorc1 gene in mice, and estimated that 9% of sites on chromosome 7 (13 Mbp, covering 300+ genes) were of introgressive origin [49].
Workflow Visualization
The following diagram illustrates the logical workflow for selecting an optimal phylogenetic approach based on dataset characteristics and research goals, incorporating the findings on locus and model choice.
Successful execution of phylogenomic studies relies on a suite of computational tools and genomic resources. The following table details key solutions used in the featured experiments and the broader field.
Table 3: Key Research Reagent Solutions for Phylogenomics
| Tool/Resource | Type | Primary Function | Application Example |
|---|---|---|---|
| BUSCO [40] | Software & Gene Sets | Assess assembly completeness & identify universal single-copy orthologs | Benchmarking gene content in new genome assemblies [40] |
| PhyloNet-HMM [49] | Software Package | Detect introgressed genomic regions using HMMs and phylogenetic networks | Identifying adaptive introgression of Vkorc1 in mice [49] |
| bpp [47] | Software Package | Full-likelihood analysis under MSC and MSci models; includes algorithms for label-switching | Inferring species history and introgression parameters [47] |
| treemix [48] | Software Package | Infer population splits and mixtures from allele frequency data | Modeling gene flow between sheep populations [48] |
| sNMF [48] | Software Package | Estimate individual ancestry coefficients | Analyzing population structure in French sheep breeds [48] |
| OrthoDB [40] | Database | Catalog of orthologous genes across the tree of life | Source of evolutionary-informed universal orthologs |
| Illumina HD BeadChip [48] | Genotyping Array | High-throughput SNP genotyping | Generating high-density genotype data for selection scans [48] |
Empirical Performance and Statistical Validation of Competing Approaches
In the field of statistical model validation, particularly within phylogenomics and research involving introgression, two powerful but philosophically distinct frameworks are often employed: Bayes Factors and Posterior Predictive Simulation (PPS). The debate between concatenation and coalescent approaches in phylogenetics provides a critical context for this comparison. Concatenation methods assume that all genetic loci share a single underlying topology, while coalescent-based models, like the multispecies coalescent (MSC), account for gene tree variation caused by mechanisms like incomplete lineage sorting (ILS) [2]. Validating which model is more appropriate for a given dataset is paramount, and this is where Bayes Factors and PPS offer complementary tools for assessing model fit and performance.
The fundamental distinction lies in their approach to inference. Bayes Factors provide a Bayesian solution for model comparison and hypothesis testing, quantifying the evidence for one statistical model over another based on the observed data [50] [51]. In contrast, Posterior Predictive Simulation is a Bayesian model-checking technique that assesses the adequacy of a chosen model by comparing the observed data to data simulated from the fitted model [2].
Theoretical Foundation and Mechanics
Bayes Factors: A Ratio of Evidential Strength
A Bayes Factor (BF) is a comparative metric that measures the relative support the data provides for two competing hypotheses or models. Mathematically, it is the ratio of the marginal likelihoods of the data under two models, H₁ and H₂ [50].
The Bayes Factor is formally defined as: BF₁₂ = P(Data | H₁) / P(Data | H₂)
A BF₁₂ greater than 1 indicates evidence in favor of H₁, while a value less than 1 supports H₂. The magnitude of the BF indicates the strength of this evidence. Unlike p-values, Bayes Factors directly compare the probabilities of the data under both hypotheses, providing a more symmetrical and intuitive measure of evidence [50] [51]. This framework allows researchers to state that "the results we observe have, for example, twice the probability under H₁ as they do under H₂" [50].
Posterior Predictive Simulation: Assessing Model Adequacy
Posterior Predictive Simulation evaluates how well a model replicates core features of the observed data. The process involves:
- Fitting a model to the observed data to obtain the posterior distribution of parameters.
- Simulating new datasets using parameter values drawn from this posterior distribution.
- Comparing the simulated data to the observed data using a discrepancy measure or test statistic.
The comparison is often summarized by a posterior predictive p-value (ppp-value), which measures the probability that a simulated dataset is more extreme than the observed data, given the fitted model. A ppp-value near 0.5 suggests the model generates data similar to the observation, while a very high or low value (e.g., <0.05 or >0.95) indicates the model is a poor fit for that particular aspect of the data [2].
Comparative Analysis in Phylogenomic Model Selection
The choice between concatenation and coalescent models is a central problem in modern phylogenomics. The following table summarizes a core evaluation based on an empirical analysis of 47 phylogenomic datasets across the tree of life, which applied both PPS and BF for model validation [2].
Table 1: Model Validation Outcomes in Phylogenomic Studies
| Validation Method | Model Evaluated | Key Finding | Implication for Model Adequacy |
|---|---|---|---|
| Posterior Predictive Simulation | Substitution Models | 44% of loci rejected the fit of common substitution models [2] | Highlights widespread violation of common sequence evolution assumptions |
| Posterior Predictive Simulation | Concatenation (TC Assumption) | 38% of loci rejected the congruent gene tree assumption [2] | Concatenation's core assumption frequently violated in multi-locus data |
| Posterior Predictive Simulation | Multispecies Coalescent (MSC) | ~11% of loci rejected the MSC model (among loci with adequate substitution model fit) [2] | MSC provides a substantially better fit to phylogenomic data than concatenation |
| Bayes Factor Comparison | MSC vs. Concatenation | "Bayesian model validation and comparison both strongly favor the MSC over concatenation across all data sets" [2] | Coalescent model is consistently a more likely generating process for phylogenomic data |
Resolving Phylogenetic Discord with PPS
The power of PPS in diagnosing model failures is exemplified in studies of the fish genus Catostomus. Researchers used PPS and other methods to test between the "Introgression Hypothesis" (discord is due to historical gene flow) and the "Convergent Evolution Hypothesis" (discord is due to morphology evolving multiple times) [52]. The PPS and related statistical tests detected extensive historical introgression, supporting the introgression hypothesis and demonstrating that the MSC model, which can account for such processes, was more adequate than simpler models that ignore gene flow [52].
