Decoding Epistasis: A Practical Guide for Evaluating Genetic Interactions in Combinatorial Mutant Libraries
This article provides a comprehensive resource for researchers and drug development professionals on the evaluation of epistatic effects in combinatorial mutant libraries.
Decoding Epistasis: A Practical Guide for Evaluating Genetic Interactions in Combinatorial Mutant Libraries
Abstract
This article provides a comprehensive resource for researchers and drug development professionals on the evaluation of epistatic effects in combinatorial mutant libraries. It covers the foundational principles of epistasis, from its molecular origins in protein active sites to its impact on evolutionary trajectories and fitness landscapes. The content details cutting-edge methodological approaches, including high-throughput experimental screens, computational design tools like FuncLib, and machine learning strategies. It further addresses key challenges in epistasis detection, such as combinatorial explosion and distinguishing specific from global epistasis, and offers troubleshooting and optimization strategies. Finally, it presents a comparative analysis of validation frameworks and discusses the translational implications of epistasis research for protein engineering and therapeutic development.
The Fundamentals of Epistasis: From Molecular Mechanisms to Rugged Fitness Landscapes
Epistasis, a concept fundamental to genetics, describes the phenomenon where the effect of a genetic mutation depends on the genetic background in which it occurs. In more precise terms, it is identified when the combined effect of two or more mutations deviates from the expected additive effect of their individual contributions [1]. In the context of protein evolution and biochemistry, epistasis arises from physical and functional interactions among amino acid residues that determine a protein's three-dimensional structure, stability, and biological activity [1]. The structure, function, and evolution of proteins are fundamentally governed by these interactions, which cause the phenotypic effect of changing an amino acid to depend on the specific sequence of the protein into which the mutation is introduced [1].
Understanding epistasis is critical for multiple scientific domains. For evolutionary biology, it determines the possible trajectories available to an evolving protein, potentially restricting paths or opening new ones to sequences and functions that would otherwise be inaccessible [1]. In protein engineering and drug development, epistasis can explain why attempts to leverage natural sequence variation or experimental observations to predict mutational effects often fail, as mutations that confer a function in one protein may have no effect or be deleterious in a related protein [2] [3]. Research has revealed that epistasis is pervasive in protein evolution, with recent studies characterizing its prevalence, biochemical mechanisms, and evolutionary impacts [1].
Classifying Epistatic Interactions: Mechanisms and Evolutionary Impacts
Epistatic interactions can be categorized into distinct types based on their underlying mechanisms and effects on evolutionary processes. Two broad classes emerge from recent research, each with different physical origins and evolutionary consequences.
Table 1: Classification of Epistasis Types and Their Characteristics
| Type of Epistasis | Mechanistic Basis | Evolutionary Impact | Common Manifestations |
|---|---|---|---|
| Specific Epistasis | Direct and indirect physical interactions between mutations that nonadditively change protein physical properties (conformation, stability, ligand affinity) [1] | Stronger effect on evolutionary rate and outcomes; imposes stricter constraints and modulates evolutionary potential more dramatically; makes evolution more contingent on historical events [1] | Positive sign epistasis (deleterious mutations become beneficial when combined); Negative sign epistasis (double mutants worse than expected) [1] |
| Nonspecific Epistasis | Mutations behave additively regarding physical properties but exhibit epistasis due to nonlinear relationships between physical properties and biological effects/fitness [1] | More moderate effect on evolutionary trajectories; arises from global nonlinearities in genotype-phenotype maps [4] | Diminishing-returns epistasis (beneficial mutations less beneficial in fitter backgrounds); Increasing-costs epistasis (deleterious mutations become more deleterious in fitter backgrounds) [4] |
Beyond these mechanistic classifications, epistasis can also be characterized by its directional effects on fitness and evolutionary accessibility:
- Sign Epistasis: Occurs when a mutation is beneficial in one genetic background but deleterious in another, potentially reversing the direction of selection [1]. This form of epistasis is particularly important as it can create evolutionary dead ends and make evolutionary outcomes dependent on the specific order of mutations.
- Magnitude Epistasis: Describes cases where the magnitude (but not the sign) of a mutation's effect changes across genetic backgrounds, without altering whether it is beneficial or deleterious [1].
The distinction between these forms of epistasis has profound implications for protein evolution. Specific epistasis with sign effects can create strong historical contingencies, where evolutionary outcomes depend critically on which mutations occurred first [1]. In contrast, global patterns of diminishing-returns epistasis appear to predictably shape adaptive landscapes by systematically reducing the benefits of mutations as fitness increases [4].
Quantitative Experimental Approaches for Measuring Epistasis
Deep Mutational Scanning and Combinatorial Libraries
Deep mutational scanning represents a powerful methodological approach for comprehensively characterizing epistatic interactions in proteins. This technique involves creating and phenotyping large libraries of protein variants, typically encompassing many or all single and double amino acid substitutions relative to a starting sequence [1]. By analyzing variants that differ by one or two amino acids from a starting protein, researchers can comprehensively characterize pairwise epistatic interactions within that protein's local sequence neighborhood [1].
In the absence of epistasis, the behavior of double mutants can be predicted with perfect accuracy by adding the effects of their constituent single mutations (R² ≈ 1). On a completely epistatic landscape, the effect of a mutation is completely independent in every background (R² ≈ 0). Experimental results typically reveal an intermediate reality: single mutants predict double mutant behavior moderately well (R² ∼ 0.65–0.75), indicating that epistasis is neither all-pervasive nor negligible [1]. Comprehensive studies, such as one conducted on protein G domain 1 (GB1), found strong deviations from additivity (by a factor >2) in approximately 5% of all pairs of mutations, while weak epistasis (<2-fold deviation) affected about 30% of pairs [1].
Table 2: Key Experimental Methods for Epistasis Research
| Method | Key Features | Applications in Epistasis Research | Representative Study Findings |
|---|---|---|---|
| Deep Mutational Scanning (DMS) | High-throughput functional characterization of large mutant libraries; assesses single and double mutants [1] | Quantifies prevalence and strength of pairwise epistasis; measures distribution of epistatic effects (negative vs. positive) [1] | Negative epistasis outnumbers positive by 3-20x; most deleterious mutations have ≥1 interacting mutation that makes them beneficial/neutral [1] |
| Combinatorial Mutagenesis (20-state) | Tests all 20 amino acid combinations at multiple targeted sites (typically 3-4 sites) [3] [5] | Dissects genetic architecture of functional specificity; identifies main effects, pairwise, and higher-order interactions [3] | In transcription factor DNA-binding domain: dense main and pairwise effects; minimal higher-order epistasis; pairwise epistasis facilitates functional evolution [3] [5] |
| Double-NIL (Double Nearly Isogenic Lines) | Two loci segregate in otherwise isogenic background; measures all 9 genotypic combinations for a QTL pair [6] | Estimates direct and interaction effects for QTL pairs; maps genetic effects to population variance components [6] | Epistasis highly variable but common; major determinant of additive genetic variance; background dependency of allelic effects [6] |
| Transposon Mutagenesis | Tracks fitness effects of insertion mutations across evolved genetic backgrounds [4] | Measures how distribution of fitness effects (DFE) changes during evolution; identifies increasing-costs epistasis [4] | In yeast: deleterious mutations tend to become more deleterious over evolution (increasing-costs epistasis), reducing mutational robustness [4] |
Ordinal Regression for Genetic Architecture Dissection
Recent methodological advances have enabled more sophisticated analysis of epistasis in comprehensive combinatorial datasets. One innovative approach applies ordinal logistic regression to directly characterize the global genetic determinants of multiple protein functions from 20-state combinatorial deep mutational scanning experiments [3] [5]. This method models a variant's genetic score as the sum of main effects (each possible amino acid at each variable site) and epistatic effects (every pair and triplet of sites), with the genetic score determining the probability of a variant falling into different functional categories through an ordinal logistic function [5].
A key advantage of this approach is that it is reference-free—model terms are encoded relative to the global functional average across all genotypes rather than a particular reference sequence [3]. This allows direct assessment of how epistasis affects the distribution of multiple functions across sequence space and their accessibility under different evolutionary scenarios. When applied to a steroid hormone receptor's DNA-binding domain, this method revealed that the genetic architecture of DNA recognition consists of a dense set of main and pairwise effects involving virtually every possible amino acid state in the protein-DNA interface, with higher-order epistasis playing only a minor role [3].