Practical Application and Experimental Protocols
A Workflow for Model Validation in Phylogenomics
The following diagram illustrates a generalized experimental protocol for applying BF and PPS to validate phylogenetic models, synthesizing methodologies from the cited research.
Key Research Reagents and Solutions
The application of these statistical frameworks relies on a suite of computational tools and reagents.
Table 2: Essential Research Reagents and Tools for Model Validation
| Research Reagent / Tool | Type | Primary Function in Validation | Example Use Case |
|---|---|---|---|
| ddRADseq Data | Genomic Data | Provides thousands of independent loci for robust multi-locus analysis [52] | Generating the input phylogenomic dataset for testing concatenation vs. coalescent models |
| PyRAD | Bioinformatics Pipeline | Processes raw sequencing reads into aligned, clustered loci for phylogenetic analysis [52] | Filtering and aligning sequence data from ddRADseq studies prior to model fitting |
| MrBayes / BEAST2 | Software Package | Bayesian phylogenetic inference allowing for PPS and complex models like MSC [2] | Fitting the multispecies coalescent model and performing posterior predictive checks |
| Phrapl | Software Package | Model selection framework using approximate likelihoods [2] | Comparing different demographic models (e.g., with and without gene flow) |
| Patterson's D-Statistic (ABBA-BABA) | Statistical Test | Tests for historical introgression by detecting specific site patterns [52] | Diagnosing the cause of model failure (e.g., introgression vs. ILS) after PPS indicates poor fit |
Strengths, Limitations, and Complementary Use
Both frameworks have distinct advantages and shortcomings, making them highly complementary.
Table 3: Comparative Advantages and Limitations of Bayes Factors and PPS
| Aspect | Bayes Factors | Posterior Predictive Simulation |
|---|---|---|
| Primary Strength | Direct, quantitative comparison of two models' relative plausibility [50] [51]. | Diagnosing how and why a single model fails to capture the data's structure [2]. |
| Key Limitation | Sensitivity to prior distributions on model parameters requires careful specification [53] [54]. | Does not provide a direct, quantitative model selection criterion like the BF; more descriptive. |
| Interpretation | "The data are 5 times more likely under the MSC model than under the concatenation model." | "The fitted MSC model generates data similar to the observed data for this test statistic (ppp-value = 0.52)." |
| Computational Demand | High, as it requires calculating marginal likelihoods, which can be unstable [54]. | High, as it requires simulating and analyzing thousands of new datasets from the posterior. |
The research clearly demonstrates their complementary nature. For instance, one can use PPS to first verify that the individual loci are adequately described by their substitution models and that the MSC model is not a poor fit. Then, BF can be used to formally compare the MSC model against the concatenation model, showing that the MSC is not just adequate, but quantitatively superior [2]. This two-step process provides a more complete picture than either method could alone.
In the context of the concatenation vs. coalescent debate, the empirical evidence strongly favors the use of model-aware frameworks like the Multispecies Coalescent. As one large-scale phylogenomic study concluded, "model comparisons are expected to consistently and more strongly favor the coalescent model over the concatenation model" for large datasets, as the core assumption of concatenation (topologically congruent gene trees) "rarely holds for phylogenomic data sets with more than 10 loci" [2].
Neither Bayes Factors nor Posterior Predictive Simulation is a silver bullet. Bayes Factors excel in providing a direct, evidence-based answer to the question "Which of these two models is better?". Posterior Predictive Simulation is unparalleled in answering the question "Is my chosen model adequate, and if not, where does it fail?". For researchers navigating complex model selection landscapes, such as those involving introgression, employing both frameworks in tandem offers the most robust strategy for statistical model validation and scientific discovery.
The multispecies coalescent (MSC) model provides a foundational framework for understanding gene tree heterogeneity due to incomplete lineage sorting (ILS) in phylogenomic analyses. This guide objectively compares the performance of coalescent-based species tree estimation methods against concatenation approaches, with a specific focus on their statistical consistency within and outside the anomaly zone—a region of tree space where the most likely gene tree topology differs from the species tree topology. We synthesize current theoretical proofs and empirical validation studies to demonstrate that coalescent-based methods remain statistically consistent under these challenging conditions where concatenation fails, providing researchers with critical insights for selecting appropriate phylogenetic methods in the presence of gene tree discordance.
Statistical consistency represents a fundamental property of phylogenetic methods, guaranteeing that as the amount of data (e.g., number of genes) increases indefinitely, the estimated tree topology converges in probability to the true species tree. The debate between concatenation and coalescent approaches centers on their respective consistency guarantees under biological realities such as incomplete lineage sorting (ILS), which causes gene trees to differ from the species tree [2]. The multispecies coalescent model has emerged as a crucial population genetics framework that describes the evolution of individual genes within a population-level species tree, modeling the stochasticity of deep coalescence events that lead to gene tree discordance [55]. Concatenation methods, which combine sequence data from multiple genes into a single supermatrix, assume topologically congruent genealogies across all loci—an assumption that is frequently violated in empirical datasets [2]. The anomaly zone presents a particular challenge for phylogenetic inference, as it creates conditions where the majority of gene trees may support an incorrect topology, making statistical consistency an essential safeguard against misleading results.
Theoretical Foundations: The Anomaly Zone and Consistency Guarantees
Defining the Anomaly Zone
The anomaly zone represents a region in tree space where the most probable gene tree topology under the MSC model does not match the species tree topology. This counterintuitive phenomenon occurs in specific topological configurations with short internal branches and large population sizes, creating conditions where incomplete lineage sorting is so extensive that the dominant gene tree pattern reflects deep coalescence rather than species relationships. Theoretical work has established that there are no anomalous rooted three-taxon species trees nor anomalous unrooted four-taxon species trees, providing a foundation for statistically consistent triplet and quartet-based methods [56]. This theoretical insight is crucial because it means that for rooted triplets and unrooted quartets, the most frequent gene tree topology will match the species tree topology, ensuring that methods based on these smaller subsets of taxa can consistently recover the true species tree even in more complex anomaly zones for larger trees.
Mechanisms of Statistical Consistency
Coalescent-based methods achieve statistical consistency through different algorithmic strategies that leverage the theoretical properties of the MSC model. Tuple-based methods operate by computing summary statistics for subsets of species (typically triplets or quartets) and then amalgamating these subsets to estimate the complete species tree [55]. The statistical consistency of these methods derives from the fact that, under the MSC model, the distribution of gene tree topologies for any subset of species contains sufficient information to reconstruct the relationships for that subset correctly. As the number of genes increases, the frequency of each possible topology for each subset converges to its expected probability under the MSC model, allowing consistent methods to identify the correct species tree topology. Table 1 summarizes the consistency properties of major coalescent-based methods, highlighting their theoretical foundations.
Table 1: Statistical Consistency Properties of Coalescent-Based Species Tree Methods
| Method | Theoretical Basis | Consistency Under MSC | Handles Anomaly Zone | Input Type |
|---|---|---|---|---|
| ASTRAL | Quartet amalgamation | Yes [55] | Yes [56] | Gene trees |
| ASTRID | Internode distances | Yes [55] | Yes | Gene trees |
| MP-EST | Pseudo-likelihood of triplets | Yes [55] [56] | Yes [56] | Gene trees |
| *BEAST | Co-estimation | Yes [55] | Yes | Sequence data |
| SVDquartets | Quartet frequencies | Yes [55] | Yes | Sequence data |
| STELAR | Triplet agreement | Yes [56] | Yes [56] | Gene trees |
| NJst | Average internode distances | Yes [55] | Yes | Gene trees |
| SNAPP | Site patterns | Yes [55] | Yes | SNP data |
Impact of Missing Data on Consistency
An important consideration for empirical studies is whether statistical consistency is maintained when gene trees miss certain taxa. Recent theoretical work has established that coalescent-based methods remain statistically consistent under realistic models of missing data, such as the Miid model (where each species is missing from each gene independently with probability p > 0) and the Mfsc model (where each subset of k species has non-zero probability of being present) [55]. This consistency holds for tuple-based methods when the calculated summary statistics are not impacted by deleting species outside the subset of interest, which is true for most summary methods like ASTRAL, MP-EST, and STELAR. The key insight is that for any subset of species, the distribution of gene trees restricted to that subset follows the MSC model for the species tree restricted to that subset, allowing consistent estimation across the taxon set despite missing data.
Empirical Performance Comparison: Coalescent vs. Concatenation
Large-Scale Model Comparison Studies
Comprehensive empirical evaluations across diverse phylogenomic datasets have demonstrated the superiority of coalescent methods over concatenation approaches. A landmark study examining 47 phylogenomic datasets across the tree of life found that 44% of loci showed a poor fit to substitution models and 38% rejected the concatenation assumption of topologically congruent gene trees [2]. In contrast, among loci adequately described by substitution models, only 11% rejected the MSC model, significantly lower than those rejecting substitution and concatenation models. Bayesian model validation strongly favored the MSC over concatenation across all datasets, with the concatenation assumption of congruent gene trees rarely holding for phylogenomic datasets with more than 10 loci. These findings indicate that model comparisons consistently and strongly favor coalescent models over concatenation for large phylogenomic datasets.
Performance Under Varying ILS Levels
Simulation studies have been instrumental in quantifying the performance of coalescent methods under controlled conditions of incomplete lineage sorting. Table 2 summarizes the performance of leading coalescent-based methods compared to concatenation under varying ILS levels, based on simulation studies across multiple research groups.
Table 2: Performance Comparison of Species Tree Methods Under Varying ILS Conditions
| Method | Low ILS | High ILS | High ILS + Missing Data (50%) | Computational Efficiency |
|---|---|---|---|---|
| ASTRAL | High accuracy | High accuracy | Maintains high accuracy [55] | Polynomial time [56] |
| ASTRID | High accuracy | High accuracy | Maintains high accuracy [55] | Polynomial time |
| MP-EST | High accuracy | High accuracy | Maintains high accuracy [55] | Computationally intensive [56] |
| STELAR | High accuracy | High accuracy | Maintains high accuracy | Polynomial time [56] |
| Concatenation | High accuracy | Inconsistent [2] | Performance degrades | Fast |
| *BEAST | Highest accuracy | Highest accuracy | Not evaluated | Computationally intensive [56] |
The experimental results consistently show that all coalescent-based species tree estimation methods improve in accuracy as the number of genes increases and often produce highly accurate species trees even when the amount of missing data is large [55]. These findings demonstrate that accurate species tree estimation is possible under a variety of conditions, even with substantial missing data, provided that appropriate coalescent-based methods are employed.
Experimental Protocols for Method Validation
Bayesian Model Validation Framework
The statistical framework for comparing coalescent and concatenation models incorporates several rigorous testing approaches:
Posterior Predictive Simulation (PPS): This Bayesian modeling approach simulates data from the posterior distribution of model parameters and compares the simulated data to the observed data. Discrepancies indicate poor model fit. In one study, PPS rejected the MSC at the level of gene trees for only four out of 25 datasets, and only 2.9% of total loci, suggesting that poor fit of the MSC is not as widespread as sometimes claimed [2].