Diagram 1: Experimental workflow for epistasis analysis using combinatorial deep mutational scanning and ordinal regression. The process begins with strategic selection of variable sites and functions to assay, proceeds through library construction and functional characterization, and culminates in genetic architecture modeling.
Evolutionary Consequences of Epistatic Interactions
Epistasis Shapes Evolutionary Trajectories and Outcomes
The presence of epistasis fundamentally alters the evolutionary dynamics and outcomes of protein evolution. When epistasis is present, a mutation may be beneficial in some genetic backgrounds but deleterious or neutral in others, reducing the number of viable evolutionary trajectories through sequence space [1]. This creates strong path dependency, where mutations fixed stochastically early in evolution determine which functional optimum an evolving protein ultimately occupies [1]. These optima may differ not only in primary sequence but also in physical and biological properties, leading to divergent evolutionary outcomes even from similar starting points.
Epistasis can create evolutionary "dead-ends" in sequence space, where a potentially beneficial mutation is not immediately accessible without first traversing through less fit intermediates [1]. In such cases, relaxation of selection or even selection for alternative protein properties may be necessary before trajectories open to superior optima. This phenomenon explains observations where approximately 95% of functional protein variants recovered in high-throughput screens would have been predicted to be nonfunctional based on the effects of single mutations alone [1]. The combinations of mutations, enabled by epistatic interactions, create functionality that would be inaccessible through single mutational steps evaluated in isolation.
Diminishing-Returns and Increasing-Costs Patterns
Microbial evolution experiments have revealed consistent macroscopic patterns of epistasis that shape adaptive landscapes. The most commonly observed form is diminishing-returns epistasis, where beneficial mutations tend to be less beneficial in fitter genetic backgrounds [4]. This pattern explains the widespread observation of declining adaptability in evolving microbial populations—populations adapt more rapidly when they start at lower fitness, with the rate of adaptation decreasing as fitness increases [4]. For example, in the E. coli long-term evolution experiment (LTEE), the rate of fitness increase has declined dramatically and reproducibly across tens of thousands of generations, primarily due to a shift in the distribution of fitness effects of beneficial mutations rather than exhaustion of beneficial mutations [4].
Conversely, increasing-costs epistasis describes the tendency for deleterious mutations to become more deleterious in fitter genetic backgrounds, effectively reducing mutational robustness as populations adapt [4]. This pattern was observed in yeast evolution experiments, where a set of 91 mostly deleterious insertion mutations became, on average, more deleterious over 10,000 generations of evolution in a laboratory environment [4]. This reduction in mutational robustness may represent a trade-off between adaptive optimization and evolutionary resilience.
Diagram 2: Contrasting patterns of global epistasis. The same mutation (X or Y) has different fitness effects depending on the genetic background, demonstrating diminishing-returns for beneficial mutations and increasing-costs for deleterious mutations.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagents and Methods for Epistasis Studies
| Reagent/Method | Function in Epistasis Research | Key Features and Applications |
|---|---|---|
| Combinatorial Mutagenesis Libraries | Systematically tests the functional effects of amino acid combinations at multiple sites [3] | Encompasses all 20 amino acids at selected positions (160,000 variants for 4 sites); reveals genetic architecture of functional specificity [3] |
| SOCoM (Structure-based Optimization of Combinatorial Mutagenesis) | Computationally designs optimized combinatorial libraries based on structural energies [2] | Uses cluster expansion to efficiently assess library-averaged energy potentials; enriches libraries in stable variants while exploring sequence diversity [2] |
| Double Barcoding System (Yeast) | Enables high-throughput phenotyping of diploid mapping populations [7] | Fuses barcodes from both haploid parents to create unique diploid identifiers; allows pooled fitness assays of ~200,000 diploid strains [7] |
| GADGETS (Genetic Algorithm for Detecting Genetic Epistasis) | Statistical method for detecting epistatic SNP-sets in genetic studies [8] | Extends to maternal-fetal genotype interactions; uses evolutionary algorithm to search large candidate SNP spaces (up to 10,000 SNPs) [8] |
| Ordinal Logistic Regression Model | Dissects genetic architecture from combinatorial DMS data [3] [5] | Reference-free modeling relative to global functional average; quantifies main, pairwise, and higher-order effects on multiple functions simultaneously [3] |
The systematic study of epistasis has transformed our understanding of protein evolution and function, revealing both constraints and opportunities that shape evolutionary trajectories. The emerging picture is one of pervasive but structured epistasis, where specific interactions between mutations create historical contingencies, while global patterns of diminishing-returns and increasing-costs epistasis create predictable shifts in adaptive landscapes [1] [4]. Crucially, recent research challenges the simplistic view that epistasis primarily constrains evolution, demonstrating instead that pairwise epistasis can facilitate functional evolution by bringing protein variants with different specificities close together in sequence space [3].
For drug development professionals, these insights have profound implications. Understanding epistatic networks can improve predictions of drug resistance evolution in pathogens and cancer, informing combination therapy strategies that preempt evolutionary escape routes. In protein therapeutic engineering, accounting for epistasis enables more rational design of stable, specific proteins by identifying combinations of mutations that work cooperatively to enhance desired properties. As structural biology and deep mutational scanning continue to advance, integrating epistatic principles into protein design pipelines promises to accelerate the development of more effective biotherapeutics and precision medicines.
In protein evolution, epistasis—the phenomenon where the effect of one mutation depends on the presence of other mutations—is a fundamental determinant of evolutionary trajectories and functional outcomes. Recent research has revealed that epistasis manifests through distinct biochemical mechanisms with different evolutionary implications. This review compares two primary categories of epistatic interactions: direct physical epistasis, resulting from specific atomic-level interactions between amino acid residues, and indirect conformational epistasis, which arises from mutations that alter the distribution of protein conformational states or globally modify biophysical properties [1] [9]. Understanding this distinction is critical for researchers interpreting combinatorial mutant libraries, predicting evolutionary pathways, and engineering proteins with novel functions.
The structure, function, and evolution of proteins are governed by complex networks of interactions among amino acids. The prevalence and strength of these interactions create a "rugged" fitness landscape where evolutionary trajectories become contingent on historical substitutions [1] [10]. This landscape topography directly influences evolutionary predictability, with rugged landscapes potentially trapping populations at suboptimal fitness peaks while smoother landscapes allow convergence on global optima [11]. Disentangling the specific mechanisms behind epistatic interactions thus provides not only fundamental insights into protein biochemistry but also practical advantages for forecasting evolutionary outcomes and rational protein design.
Theoretical Foundations and Key Concepts
Direct Physical Epistasis (Specific Epistasis)
Direct physical epistasis, also termed specific epistasis, occurs when one mutation directly influences the phenotypic effect of another through physical interactions within the protein structure. This form of epistasis typically involves residues in close spatial proximity and operates through atomic-level interactions that nonadditively change the protein's physical properties, including local conformation, binding affinity for specific ligands, or catalytic efficiency [1] [9]. These interactions are often strongly mediated by the protein's three-dimensional architecture, with direct contacts between side chains creating highly specific dependencies.
The evolutionary impact of direct physical epistasis is particularly significant because it can impose strict constraints on accessible evolutionary paths and dramatically modulate evolutionary potential [1]. This makes evolution more contingent on historical events and leaves distinctive marks on protein families. Specific epistasis often manifests as sign epistasis, where a mutation that is beneficial in one genetic background becomes deleterious in another, potentially creating evolutionary traps or dead-ends in sequence space [1] [12].
Indirect Conformational Epistasis (Global Epistasis)
Indirect conformational epistasis, increasingly referred to as global epistasis, describes a phenomenon where mutations modify the effect of many other mutations through nonlinear relationships between physical properties and their biological effects. These interactions typically behave additively with respect to the fundamental physical properties of a protein but exhibit epistasis due to nonlinear mapping from these properties to observable phenotypes or fitness [1] [9]. For example, multiple mutations might contribute additively to protein stability, but their combined effect on fitness becomes nonadditive because stability itself relates nonlinearly to functional output—particularly near stability thresholds [1].