Bayes Factor Comparison: This method directly compares the marginal likelihood of the data under competing models (MSC vs. concatenation). The study of 47 phylogenomic datasets found that Bayes factors consistently favored the MSC model over concatenation [2].
Tests of Congruence: These evaluate the concatenation assumption of topologically congruent gene trees across loci. The widespread rejection of this assumption (38% of loci across datasets) provides indirect support for models like the MSC that explicitly account for gene tree heterogeneity [2].
Simulation Study Design
Robust evaluation of species tree methods requires carefully controlled simulation studies that incorporate biological realism:
Species Tree Simulation: Generate species trees under birth-death processes with varying branching patterns to include potential anomaly zone conditions.
Gene Tree Simulation: Simulate gene trees within the species tree under the MSC model using tools like MS or Seq-Gen, incorporating parameters for population sizes and branch lengths to control ILS levels.
Sequence Evolution: Evolve DNA sequences along gene trees under appropriate substitution models (e.g., GTR+Γ) with varying rates and patterns to mimic empirical data.
Taxon Deletion: Introduce missing data under specific models (e.g., Miid or Mfsc) to evaluate robustness to incomplete genes [55].
Method Application: Apply both coalescent and concatenation methods to the simulated data and compare accuracy using metrics such as Robinson-Foulds distance to the true species tree.
This experimental protocol allows researchers to systematically evaluate method performance under controlled conditions where the true species tree is known, providing insights into consistency and accuracy across different challenging scenarios.
Visualization of Key Concepts and Workflows
The Anomaly Zone Concept
Diagram Title: Gene Tree Discordance in the Anomaly Zone Under MSC
Coalescent Method Workflow
Diagram Title: Coalescent-Based Species Tree Estimation Workflow
Table 3: Research Reagent Solutions for Coalescent-Based Phylogenomics
| Tool/Resource | Function | Application Context |
|---|---|---|
| ASTRAL | Species tree estimation from gene trees via quartet amalgamation | Genome-scale species tree estimation [55] [56] |
| STELAR | Species tree estimation by maximizing triplet consistency | Scalable species tree estimation from gene trees [56] |
| MP-EST | Maximum pseudo-likelihood estimation from triplet frequencies | Species tree estimation with branch lengths [55] [56] |
| *BEAST | Bayesian co-estimation of gene and species trees | Detailed parameter estimation with credible intervals [55] [56] |
| SVDquartets | Species tree estimation directly from sequence data | Analysis without prior gene tree estimation [55] |
| PhyloNet | Phylogenetic network inference | Detecting hybridization and introgression |
- ASTRAL: This method estimates species trees by searching for the tree that agrees with the largest number of quartets induced by the input gene trees. Its polynomial time complexity and statistical consistency make it suitable for large datasets with hundreds of taxa [55] [56].
STELAR: A recently developed method that solves the Constrained Triplet Consensus problem by finding a species tree that maximizes agreement with triplets induced from gene trees. It provides statistical consistency under the MSC model with polynomial time complexity [56].
*BEAST: A Bayesian implementation that co-estimates gene trees and species trees simultaneously. While considered highly accurate, it is computationally intensive and may be impractical for very large datasets [56].
The anomaly zone presents challenging conditions for species tree inference where coalescent-based methods demonstrate their critical advantage over concatenation approaches. Theoretical proofs and empirical validation studies collectively establish that coalescent methods remain statistically consistent even in anomaly zones and under realistic conditions of missing data, while concatenation methods are statistically inconsistent under these conditions. For researchers investigating species relationships where rapid radiations, incomplete lineage sorting, or gene tree discordance are concerns, coalescent-based approaches like ASTRAL, STELAR, and MP-EST provide robust frameworks for accurate phylogenetic inference. As phylogenomic datasets continue to grow in size and complexity, the statistical consistency guarantees of coalescent methods make them indispensable tools for resolving challenging evolutionary relationships.
| Study System (Clade) | Primary Source of Discordance | Impact on Concatenation | Reference |
|---|---|---|---|
| Giant Cockroaches (Blaberidae) | Incomplete Lineage Sorting (ILS) in rapid radiation | Produced anomalous species tree; coalescent method required | [57] |
| Asian Lappula (Boraginaceae) | Hybridization & ILS | Led to polyphyly and significant gene tree conflict | [58] |
| Allium subg. Cyathophora (Plants) | ILS & Reticulate Evolution | Caused extensive conflict between nuclear and plastid trees | [59] |
| Tulipa (Tulips) | Pervasive ILS & Introgression | Prevented reliable resolution of deep relationships | [10] |
| Oak Family (Fagaceae) | Ancient Hybridization & ILS | Caused sharp cyto-nuclear discordance | [18] |
The fundamental assumption of the concatenation approach in phylogenomics—that all genes share an identical evolutionary history—is increasingly being rejected by empirical evidence across diverse lineages. Concatenation combines sequence data from multiple genes into a single "supermatrix" for analysis, implicitly assuming that gene tree-species tree discordance is negligible. Recent studies consistently demonstrate that this assumption is frequently violated due to pervasive biological processes like Incomplete Lineage Sorting (ILS) and hybridization, leading to erroneous phylogenetic inferences when concatenation is applied [57] [18].
Quantitative Evidence of Discordance Drivers
Table: Relative Contributions to Gene Tree Variation in Fagaceae (Oak Family)
| Source of Variation | Contribution (%) | Explanation |
|---|---|---|
| Gene Tree Estimation Error (GTEE) | 21.19% | Error from analytical processes and limited phylogenetic signal |
| Incomplete Lineage Sorting (ILS) | 9.84% | Retention of ancestral polymorphisms during rapid speciation |
| Gene Flow (Hybridization) | 7.76% | Introgression of genetic material between lineages |
This decomposition analysis, conducted on nuclear gene trees from the oak family, reveals that while analytical error is a significant factor, biological processes (ILS and gene flow) collectively account for nearly a fifth of all gene tree variation, fundamentally violating concatenation's core assumption [18].