This form of epistasis frequently emerges in allosteric proteins where mutations affect the distribution of conformational states or alter the intricate networks of states inherent to allosteric function [13]. The biophysical basis often involves mutations that collectively shift the equilibrium between protein conformations or affect global properties like folding stability, which subsequently influences the observed activity across multiple sites. Unlike direct epistasis, indirect epistasis tends to produce consistent, predictable patterns such as diminishing-returns epistasis, where beneficial mutations provide smaller advantages in already-optimized backgrounds [4].
Table 1: Comparative Features of Direct and Indirect Epistasis
| Feature | Direct Physical Epistasis | Indirect Conformational Epistasis |
|---|---|---|
| Primary Mechanism | Atomic-level physical interactions between specific residues | Nonlinear mapping from biophysical properties to function; altered conformational equilibria |
| Structural Basis | Residues in close spatial proximity; direct contacts | Distributed effects; often mediated by global properties like stability |
| Interaction Specificity | Highly specific; one mutation affects few others | Nonspecific; one mutation affects many others |
| Evolutionary Impact | Strong path dependency; restrictive constraints | General shifts in distribution of mutation effects |
| Detection Methods | Resample and Reorder (R&R) rank statistics [9] | Global epistasis models; nonlinear regression |
| Prevalence in Proteins | ~5% strong epistasis; ~30% weak epistasis [1] | Widespread; emerges consistently across systems |
Experimental Approaches and Methodologies
High-Throughput Mutagenesis and Deep Mutational Scanning
Deep Mutational Scanning (DMS) has revolutionized the empirical study of epistasis by enabling high-throughput characterization of thousands to millions of protein variants [1] [9]. The core methodology involves creating saturated mutant libraries, expressing these variants, applying functional selections, and using high-throughput sequencing to quantify variant frequencies before and after selection.
Key Experimental Protocol:
- Library Construction: Generate comprehensive mutant libraries using codon-based mutagenesis targeting specific protein regions. For example, studies of GB1 domain examined all 160,000 possible variants across four sites [12], while folA (DHFR) studies characterized ~260,000 variants in a 9-bp region [11].
- Functional Selection: Apply selection pressure relevant to protein function—for enzyme studies, this often involves growth under antibiotic pressure (e.g., trimethoprim for DHFR [11]); for binding proteins like GB1, selection may use affinity capture [12].
- Variant Quantification: Use Illumina sequencing to count variant frequencies pre- and post-selection, enabling fitness calculation based on enrichment/depletion [12] [11].
- Fitness Calculation: Compute relative fitness as log2(frequencypost/frequencypre) normalized to wild-type [12] [11].
This approach generates the comprehensive datasets needed to distinguish direct from indirect epistasis by examining how mutational effects change across diverse genetic backgrounds.
Distinguishing Direct from Indirect Epistasis
The Resample and Reorder (R&R) method provides a powerful statistical framework for identifying direct physical epistasis in the presence of global epistasis [9]. This approach exploits the observation that global epistasis preserves the rank order of mutational effects across genetic backgrounds when the underlying nonlinearity is monotonic.
R&R Protocol:
- Rank Comparison: For each pair of mutations A and B, compare their fitness effects across multiple genetic backgrounds.
- Rank Preservation Test: Under pure global epistasis, the rank order between A and B should remain consistent; significant rank reversals indicate specific epistasis.
- Resampling: Account for measurement noise through statistical resampling to establish significance thresholds.
- Heteroskedasticity Adjustment: Adjust for varying measurement precision across fitness ranges common in sequencing-based assays [9].
This method successfully identifies residue pairs in direct physical contact with accuracy comparable to more complex procedures, without requiring assumptions about the precise form of global epistasis [9].
Biophysical Modeling for Allosteric Proteins
For allosteric proteins like LacI, biophysical modeling offers a superior framework for understanding epistasis by explicitly accounting for conformational equilibria [13].
Key Protocol Elements:
- Dose-Response Profiling: Precisely measure dose-response curves for hundreds of variants with overlapping mutation combinations [13].
- Multi-State Modeling: Parameterize models that account for multiple conformational states (e.g., DNA-bound vs. unbound).
- Parameter Comparison: Contrast biophysical models with phenomenological approaches (e.g., Hill equation models) for fitting accuracy and parameter epistasis [13].
Studies of LacI demonstrate that biophysical models fit extensive mutational data more parsimoniously, with significantly less epistasis required in model parameters compared to phenomenological approaches [13].
Table 2: Key Research Reagents and Solutions for Epistasis Studies
| Reagent/Solution | Function | Example Application |
|---|---|---|
| Codon-Mutagenized Libraries | Comprehensive coverage of sequence space | GB1 (160,000 variants) [12]; folA (~260,000 variants) [11] |
| mRNA Display Systems | In vitro selection of functional variants | Protein binding affinity measurements [12] |
| Illumina Sequencing Reagents | High-throughput variant quantification | Fitness measurement from pre-/post-selection frequencies [12] [11] |
| Trimethoprim Selection Media | Selective pressure for DHFR function | folA (E. coli dihydrofolate reductase) studies [11] |
| IgG-Fc Coated Surfaces | Affinity selection for binding proteins | GB1 domain binding experiments [12] |
| Biophysical Assay Buffers | Precise dose-response measurements | LacI allosteric function profiling [13] |
Empirical Evidence and Comparative Analysis
Case Study: GB1 Domain (Protein G)
The GB1 immunoglobulin-binding domain represents a paradigmatic system for quantifying epistatic interactions. A comprehensive study examining all 204 (160,000) amino acid variants at four sites (V39, D40, G41, V54) revealed extensive epistasis that profoundly influences evolutionary accessibility [12].
Key Findings:
- Rugged Fitness Landscape: The landscape contained numerous fitness peaks, with reciprocal sign epistasis blocking many direct adaptive paths [12].
- Indirect Paths Enable Adaptation: Despite pervasive epistasis, 95% of functional variants required gain and subsequent loss of mutations, demonstrating how indirect paths circumvent evolutionary traps [12].
- Prevalence of Epistasis: Approximately 5% of mutation pairs showed strong epistasis (>2-fold deviation from additivity), while ~30% exhibited weaker epistasis [1].
This high-dimensional analysis demonstrated that evolutionary accessibility depends critically on considering indirect paths through sequence space that would be inaccessible when examining only direct routes.
Case Study: folA (Dihydrofolate Reductase) in E. coli
Analysis of a 9-bp region in folA encompassing ~260,000 variants revealed a highly rugged landscape with 514 fitness peaks, though incorporating experimental error reduced this to 127 significant peaks, all in high-fitness regions [11].
Fluid Epistasis Observation:
- The nature of pairwise epistasis changed dramatically across genetic backgrounds, with higher-order interactions making pairwise relationships "fluid" [11].
- In functional backgrounds, mutation pairs exhibited no epistasis in ~57% of cases, positive epistasis in ~21%, negative epistasis in ~11%, and various forms of sign epistasis in the remainder [11].
- This fluidity highlights the background dependence of epistatic interactions and challenges simplified models of protein evolution.
Case Study: LacI Allosteric Regulation
Systematic analysis of 164 LacI variants with overlapping missense mutations compared biophysical and phenomenological models for explaining epistasis in allosteric proteins [13].
Comparative Findings:
- Biophysical Models: Provided more parsimonious fits with significantly less epistasis in parameters by explicitly accounting for conformational states and binding equilibria [13].
- Phenomenological Models: Offered slightly better predictive accuracy from single-mutation effects alone but required more extensive parameter epistasis [13].
- Implication: The multi-state, multi-dimensional nature of allosteric function fundamentally shapes epistatic interactions, favoring mechanistic models over correlative approaches.
Table 3: Quantitative Comparison of Epistasis Across Protein Systems
| Protein System | Experimental Scale | Direct Epistasis Prevalence | Indirect Epistasis Patterns | Evolutionary Outcomes |
|---|---|---|---|---|
| GB1 Domain | 160,000 variants (4 sites) | ~5% strong; ~30% weak pairwise epistasis [1] | Reciprocal sign epistasis blocks direct paths [12] | Indirect paths enable adaptation (95% variants) [12] |
| folA (DHFR) | ~260,000 variants (9-bp) | Fluid epistasis with background-dependent categories [11] | Highly rugged landscape with 127 fitness peaks [11] | Broad basins of attraction maintain accessibility [11] |
| LacI | 164 variants with overlapping mutations | Reduced in biophysical model parameters [13] | Dominant in phenomenological models [13] | Allosteric regulation shapes epistatic network [13] |
Implications for Protein Evolution and Engineering
The distinction between direct and indirect epistasis has profound consequences for understanding evolutionary dynamics and developing protein engineering strategies.
Evolutionary Contingency and Predictability
Direct physical epistasis creates stronger evolutionary contingency, making outcomes dependent on historically specific substitutions [1] [10]. This "epistatic drift" causes homologs diverging from common ancestors to gradually accumulate different constraints, changing which subsequent mutations are accessible in each lineage [10]. In contrast, patterns of global epistasis—particularly diminishing-returns relationships—impose statistical predictability on evolutionary processes, even when individual mutational effects remain unpredictable [11] [4].
Engineering Implications
For protein engineers, recognizing these distinct epistatic mechanisms informs library design and screening strategies:
- Direct Epistasis: Requires coupled mutations be introduced together, suggesting strategies like site-saturation mutagenesis at interacting positions.
- Indirect Epistasis: Can be leveraged by focusing on mutations that optimize global properties like stability before introducing functional mutations.
The systematic analysis of combinatorial mutant libraries reveals that evolutionary accessibility depends critically on high-dimensional paths through sequence space, providing optimism for protein engineering despite pervasive epistasis [12] [11]. By understanding and distinguishing these epistatic mechanisms, researchers can better predict evolutionary outcomes, design more effective protein engineering strategies, and interpret natural protein variation across diverse biological systems.
The concept of the fitness landscape, introduced by Sewall Wright in 1931, provides a powerful metaphor for understanding evolution. It defines the relationship between genotypes and their reproductive success in a given environment [14]. Within these landscapes, epistasis—the phenomenon where the effect of one mutation depends on the presence of other mutations—creates a complex topography of peaks, valleys, and ridges that fundamentally constrains evolutionary paths [14] [15]. When extensive, epistasis generates a rugged fitness landscape, characterized by multiple fitness peaks separated by valleys of lower fitness, as opposed to a smooth, single-peaked landscape where gradual improvement is always possible [16].
Understanding the structure of these landscapes is not merely an academic exercise; it is crucial for predicting evolutionary outcomes in diverse fields, from the development of antibiotic resistance to the engineering of novel proteins for therapeutic applications [14] [17]. This review compares how rugged fitness landscapes, shaped by epistasis, constrain evolutionary trajectories across different biological systems and experimental approaches, providing a framework for researchers navigating the complex genetics of drug development and protein engineering.
Empirical Insights into Rugged Landscapes
Experimental Mapping of Genotypic Landscapes
The most direct approach to understanding epistasis involves constructing combinatorial mutant libraries and measuring the fitness of each variant. This protocol typically involves: (1) identifying a set of L mutations of interest; (2) generating a library of genotypes containing all possible combinations of these mutations (a 2^L library for binary combinations); and (3) precisely measuring the fitness of each genotype in a relevant environment [14].
A landmark study by Weinreich et al. exemplified this approach, exploring five mutations conferring antibiotic resistance in Escherichia coli. They demonstrated that the ruggedness of the landscape severely constrained viable evolutionary paths to the fitness maximum, with only a few mutational trajectories accessible to natural selection [14]. This finding highlighted the predictive power of empirical landscape mapping.
Table 1: Characteristics of Empirical Fitness Landscapes from Diverse Biological Systems
| Biological System | Key Finding on Ruggedness/Epistasis | Implication for Evolutionary Trajectories |
|---|---|---|
| Transcriptional Repressors (LacI/GalR) | Extremely rugged landscape due to high epistasis, enabling rapid specificity switching [16]. | Constrains paths to prevent adverse regulatory crosstalk (promiscuity) [16]. |
| Antibiotic Resistance (E. coli) | Ruggedness renders only very few mutational paths to the fitness maximum accessible [14]. | Limits predictability; populations may become trapped on local peaks. |
| CRISPR-Cas9 (SaCas9) | Machine learning reveals pervasive epistasis; activity-enhancing mutations are context-dependent [17]. | Rational protein engineering must account for background-dependent effects. |
| Aspergillus niger | Empirical landscapes from random mutations are more rugged than those from selected mutations [14]. | Experimental protocol for obtaining mutations influences observed landscape structure. |
A Case Study in Ruggedness: Transcriptional Repressors
A comprehensive analysis of the LacI/GalR transcriptional repressor family provided a striking example of an extremely rugged landscape. Researchers characterized 1,158 extant and ancestral sequences, revealing that the landscape was not smooth but instead marked by high levels of epistasis [16]. This ruggedness manifested as rapid switches in DNA-binding specificity, even between closely related sequences.
Notably, this ruggedness is not a mere evolutionary impediment; it serves a crucial biological function. The study concluded that the rugged landscape minimizes promiscuity—undesired off-target regulatory crosstalk—in the evolution of new repressors. The landscape is shaped to favor mutations that simultaneously achieve specificity for asymmetric DNA operators and disfavor interactions with other targets, a constraint that inherently creates a complex fitness topography [16].
Diagram 1: Rugged landscape of transcriptional repressors. Epistatic constraints create a landscape where paths leading to promiscuous variants (Mutation C) are blocked by low fitness, funneling evolution toward stable, specific solutions and minimizing off-target effects.
Computational and Analytical Tools for Navigating Epistasis
The Statistical Challenge of Detecting Epistasis
The search for epistasis in genome-wide association studies (GWAS) presents a massive computational challenge due to combinatorial explosion; the number of potential interactions increases exponentially with the number of genetic variants considered [18] [19]. This has led to the development of specialized tools and frameworks.
BiForce is one such tool designed for high-throughput analysis of epistasis. It performs a full pairwise genome scan using efficient computational strategies [18]:
- Bitwise data structures: SNP genotype data are converted into Boolean bit values for memory-efficient storage.
- Boolean bitwise operations: Logical operations (e.g., AND) on bit arrays enable extremely fast calculation of SNP interactions.
- Multithreaded parallelization: This allows for feasible genome-wide scans on single workstations or computer clusters.
Despite these tools, a fundamental challenge remains: most models must make assumptions about the mathematical form of epistatic interactions (e.g., limiting searches to pairwise or three-way interactions) to make the problem tractable. Overcoming these limitations is an active area of research, with emerging approaches using deep neural networks (DNNs) that can, in theory, approximate arbitrary functional relationships without a pre-defined model [19].
A Phenotypic Framework: Fisher's Geometric Model
To infer the properties of underlying fitness landscapes from small empirical samples, researchers often turn to phenotypic models. Fisher's geometric model is a prominent example that projects the vast genotypic space onto a simpler continuous phenotypic space [14]. It assumes phenotypes are under stabilizing selection toward an optimum, that mutational effects are Gaussian in phenotypic space, and that mutations combine additively in this space.
This model solves the problem of high dimensionality and has successfully predicted experimental quantities like the distribution of epistasis coefficients between pairs of mutations [14]. However, a rigorous survey of 26 empirical landscapes across nine biological systems revealed that Fisher's model could only fully explain the landscape structure in three of those systems [14]. This indicates that while highly useful, no single model can universally capture the constraining nature of epistasis across all biological contexts.
Engineering in the Face of Epistatic Constraint
Structure-Based and Machine Learning Approaches
The pervasive nature of epistasis presents a significant challenge for protein engineering. Exploring all possible combinations of mutations even in a focused library is experimentally infeasible. This has spurred the development of computational methods to navigate rugged landscapes more efficiently.
Structure-based Optimization of Combinatorial Mutagenesis (SOCoM) is a method that optimizes libraries directly based on the structural energies of their constituents. It uses a cluster expansion (CE) to transform structure-based energy evaluations into a function that can be efficiently computed and optimized over vast combinatorial spaces, choosing both positions and substitutions to maximize the library's average quality [2].