Detailed Experimental Protocols from Key Studies
Protocol: Resolving Rapid Radiation in Giant Cockroaches
- Research Objective: To evaluate the prevalence of gene tree-species tree discordance in the rapidly radiating Blaberidae cockroach family and compare the performance of concatenation versus coalescent methods [57].
- Dataset: Genomic data from multiple species of Blaberidae.
- Gene Tree Inference: For each individual gene, maximum likelihood trees were inferred under three different evolutionary models of varying complexity:
- GTR: The General Time Reversible nucleotide model.
- FMutSel0: A codon model that uses a single parameter (omega) to model selection.
- SelAC: A more complex, selection-based codon model that explicitly models stabilizing selection for optimal amino acid sequences [57].
- Species Tree Inference:
- Concatenation: All genes were combined into a single supermatrix and analyzed using RAxML.
- Coalescent: Individual gene trees were used as input for the coalescent-based species tree inference in ASTRAL, which explicitly accounts for ILS [57].
- Key Finding: Despite moderate to low levels of gene tree discordance, the concatenation approach failed to recover the correct species tree for the anomalous radiation. The coalescent-based species tree was less discordant with the individual gene trees [57].
Protocol: Dissecting Cyto-Nuclear Discordance in Oaks (Fagaceae)
- Research Objective: To tease apart the biological and analytical factors causing phylogenetic discordance among nuclear, chloroplast (cpDNA), and mitochondrial (mtDNA) genomes [18].
- Dataset:
- Nuclear: 2,124 nuclear loci from 122 individuals (90 species).
- Chloroplast: Data from a previous study.
- Mitochondrial: A newly assembled reference genome for Castanopsis eyrei, used to call mitochondrial SNPs from sequencing reads [18].
- Phylogenetic Analysis:
- Separate phylogenies were reconstructed for each genome (nuclear, cpDNA, mtDNA) using both Maximum Likelihood (IQ-TREE) and Bayesian Inference (MrBayes).
- Incongruences between the resulting trees were identified.
- Discordance Decomposition:
- The relative contributions of Gene Tree Estimation Error (GTEE), Incomplete Lineage Sorting (ILS), and gene flow to nuclear gene tree variation were quantified.
- Genes were categorized as "consistent" or "inconsistent" based on their phylogenetic signals.
- Key Finding: The cpDNA and mtDNA trees divided Fagaceae into New World and Old World clades, a pattern that sharply conflicted with the nuclear genome tree. This cyto-nuclear discordance was attributed to ancient hybridization. The study quantified that gene flow and ILS were significant contributors to gene tree variation [18].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table: Key Reagents and Tools for Phylogenomic Conflict Research
| Item Name | Function / Application | Example Use Case |
|---|---|---|
| ASTRAL | Coalescent-based species tree inference from gene trees. Infers a species tree that minimizes deep coalescences, accounting for ILS. | Inferring the species tree for giant cockroaches despite gene tree discordance [57]. |
| D-Statistics (ABBA-BABA) | Test for gene flow (introgression) between taxa by quantifying allele sharing patterns. | Detecting ancient hybridization among Tulipa, Amana, and Erythronium genera [10]. |
| Quartet Sampling (QS) | Assesses branch support and distinguishes among biological (ILS, gene flow) and analytical causes of discordance. | Evaluating support and conflict for relationships in Lappula and Allium [59] [58]. |
| Phylogenetic Networks (e.g., MSCquartets) | Reconstructs evolutionary relationships that are not strictly tree-like, visualizing hypothesized hybridization events. | Inferring allopolyploid hybridization events in Lappula [58]. |
| SelAC / FMutSel0 Models | Complex codon models that incorporate selection into phylogenetic estimation, potentially reducing systematic error in gene trees. | Testing if biologically realistic models improve gene tree estimation in cockroaches [57]. |
| HybPiper / Easy353 | Bioinformatics pipelines for target sequence capture data assembly, used to recover hundreds of nuclear loci for phylogenomics. | Assembling 262-353 single-copy nuclear genes for Lappula phylogeny [58]. |
Workflow for Phylogenomic Conflict Analysis
The following diagram illustrates the general experimental and analytical workflow for investigating phylogenetic discordance, as applied in the cited studies:
Workflow for Investigating Phylogenomic Conflict. This diagram summarizes the key steps for evaluating sources of phylogenetic tree discordance, from data generation to diagnosis of biological causes like ILS and introgression.
Comparative Analysis of Coalescent vs. Concatenation Performance
Empirical studies consistently demonstrate that the multi-species coalescent model, which explicitly accounts for ILS, often provides a more biologically realistic framework than concatenation when analyzing genomic-scale data.
- Anomaly Zones and Rapid Radiations: In groups like giant cockroaches (Blaberidae), phylogenies contain "anomaly zones"—regions of parameter space where the most likely gene tree differs from the species tree due to ILS. One study identified an anomaly zone spanning 10 backbone nodes. Within these zones, concatenation is statistically inconsistent and can converge on an incorrect species tree with high support, whereas coalescent methods remain consistent [57].
- Hybridization and Reticulate Evolution: In plant genera like Allium, Lappula, and Tulipa, phylogenomic analyses reveal that extensive gene tree incongruence is driven by a combination of ILS and hybridization [59] [58] [10]. Concatenation forces this complex history into a single, bifurcating tree, obscuring the true reticulate evolutionary history. Coalescent-based networks or methods that test for introgression (like D-statistics) are required to identify and confirm these hybridization events.
- The "Concatalescence" Pitfall: A critical methodological issue arises when exons from the same gene, separated by long intronic regions where recombination can occur, are concatenated before coalescent analysis. This "concatalescence" approach violates the coalescent assumption of free recombination between loci and no recombination within them, potentially leading to conflicting and unreliable results [60]. Proper locus selection is therefore crucial for accurate coalescent analysis.
The accurate reconstruction of species trees represents a fundamental goal in evolutionary biology, with profound implications for understanding biodiversity, tracing adaptation, and informing conservation. For decades, the concatenation approach—which combines multiple genes into a single supermatrix for analysis—dominated phylogenetic inference. This method assumes that a single evolutionary history underlies all genes, an assumption frequently violated by pervasive biological processes like introgression and incomplete lineage sorting (ILS). Coalescent model-based approaches provide a statistical framework that explicitly accounts for the fact that different genes can have distinct evolutionary histories. While introgression, the transfer of genetic material between species through hybridization, was once considered a confounding anomaly, modern genomic studies reveal it to be a widespread force shaping genomes across the tree of life, from plants to bacteria [61] [11].
This guide provides a comparative analysis of concatenation versus coalescent methods for species tree inference in the presence of introgression. We synthesize current experimental evidence and methodological advances to demonstrate how coalescent models, coupled with new analytical tools, enable researchers to disentangle complex evolutionary signals and reconstruct more accurate species histories, even when gene flow has occurred.
Mechanistic Foundations: How Introgression and ILS Shape Genomic Landscapes
Biological Processes Creating Phylogenetic Discordance
- Incomplete Lineage Sorting (ILS): A phenomenon where ancestral genetic polymorphisms persist through multiple speciation events, causing closely related species to share alleles not by recent gene flow but by deep ancestral descent. ILS is prevalent in rapid radiations with short internodes and large ancestral populations [10] [62].
- Introgression: The transfer of genetic material between distinct species through hybridization and backcrossing. This process introduces alleles with foreign evolutionary histories into a recipient genome, creating phylogenetic patterns that conflict with the majority of the genome [61] [36].
- Deep Coalescence: When gene lineages fail to coalesce (find a common ancestor) within the time frame of successive speciation events, resulting in gene trees that differ from the species tree [12].
While both ILS and introgression cause gene tree-species tree discordance, they stem from fundamentally different mechanisms. ILS involves the passive retention of ancestral variation, whereas introgression requires active transfer between diverged lineages. Disentangling their effects is crucial for accurate phylogenetic inference.
Visualizing Phylogenetic Discordance Mechanisms
The following diagram illustrates how ILS and introgression create conflicting signals between gene trees and the species tree.
Methodological Comparison: Concatenation vs. Coalescent Approaches
Core Methodological Differences
The debate between concatenation and coalescent methods centers on how they handle gene tree heterogeneity. Concatenation combines all aligned sequences into a single supermatrix, assuming a common evolutionary history across all loci. This approach can be misled by heterogeneous phylogenetic signals, treating discordance as noise rather than meaningful biological signal. In contrast, multispecies coalescent (MSC) models estimate individual gene trees first, then reconcile them into a consensus species tree while accounting for ILS. This approach explicitly models gene tree heterogeneity, providing a more realistic representation of evolutionary history [12] [10].
Modern implementations of coalescent-based phylogenetics include methods like STAR-BEAST for phylogenetic reconstruction and ASTRAL for species tree estimation from gene trees. These tools incorporate population genetic parameters that enable them to distinguish between shared ancestral polymorphisms (ILS) and recent gene flow (introgression) [12] [10].
Experimental Evidence from Empirical Studies
Recent phylogenomic studies across diverse taxa provide compelling evidence for the superiority of coalescent approaches when introgression is present:
- Wingnut Trees (Pterocarya): Genomic analyses of three Chinese wingnut species revealed substantial past introgression between P. hupehensis and P. macroptera, with introgressed regions containing genes related to environmental adaptation (TPLC2, CYCH;1, LUH) and exhibiting lower genetic load with higher genetic diversity [61].
- Brake Ferns (Pteris): Multispecies coalescent analysis of chloroplast SNPs uncovered deep coalescence and incomplete lineage sorting within this genus, with statistical tests (D-statistics) confirming genetic admixture and introgression that confounded simpler concatenation approaches [12].
- Tulips (Tulipeae): Transcriptome sequencing of 46 species revealed pervasive ILS and reticulate evolution among Amana, Erythronium, and Tulipa genera. Coalescent simulations combined with D-statistics and QuIBL analyses demonstrated that neither ILS nor introgression alone could explain the observed phylogenetic discordance [10].
- Goldenhorn (Xanthoceras): Investigation of pronounced cyto-nuclear discordance in Sapindaceae revealed ancient introgression approximately 16% of genetic material from ancestral Sapindoideae lineages into Xanthoceras, occurring during the Paleogene (~57.9 Mya). Coalescent simulations determined ILS alone was insufficient to explain the observed conflict [36].
Table 1: Quantitative Comparison of Concatenation vs. Coalescent Performance in Empirical Studies
| Study System | Data Type | Concatenation Resolution | Coalescent Resolution | Key Discordance Factor |
|---|---|---|---|---|
| Pterocarya (Wingnuts) [61] | Whole-genome | Incomplete, masked introgression | Identified adaptive introgressed regions | Ancient introgression between species |
| Pteris (Brake Ferns) [12] | Chloroplast SNPs (matK, rbcL) | Incongruent gene trees | Resolved deep coalescence | ILS and genetic admixture |
| Tulipeae (Tulips) [10] | Transcriptome (2,594 nuclear genes) | Unresolved polytomies | Confirmed subgeneric monophyly | Pervasive ILS and reticulation |
| Xanthoceras (Goldenhorn) [36] | Transcriptome & plastomes | Cyto-nuclear discordance | Detected ancient cross-subfamily introgression | Ancient introgression (~16% genome) |
| Aspidistra [62] | Transcriptome | Morphological incongruence | Identified convergent evolution | ILS and positive selection |
Analytical Framework: Detecting and Interpreting Introgression
Methodological Toolkit for Introgression Research
Advanced statistical methods have been developed specifically to detect and quantify introgression from genomic data:
- D-Statistics (ABBA-BABA Tests): Used to detect signatures of introgression by measuring allele sharing patterns between species beyond what is expected from ancestral polymorphism [12] [10].