More recently, Machine Learning (ML)-coupled approaches have shown remarkable efficacy. In one study aimed at engineering the CRISPR-Cas9 genome editor, researchers demonstrated that an ML-guided approach could reduce the experimental screening burden by up to 95% while enriching top-performing variants by approximately 7.5-fold compared to random screening [17]. The workflow involves training a model on a small, experimentally characterized subset of a combinatorial library, then using the model to predict the fitness of all virtual variants in silico, prioritizing the best candidates for further testing.
Table 2: Computational Strategies for Engineering on Rugged Landscapes
| Method | Core Principle | Key Advantage | Application Example |
|---|---|---|---|
| SOCoM [2] | Optimizes libraries based on averaged structural energies of variants using Cluster Expansion. | Focusses experimental effort on libraries enriched for stable, folded proteins. | Engineering GFP, β-lactamase, and lipase A for stability. |
| ML-guided Engineering [17] | Uses machine learning to predict variant fitness from a small training set, enabling in-silico library screening. | Drastically reduces experimental burden (e.g., by 95%) while enriching for high-fitness variants. | Optimizing Cas9 nuclease and base editor activity in human cells. |
| Approximate Bayesian Computation [14] | Statistical framework to fit phenotypic landscape models (e.g., Fisher's) to empirical data, accounting for sampling bias. | Allows inference of underlying landscape properties and meaningful cross-study comparison. | Comparing landscape structure across 26 datasets from nine biological systems. |
Diagram 2: Machine learning-coupled engineering workflow. This resource-efficient approach uses a small amount of experimental data to guide the exploration of vast combinatorial libraries, overcoming the constraint of directly testing all epistatic interactions.
The Genetic and Molecular Toolkit for Epistasis Research
Table 3: Research Reagent Solutions for Epistasis and Fitness Landscape Studies
| Reagent / Solution | Function in Research | Key Application Note |
|---|---|---|
| Combinatorial Mutagenesis Libraries | Systematically tests the fitness effects of mutations in combination. | Essential for empirically mapping epistatic interactions; library design (random vs. focused) impacts findings [14] [2]. |
| Model Organisms (E.g., E. coli, Yeast, Drosophila) | Provides a tractable, genetically manipulable system for high-throughput fitness assays. | Enables large-scale studies of gene-gene interactions; e.g., Drosophila Genetic Reference Panel for behavioral genetics [15]. |
| Software: BiForce [18] | Enables high-throughput pairwise epistasis scans in GWAS data. | Uses bitwise operations for speed; critical for handling combinatorial explosion in genome-wide data. |
| Software: MLDE [17] | Machine Learning-assisted Directed Evolution package for protein engineering. | Reduces experimental screening burden by predicting high-fitness protein variants from limited data. |
| Phenotypic Landscape Models (E.g., Fisher's Model) [14] | Provides a theoretical framework to infer global landscape properties from limited empirical data. | Useful for cross-system comparisons but may not capture full epistatic structure in all biological systems. |
Discussion: Implications for Evolutionary Trajectories and Protein Engineering
The evidence from diverse biological systems consistently demonstrates that epistasis is not a minor nuisance but a fundamental constraint that shapes evolutionary landscapes. Ruggedness, arising from pervasive epistatic interactions, limits the number of accessible evolutionary paths, increases the predictability of some outcomes while trapping populations in local fitness maxima, and can itself be an evolved property to maintain functional specificity, as seen in transcriptional repressors [14] [16].
For researchers in drug development and protein engineering, these principles are paramount. The success of engineering a therapeutic protein or understanding the evolution of drug resistance hinges on acknowledging that mutation effects are not additive but context-dependent. The failure of Fisher's geometric model to explain all landscape structures underscores the necessity of system-specific validation [14]. The most promising path forward lies in leveraging combinatorial mutagenesis with advanced computational methods like structure-based design and machine learning. These approaches provide a "navigational chart" for the rugged fitness landscape, allowing scientists to identify optimal genotypes with resource efficiency, turning a fundamental evolutionary constraint into a manageable engineering parameter.
The quest to understand the genetic architecture of complex traits has evolved from a primary focus on additive genetic effects to a more nuanced appreciation for the pervasive role of genetic interactions, or epistasis. While genome-wide association studies (GWAS) have successfully identified thousands of loci associated with traits and diseases, they have traditionally captured only a fraction of the heritability, leaving a significant portion unexplained. Epistasis—defined as non-additive interactions between genetic loci where the effect of one variant depends on the presence of other variants—represents a crucial component of this missing heritability. The pervasiveness of epistatic interactions challenges the simple additive model of genetic inheritance and has profound implications for understanding evolutionary trajectories, complex disease risk, and protein engineering.
Historically, the detection of epistasis in genome-wide studies has been hampered by formidable computational and statistical challenges. With millions of genetic markers typically assayed in modern studies, testing all possible pairwise or higher-order interactions requires an intractable number of statistical tests, creating a massive multiple testing burden that severely reduces statistical power. Furthermore, traditional exhaustive search methods for detecting epistasis scale poorly with biobank-scale datasets comprising hundreds of thousands of individuals. This review examines how next-generation computational frameworks and experimental approaches in model organisms are overcoming these limitations to reveal the extensive role of epistatic interactions in shaping phenotypic variation, with particular emphasis on comparative performance metrics and methodological innovations driving this paradigm shift.
Methodological Innovations in Detecting Genome-wide Epistasis
Computational Frameworks for Large-scale Epistasis Detection
The evolution of epistasis detection methods has progressed from exhaustive pairwise testing toward more sophisticated statistical frameworks that balance computational efficiency with statistical power. Three principal approaches have emerged as frontrunners in analyzing biobank-scale data:
The Sparse Marginal Epistasis (SME) Test represents a significant advancement by concentrating statistical power on biologically relevant genomic regions. By incorporating functional genomic annotations (e.g., DNase I-hypersensitivity sites, chromatin accessibility data), SME restricts interaction testing to variants within functionally enriched regions, dramatically reducing the multiple testing burden. This method employs a linear mixed model where the combined pairwise interaction effects between a focal SNP and all other variants are estimated simultaneously, using an indicator function to mask interactions outside predefined functional domains [20].
The Marginal Epistasis Test (MAPIT) framework estimates the likelihood of a SNP being involved in any interaction without requiring identification of specific interacting partners. Formulated as a linear mixed model with random effects, MAPIT uses method-of-moments algorithms for variance component estimation. While effectively reducing multiple testing concerns by testing one SNP at a time, its computational complexity scales quadratically with sample size, creating limitations for biobank-scale applications [20].
The Fast Marginal Epistasis Test (FAME) incorporates computational improvements including stochastic trace estimators and optimized matrix multiplication to accelerate the MAPIT framework. Despite these advancements, FAME still requires substantial computational resources for genome-wide applications in large datasets, prompting the development of more efficient alternatives like SME [20].
Table 1: Comparison of Computational Methods for Epistasis Detection
| Method | Statistical Approach | Computational Complexity | Key Advantages | Limitations |
|---|---|---|---|---|
| SME Test | Sparse linear mixed model | 10-90x faster than alternatives | Incorporates functional annotations; Reduced multiple testing | Dependent on quality of functional annotations |
| MAPIT | Linear mixed model | O(JN²) for J SNPs, N individuals | Identifies epistatic SNPs without partner identification | Computationally intensive for biobank data |
| FAME | Stochastic linear mixed model | Moderate improvement over MAPIT | Efficient matrix operations; Stochastic estimation | Still challenging for genome-wide studies |
| Exhaustive Pairwise | Fixed-effects regression | O(J²N) for J SNPs, N individuals | Comprehensive; Identifies specific interacting pairs | Prohibitive multiple testing burden |
Experimental Approaches in Model Organisms
Model organisms provide controlled genetic backgrounds and environmental conditions that facilitate the detection of epistatic interactions often obscured in human studies by greater heterogeneity:
Deep Mutational Scanning (DMS) enables comprehensive functional characterization of combinatorial mutations by systematically assaying all possible amino acid combinations at targeted sites. In a landmark study of the steroid hormone receptor DNA-binding domain, researchers performed a complete scan of 160,000 combinations across four sites, categorizing variants as null, weak, or strong activators on two different DNA response elements. This approach revealed that genetic architecture consists predominantly of main and pairwise effects with minimal higher-order epistasis [21].