- PhyloNetworks Modeling: Implements network-based approaches to visualize and quantify reticulate evolutionary relationships, representing species histories as networks rather than strictly bifurcating trees [10] [36].
- Site Concordance Factors (sCF): Measures the percentage of decisive alignment sites supporting a particular branch in the phylogeny, helping to identify regions of the genome with conflicting evolutionary histories [10].
- Gene Genealogy Interrogation (GGI): A hypothesis-testing framework for phylogenomics that systematically evaluates conflict between gene trees and species trees [62].
Integrated Workflow for Coalescent-Based Phylogenetics
A modern phylogenomic analysis investigating introgression typically follows an integrated workflow that combines multiple analytical approaches, as diagrammed below.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2: Key Research Reagents and Computational Tools for Coalescent-Based Phylogenetics
| Tool/Reagent | Category | Primary Function | Application Example |
|---|---|---|---|
| BEAST/STAR-BEAST [12] | Software Package | Bayesian phylogenetic analysis using coalescent models | Multispecies coalescent analysis of Pteris phylogeny |
| ASTRAL | Software Package | Species tree estimation from gene trees | Handling incomplete lineage sorting in large datasets |
| PhyloNet [36] | Software Package | Inference and visualization of phylogenetic networks | Detecting ancient introgression in Xanthoceras |
| HyDe [36] | Software Package | Detection of hybridization and introgression | Testing for ghost introgression in Sapindaceae |
| Transcritome Sequencing | Wet Lab Method | Genome-wide gene expression data for non-model organisms | Phylogenomic analysis of Tulipa and Aspidistra [10] [62] |
| Universal Angiosperms353 | Probe Set | Targeted sequencing of conserved nuclear genes | Broad phylogenetic sampling of Sapindaceae [36] |
| D-Statistics | Analytical Method | Test for introgression using allele patterns | Detecting admixture in Pteris and Tulipeae [12] [10] |
| PhyParts [36] | Software Package | Analysis of gene tree concordance and conflict | Quantifying cyto-nuclear discordance in Sapindaceae |
The integration of coalescent models with genomic data has fundamentally transformed our ability to reconstruct species trees in the face of widespread introgression. While concatenation approaches remain useful for initial phylogenetic estimations, particularly in data-rich contexts with minimal discordance, coalescent-based methods provide a more statistically rigorous framework for accommodating the heterogeneous evolutionary histories that characterize real genomic datasets.
Future methodological developments will likely focus on integrating summary statistics, probabilistic modeling, and supervised learning approaches to further enhance the detection of introgressed loci [63]. As genomic datasets continue expanding across diverse taxa, coalescent models will play an increasingly vital role in deciphering the complex interplay between diversification, introgression, and adaptation that has shaped the patterns of biodiversity we observe today.
For researchers investigating non-model organisms with complex evolutionary histories, transcriptome sequencing combined with multispecies coalescent analysis represents a particularly powerful approach, as demonstrated in studies of Tulipa, Aspidistra, and Xanthoceras [10] [62] [36]. This methodology provides the necessary genomic depth while remaining computationally tractable, offering insights into both phylogenetic relationships and the functional genomic basis of adaptation.
Conclusion
The paradigm in phylogenomics has decisively shifted toward model-based approaches that explicitly account for population-level processes. Empirical evidence consistently shows that the multispecies coalescent model provides a more adequate fit for genomic data than concatenation, which is frequently rejected due to its unrealistic assumption of topologically congruent gene trees. While both ILS and introgression are pervasive sources of conflict, the MSC framework, especially when extended to phylogenetic networks, offers a powerful and statistically consistent path to the species tree. Future directions involve the integration of additional biological complexities, such as continuous gene flow and selection, into coalescent-based models. For biomedical research, adopting these robust phylogenetic methods is crucial for accurately reconstructing the evolutionary history of gene families, pathogen lineages, and host populations, thereby providing a reliable foundation for comparative genomics and drug discovery.