Ordinal Logistic Regression Modeling provides a reference-free framework for dissecting 20-state sequence-function relationships from combinatorial DMS data. This method quantifies the main effect of every possible amino acid at each variable site plus epistatic effects for all pairs and triplets, with variant genetic scores determining activation probabilities through an ordinal logistic function. Applied to the steroid receptor DMS data, this approach demonstrated that pairwise epistasis facilitates evolutionary innovation by expanding functional sequence space and enabling specificity switching [21].
Genetic Interaction Mapping in Drosophila utilizes the FlyBase Interactions Browser to visualize enhancement (green) and suppression (red) relationships between genes and alleles. This tool displays interaction networks with query genes shown in brown, direct interactors in dark blue, and secondary interactors in light blue, allowing researchers to explore how genetic interactions shape phenotypic outcomes in a well-characterized model organism [22].
Table 2: Experimental Approaches for Epistasis Detection in Model Organisms
| Method | Organism | Throughput | Key Insights | References |
|---|---|---|---|---|
| Combinatorial DMS | Yeast, mammalian cells | 160,000 variants | Pairwise effects dominate genetic architecture; Higher-order epistasis minimal | [21] |
| Ordinal Regression Modeling | In silico analysis | All 20 amino acids at 4 sites | Epistasis facilitates evolutionary paths; Enables specificity switching | [21] |
| FlyBase Interaction Browser | Drosophila melanogaster | Network-based | Visualizes enhancement/suppression relationships; Organizes genetic interaction data | [22] |
| Protein Fitness Landscapes | Diverse orthologs | Limited by structural data | Epistasis creates rugged fitness landscapes; Constrains evolutionary trajectories | [23] |
Performance Comparison: Quantitative Benchmarks
Computational Efficiency and Scalability
Direct performance comparisons reveal substantial differences in computational efficiency between epistasis detection methods. The SME test demonstrates remarkable speed improvements, operating 10-90 times faster than state-of-the-art alternatives like MAPIT and FAME when analyzing UK Biobank-scale data comprising 349,411 individuals and millions of genetic variants. This efficiency stems from SME's sparse modeling approach, which leverages functional enrichment data to restrict the search space and employs innovative approximations to stochastic trace estimators [20].
The computational advantage of SME becomes increasingly pronounced with larger sample sizes. While MAPIT scales quadratically with sample size (O(N²)), SME's sparse formulation reduces this dependency significantly, making genome-wide epistasis detection feasible in biobank datasets. This scalability enables researchers to detect interactions with smaller effect sizes that would be underpowered in smaller studies, addressing a critical limitation in epistasis research [20].
Statistical Power and Detection Sensitivity
Statistical power represents a crucial metric for evaluating epistasis detection methods. SME demonstrates enhanced power compared to previous approaches by concentrating statistical resources on biologically plausible interactions. In simulation studies, SME maintained appropriate type I error control while detecting a greater proportion of true epistatic interactions compared to MAPIT and FAME, particularly for interactions involving regulatory genomic elements [20].
The ordinal regression approach applied to DMS data achieved exceptional classification accuracy, with >97% concordance in activation class assignment between experimental replicates. This high reproducibility substantially exceeds the correlation of continuous fluorescence measurements (R² = 0.62 for functional variants), highlighting the importance of analytical method selection for detection sensitivity [21].
Molecular Mechanisms and Biological Implications
Structural Basis of Epistatic Interactions
Epistatic interactions originate from fundamental biophysical principles governing protein structure and function. In densely packed protein active sites, mutations often exhibit direct epistasis through physical contacts including electrostatics and van der Waals interactions. For example, a large-to-small mutation may improve substrate fit while creating destabilizing cavities, necessitating compensatory small-to-large mutations at contacting positions [23].
Indirect conformational epistasis occurs when mutations alter protein dynamics or backbone positioning, affecting residues distant from the mutation site. A notable example exists in mammalian hemoglobins, where a histidine-to-proline mutation eliminates a hydrogen bond to a nearby helix, reorienting protein subunits and increasing oxygen affinity. Such long-range epistasis demonstrates how mutations outside active sites can profoundly influence function through allosteric mechanisms [23].
Evolutionary Consequences of Epistasis
Epistasis fundamentally shapes evolutionary trajectories by creating rugged fitness landscapes where optimal genotypes may be separated by valleys of reduced fitness. In the steroid receptor system, research revealed that pairwise epistasis massively expands the number of opportunities for single-residue mutations to switch specificity between DNA targets. Rather than constraining evolution, these interactions facilitate functional innovation by bringing variants with different functions closer together in sequence space [21].
The pervasiveness of epistatic interactions helps explain why intermediate evolutionary forms often appear nonfunctional in ancestral backgrounds. Studies of triosephosphate isomerase demonstrated that disease-causing mutations in humans (Gly122Arg) create unfavorable steric clashes that are tolerated in bacterial orthologs with different compensatory backgrounds (Trp90Lys), illustrating how epistatic relationships shape species-specific genetic vulnerability [23].
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents and Resources for Epistasis Studies
| Resource | Type | Function | Access |
|---|---|---|---|
| SME Test Implementation | Software package | Genome-wide epistasis detection with functional enrichment | [20] |
| FlyBase Interactions Browser | Database with visualization | Genetic and physical interaction data for Drosophila | [22] |
| SBOL Visual Standards | Graphical notation | Standardized symbols for genetic design communication | [24] |
| UCSC Gene Interactions Track | Curated database | Protein-protein and genetic interactions from multiple sources | [25] |
| Ordinal Regression Framework | Analytical method | Dissection of genetic architecture from DMS data | [21] |
| Combinatorial DMS Libraries | Experimental resource | Comprehensive variant libraries for functional assays | [21] |
Discussion and Future Directions
The accumulating evidence from genome-wide studies firmly establishes the pervasiveness of genetic interactions across biological systems. The SME framework demonstrates that epistatic contributions to complex traits become detectable when statistical power is enhanced through functional enrichment and computational efficiency. Similarly, DMS experiments in model organisms reveal that pairwise epistasis represents a fundamental organizing principle of protein genetic architecture, with higher-order interactions playing a comparatively minor role.
These findings have transformative implications for therapeutic development. The rugged fitness landscapes created by epistatic interactions suggest that many disease-causing mutations may be context-dependent, potentially explaining why interventions targeting single pathways often show limited efficacy. Understanding epistatic networks could enable identification of compensatory therapeutic targets that rescue disease phenotypes without directly modifying primary genetic lesions.
Future methodological developments will likely focus on integrating multi-omics data streams to further refine epistasis detection, incorporating protein structural information to predict interaction sites, and developing machine learning approaches that generalize across diverse biological contexts. As these methods mature, their application to increasingly diverse populations and model systems will provide a more comprehensive understanding of how genetic interactions shape biological complexity and disease susceptibility.
The consistent observation that epistasis facilitates rather than constrains evolutionary innovation suggests tremendous untapped potential for engineering proteins with novel functions. By strategically navigating epistatic relationships, protein engineers may bypass evolutionary constraints that have limited natural exploration of sequence space, opening new frontiers in therapeutic protein design and synthetic biology.
Advanced Tools and Workflows: From Library Generation to AI-Driven Analysis
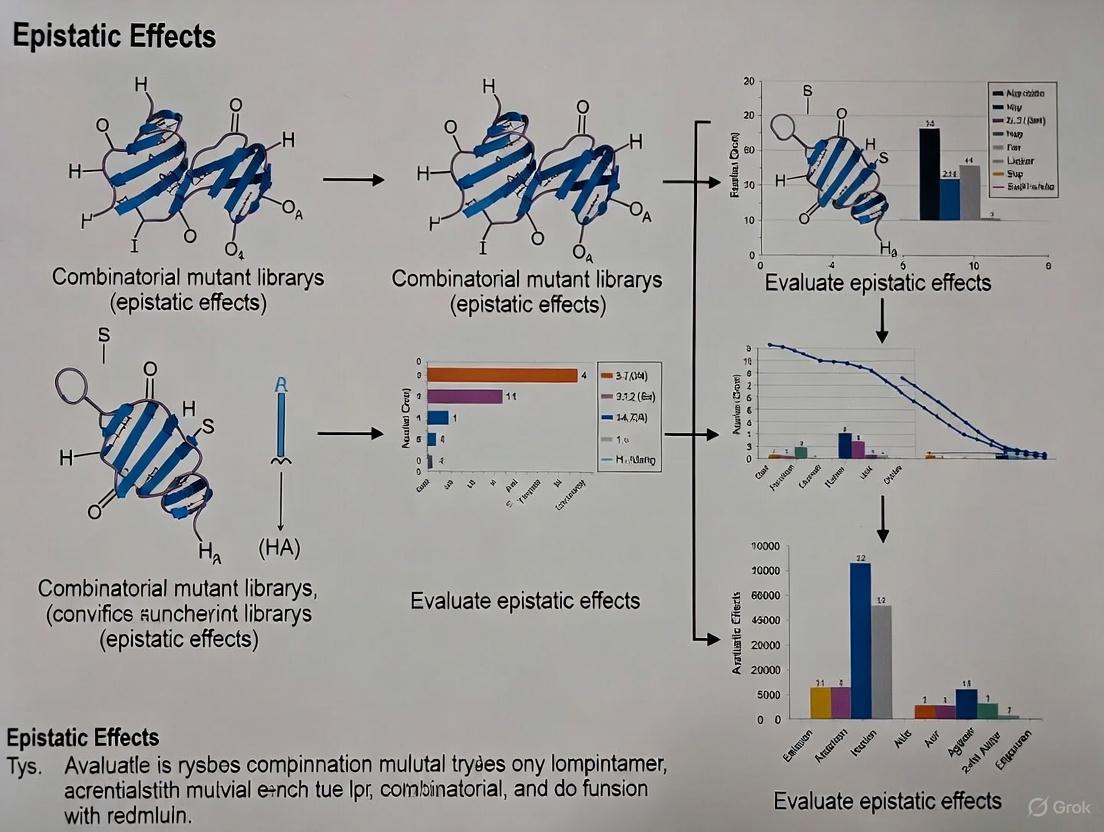
In the field of protein engineering and functional genomics, the construction of high-quality mutant libraries has become a critical component for large-scale functional screening, particularly for studying epistatic effects—the non-additive interactions between mutations that define the ruggedness of fitness landscapes [26] [27]. As synthetic biology advances toward precise design, researchers require methods that offer controlled mutagenesis, comprehensive coverage, high throughput, and operational simplicity to effectively map these complex genetic interactions [28]. The ideal mutagenesis library should possess high mutation coverage, diverse mutation profiles, and uniform variant distribution to enable deep functional phenotyping and reliable detection of epistatic relationships [28].
Understanding epistasis is fundamental to protein engineering, evolutionary biology, and therapeutic development. Epistatic interactions are often observed between mutations in close structural proximity and are enriched at binding surfaces or enzyme active sites due to direct interactions between residues, substrates, and/or cofactors [26]. These interactions can pose substantial challenges for directed evolution campaigns, as beneficial mutations in the context of an initial sequence may not be beneficial in combination with other mutations [26]. This review comprehensively compares modern library construction techniques, with particular emphasis on nicking mutagenesis and its application in generating combinatorial libraries for epistasis research.
Comparison of Library Construction Methodologies
Technical Approaches and Their Applications
Multiple molecular biology techniques have been developed for constructing mutant libraries, each with distinct advantages, limitations, and optimal use cases in epistasis studies.
Nicking Mutagenesis enables construction of combinatorial libraries where multiple user-defined mutations are encoded at defined positions in a sequence [29]. This template-based method utilizes oligonucleotides containing mismatches with the parental DNA sequence that anneal to an ssDNA plasmid template. The protocol can create large combinatorial libraries with near-complete (>99%) coverage of combinatorial mutations at up to 14 different positions (a library size of 2^14 or 16,384 variants) with low carry-over of wild-type parental DNA [29]. Its particular strength lies in circumstances where the desired combinatorial library contains one or two user-defined mutations per codon, making it invaluable for exploring epistatic interactions between known beneficial mutations [29].
Chip-Based Oligonucleotide Synthesis represents a high-throughput, precisely controlled method for constructing mutagenesis libraries [28]. Using array-based DNA synthesis, this approach enables cost-effective and scalable production of diversified oligonucleotide pools that can be assembled into full-length genes. In a demonstration using PSMD10 as a model, researchers constructed a full-length amber codon scanning mutagenesis library with 93.75% mutation coverage [28]. Systematic evaluation of five high-fidelity DNA polymerases revealed that KAPA HiFi HotStart, Platinum SuperFi II, and Hot-Start Pfu DNA Polymerase demonstrated higher amplification efficiency and lower chimera formation rates, making them preferred enzymes for optimized library construction [28].
Error-Prone PCR (epPCR) employs low-fidelity DNA polymerase to introduce random mutations during PCR amplification of a target gene [28] [30]. This method introduces mutations by increasing polymerase error rate, predominantly generating point mutations such as base substitutions, but is inefficient at producing more complex types like insertions or deletions [28]. Although simple to perform, its low and poorly controlled mutation frequency limits both diversity and representativeness, and it exhibits significant mutational preference due to the degeneracy of the genetic code and inherent characteristics of the employed polymerase [28].
Saturation Mutagenesis is a targeted library creation technique designed to systematically replace amino acids at one or more specific positions using synthetic oligonucleotides containing randomized codons flanked by wild-type sequences [28]. While conventional degenerate codons (NNK, where N is A/C/G/T and K is G/T) reduce redundancy from 64 to 32 codons and exclude two of the three stop codons compared to fully degenerate NNN mixtures, they still generate libraries with inherent limitations including residual codon redundancy and uneven amino acid representation [28].
Table 1: Comparison of High-Throughput Mutagenesis Techniques
| Method | Key Features | Library Coverage | Epistasis Applications | Technical Limitations |
|---|---|---|---|---|
| Nicking Mutagenesis | Template-based with mutagenic oligonucleotides; cost-effective; 2-day protocol | >99% for up to 14 positions (16,384 variants) [29] | Combining beneficial mutations; studying pairwise interactions [29] | Limited to ~8 positions with single plasmid; efficiency depends on primer-template mismatches [29] |
| Chip-Based Oligonucleotide Synthesis | High-throughput array synthesis; precise control; PCR assembly | 93.75% mutation coverage demonstrated [28] | Deep mutational scanning; full-length gene variant libraries [28] | Oligonucleotide synthesis errors; chimeric sequence formation during PCR [28] |
| Error-Prone PCR | Simple "sloppy" PCR; requires minimal design | Limited diversity; biased mutation spectrum [28] | Initial diversification; random mutagenesis campaigns [30] | Primarily point mutations; high bias; limited coverage of sequence space [28] |
| Saturation Mutagenesis | Targeted positions; systematic amino acid replacement | Varies with degenerate codon strategy [28] | Active site profiling; single-position comprehensive mutagenesis [28] | Amino acid bias; redundancy; screening burden for multiple sites [28] |
Experimental Performance Metrics
Recent systematic evaluations provide quantitative performance data for various mutagenesis approaches. In nicking mutagenesis, the expected frequency per number of mutations relative to the parental sequence follows a predictable distribution, with the method demonstrating even mutation incorporation across targeted positions [29]. For oligonucleotide-based methods, the efficiency depends critically on polymerase selection, with KAPA HiFi HotStart, Platinum SuperFi II, and Hot-Start Pfu DNA Polymerase demonstrating superior performance in both construction efficiency and chimera formation rate [28].
Analysis of unmapped reads in chip-synthesized libraries highlights key technical factors affecting performance, including oligonucleotide synthesis errors and chimeric sequence formation caused by incomplete extension of DNA polymerase or synthesis across discontinuous templates during PCR [28]. To improve efficiency and fidelity, researchers recommend refining PCR conditions and strengthening oligo synthesis quality control [28].