References
- 1. Gene tree species tree incongurence [https://theg-cat.com/tag/gene-tree-species-tree-incongurence/]
- 2. The Multispecies Coalescent Model Outperforms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7302055/]
- 3. Dealing with incongruence in phylogenomic analyses - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2607408/]
- 4. Gene trees and species trees: irreconcilable differences - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3526438/]
- 5. The Implications of Incongruence between Gene Tree and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9366463/]
- 6. Complex Models of Sequence Evolution Improve Fit, But ... [https://pubmed.ncbi.nlm.nih.gov/39392926/]
- 7. Incomplete lineage sorting [https://en.wikipedia.org/wiki/Incomplete_lineage_sorting]
- 8. Importance of incomplete lineage sorting and introgression ... [https://www.nature.com/articles/hdy201672]
- 9. Reticulate evolutionary history and extensive introgression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5146328/]
- 10. Incomplete lineage sorting and introgression among genera ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12039212/]
- 11. Introgression impacts the evolution of bacteria, but species ... [https://www.nature.com/articles/s41467-025-64947-1]
- 12. A reappraisal of chloroplast genetic SNPs in phylogeny ... [https://www.sciencedirect.com/science/article/abs/pii/S2452014425000974]
- 13. Chapter 4 Phylogenetic Tree Visualization [https://yulab-smu.top/treedata-book/chapter4.html]
- 14. 4 Visualization and annotation of phylogenetic trees: ggtree [https://guangchuangyu.github.io/ggtree-book/chapter-ggtree.html]
- 15. Adaptive Introgression as an Evolutionary Force: A Meta ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12181397/]
- 16. Plant chromosome engineering - past, present and future [https://pubmed.ncbi.nlm.nih.gov/37984056/]
- 17. Common Methods for Phylogenetic Tree Construction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11117635/]
- 18. Teasing apart the sources of phylogenetic tree discordance ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-025-06963-3]
- 19. Concatenation and model testing [https://evomics.org/wp-content/uploads/2024/01/partitioning_and_concatenation.html]
- 20. Multispecies coalescent and its applications to infer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8692950/]
- 21. A multispecies coalescent model for quantitative traits [https://elifesciences.org/articles/36482]
- 22. Challenges in Species Tree Estimation Under the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5161269/]
- 23. Multispecies coalescent process [https://en.wikipedia.org/wiki/Multispecies_coalescent_process]
- 24. Incomplete lineage sorting and introgression among genera ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-025-02204-z]
- 25. Implementing and testing the multispecies coalescent model [https://www.sciencedirect.com/science/article/pii/S1055790315003309]
- 26. The performance of coalescent-based species tree estimation ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-4619-8]
- 27. Resolving phylogenetic conflicts in Pandanales: the dual ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1511582/full]
- 28. Detecting Introgression in Shallow Phylogenies [https://pmc.ncbi.nlm.nih.gov/articles/PMC12485364/]
- 29. Introgression detection - Evolution and Genomics [https://evomics.org/learning/population-and-speciation-genomics/2025-population-and-speciation-genomics/introgression-detection/]
- 30. Phylogenomic analyses re-evaluate the backbone of ... [https://www.sciencedirect.com/science/article/abs/pii/S1055790325000107]
- 31. Teasing apart the sources of phylogenetic tree discordance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12269116/]
- 32. Molecular phylogeny and biogeography of the fern genus Pteris ... [https://pubmed.ncbi.nlm.nih.gov/24908681/]
- 33. PhyloScape: interactive and scalable visualization platform for ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06203-3]
- 34. Coalescence vs. concatenation: Sophisticated analyses vs. ... [https://www.sciencedirect.com/science/article/abs/pii/S1055790315001487]
- 35. Phylogenomic approach reveals strong signatures of ... [https://www.sciencedirect.com/science/article/pii/S1055790321001330]
- 36. Unraveling the Ancient Introgression History of ... [https://www.mdpi.com/1422-0067/26/4/1581]
- 37. Improved robustness to gene tree incompleteness, ... [https://pubmed.ncbi.nlm.nih.gov/40000439/]
- 38. Robust regression rescues poor phylogenetic decisions [https://bmcecolevol.biomedcentral.com/articles/10.1186/s12862-025-02451-2]
- 39. Coalescence vs. concatenation: Sophisticated analyses vs. ... [https://pubmed.ncbi.nlm.nih.gov/26002829/]
- 40. Universal orthologs infer deep phylogenies and improve ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-025-02328-2]
- 41. Disentangling Sources of Gene Tree Discordance in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7875436/]
- 42. Phylogenomic Discordance is Driven by Wide-Spread ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11906154/]
- 43. Localized Phylogenetic Discordance Among Nuclear Loci ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.850521/full]
- 44. Incomplete lineage sorting and long-branch attraction ... [https://www.frontiersin.org/journals/ecology-and-evolution/articles/10.3389/fevo.2024.1243221/full]
- 45. Leveraging shared ancestral variation to detect local ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1010155]
- 46. Extensive Genome-Wide Phylogenetic Discordance Is Due to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8233498/]
- 47. Estimation of Cross-Species Introgression Rates Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9087891/]
- 48. Revealing the selection history of adaptive loci using genome ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-4447-x]
- 49. An HMM-Based Comparative Genomic Framework for ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003649]
- 50. Bayes factors vs. P-values [https://www.sciencedirect.com/science/article/abs/pii/S0261517717302601]
- 51. Bayes Factor and Posterior Probability [https://pubmed.ncbi.nlm.nih.gov/26212520/]
- 52. Unraveling historical introgression and resolving phylogenetic ... [https://bmcecolevol.biomedcentral.com/articles/10.1186/s12862-018-1197-y]
- 53. Category Archives: Bayes-Factors [https://replicationindex.com/category/bayes-factors/]
- 54. Bayesian model selection using test statistics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2760999/]
- 55. The performance of coalescent-based species tree ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5998899/]
- 56. STELAR: a statistically consistent coalescent-based species ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6519-y]
- 57. Concatenation fails to describe the anomalous radiation of ... [https://bmcecolevol.biomedcentral.com/articles/10.1186/s12862-025-02409-4]
- 58. Phylogenomics provides new insight into the phylogeny ... [https://www.sciencedirect.com/science/article/pii/S1055790325000788]
- 59. Phylogenomic Analyses Reveal Species Relationships and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12252102/]
- 60. Concatenation versus coalescence versus “concatalescence” [https://pmc.ncbi.nlm.nih.gov/articles/PMC3612616/]
- 61. Genomic introgression underlies environmental adaptation ... [https://www.sciencedirect.com/science/article/pii/S2468265925000563]
- 62. Impact of incomplete lineage sorting and natural selection on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12528535/]
- 63. Review Decoding genomic landscapes of introgression [https://www.sciencedirect.com/science/article/pii/S0168952525001660]