Table 2: Experimental Performance Data for Library Construction Methods
| Method | Mutation Efficiency | Key Quality Metrics | Optimal Enzymes/Reagents |
|---|---|---|---|
| Nicking Mutagenesis | High incorporation with 5:1 oligo:template ratio [29] | Low wild-type carryover; even mutation distribution [29] | Nt.BbvCI/Nb.BbvCI nicking enzymes; Phusion High-Fidelity Polymerase [29] |
| Chip-Based Oligonucleotide Synthesis | 93.75% coverage in PSMD10 model [28] | Mapping efficiency; dropout variants; chimera formation [28] | KAPA HiFi HotStart, Platinum SuperFi II, Hot-Start Pfu DNA Polymerase [28] |
| Enhanced QuikChange Protocol | Significantly improved over standard method [31] | Full-length plasmid synthesis; transformation efficiency [31] | Primers with extended non-overlapping 3' ends; Pfu DNA polymerase [31] |
Nicking Mutagenesis: Detailed Workflow and Protocol
Experimental Workflow
The nicking mutagenesis protocol enables efficient construction of combinatorial libraries through a series of enzymatic steps that introduce mutations at predefined positions. The method is an extension of multi-site nicking mutagenesis, conceptually similar to Kunkel mutagenesis, wherein mutations are encoded using oligonucleotides containing mismatches with the parental DNA sequence [29].
Nicking Mutagenesis Experimental Workflow
Detailed Protocol and Conditions
Parental DNA Preparation: The protocol begins with plasmid preparation from a dam+ Escherichia coli strain using a commercial miniprep kit. The parental plasmid must contain a BbvCI site (Nt.BbvCI - CCTCAGC; Nb.BbvCI - GCTGAGG), and it is acceptable for the plasmid to contain multiple BbvCI sites only if all are in the same orientation. For each parental sequence, 0.76 pmol (typically 2-3 μg) of dsDNA plasmid must be freshly prepared [29].
Mutagenic Oligonucleotide Design: Mutagenic oligonucleotides contain degenerate codons that allow for either the parental sequence residue(s) or user-defined mutation(s) to be encoded at specific positions. Residues close together (less than 30bp apart) should be incorporated into one oligonucleotide, while residues 30bp or greater apart should be incorporated in different oligonucleotides. Primers should be designed to have 30bp homology arms where possible, with total oligo length not exceeding 100 nucleotides [29].
Template Preparation and Enzymatic Reactions: ssDNA template is prepared through enzymatic degradation of dsDNA using nicking enzymes. The process employs a molar ratio of 5:1 mutagenic oligonucleotides to template, allowing multiple primers to anneal simultaneously. After generation of the complementary strand containing mutations, the ssDNA template is selectively nicked and degraded. The complement of the mutagenic strand is then regenerated, leaving mutagenic plasmid dsDNA. Critical enzymes include Nt.BbvCI and Nb.BbvCI nicking enzymes, exonuclease III, Phusion High-Fidelity DNA Polymerase, and Taq DNA ligase [29].
Transformation and Library Validation: The final product is treated with DpnI to destroy residual parental methylated DNA before transformation into E. coli cells such as XL1-Blue high-efficiency electrocompetent cells. Library quality is assessed by sequencing, with successful implementation yielding >99% coverage of the intended combinatorial mutations [29].
Research Reagent Solutions
Table 3: Essential Research Reagents for Nicking Mutagenesis
| Reagent/Kit | Manufacturer | Function in Protocol | Key Features |
|---|---|---|---|
| Nt.BbvCI & Nb.BbvCI | New England Biolabs | Site-specific nicking of DNA strands | Creates targeted ssDNA templates for mutagenesis [29] |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | Complementary strand synthesis | High fidelity synthesis of mutated DNA strands [29] |
| Taq DNA Ligase | New England Biolabs | Ligation of nicked DNA | Seals nicks in the newly synthesized DNA strands [29] |
| Exonuclease III | New England Biolabs | Degradation of nicked template | Selectively removes original template strands [29] |
| XL1-Blue Electrocompetent Cells | Agilent | Library transformation | High-efficiency transformation of mutant libraries [29] |
| Archer Reveal ctDNA 28 Kit | ArcherDx | Targeted sequencing library prep | UMI incorporation for accurate variant calling [32] |
| NEBNext Direct Cancer HotSpot Panel | New England Biolabs | Targeted enrichment | Hybrid capture/PCR for mutation detection [32] |
| QIAseq Human Actionable Solid Tumor Panel | Qiagen | Targeted sequencing | High library complexity; superior on-target rates (52%) [32] |
Applications in Epistasis Research and Functional Studies
Mapping Genetic Interactions
Combinatorial mutagenesis libraries constructed via nicking mutagenesis and related methods have proven invaluable for quantitative evaluation of epistasis and addressing fundamental questions in molecular evolution [29]. Recent research demonstrates that epistasis plays a facilitating role in functional evolution by increasing the number of functional genotypes and bringing genotypes with different functions closer together in sequence space [27]. This finding counters the traditional view that epistasis primarily constrains evolutionary paths.
In a significant study of an ancient transcription factor, researchers used complete combinatorial variant libraries to demonstrate that changes in function are largely attributable to pairwise rather than higher-order interactions, and that epistasis potentiates, rather than constrains, evolutionary paths [27]. These findings were made possible by reference-free analysis of a 20-state combinatorial dataset, which revealed that epistasis brings genotypes with different functions closer in sequence space and expands the total number of functional sequences [27].
Machine Learning Integration
The generation of high-quality combinatorial libraries has enabled the development of machine learning-assisted directed evolution (MLDE) strategies that can identify high-fitness protein variants more efficiently than typical directed evolution approaches [26]. Systematic analysis of multiple MLDE strategies across 16 diverse protein fitness landscapes revealed that MLDE offers greater advantages on landscapes that are more challenging for directed evolution, especially when focused training is combined with active learning [26].
These MLDE approaches utilize supervised machine learning models trained on sequence-fitness data from combinatorial libraries to capture non-additive epistatic effects. The trained models can then predict high-fitness variants across the entire landscape in a single evaluation round or iteratively in an active-learning fashion [26]. The quality of the training data derived from combinatorial libraries significantly influences model performance, with focused training using zero-shot predictors consistently outperforming random sampling for both binding interactions and enzyme activities [26].
High-throughput library construction techniques, particularly nicking mutagenesis and chip-based oligonucleotide synthesis, have revolutionized our ability to study epistatic interactions in proteins. These methods enable researchers to construct comprehensive combinatorial libraries with high coverage and precision, providing the essential foundation for mapping fitness landscapes and understanding how genetic interactions shape protein evolution and function.
As the field advances, integration of these experimental approaches with machine learning methodologies promises to further accelerate protein engineering efforts and enhance our understanding of sequence-function relationships. The continued refinement of library construction protocols, coupled with appropriate polymerase selection and quality control measures, will ensure researchers can reliably generate the high-quality data needed to unravel the complex epistatic networks that underlie protein function and evolution.
Deep Mutational Scanning (DMS) has emerged as a transformative technology that systematically links genetic variations to phenotypic outcomes, enabling researchers to quantify the effects of thousands of protein variants in a single, highly parallel assay [33]. By combining comprehensive mutant library generation, high-throughput functional screening, and deep sequencing, DMS provides unprecedented resolution in understanding sequence-function relationships [34]. This capability is particularly valuable for investigating epistatic effects—where the functional consequence of one mutation depends on the presence of other mutations—in combinatorial mutant libraries [33]. The application of DMS spans diverse research areas including protein engineering, clinical variant interpretation, vaccine design, and fundamental studies of protein evolution [35] [36]. This guide objectively compares the experimental frameworks and analytical tools for phenotyping combinatorial variants, with emphasis on their utility for epistasis research in therapeutic development contexts.
Core Methodological Components of DMS
A typical DMS experiment comprises three integrated phases: library generation, functional screening, and data analysis [33] [34]. Each phase involves critical decisions that influence the quality and interpretability of the resulting epistasis data.
Library Generation Strategies
The construction of mutant libraries with sufficient diversity and coverage forms the foundation of any DMS study. Current methods offer different trade-offs between completeness, bias, and technical feasibility.
Table 1: Comparison of Mutant Library Generation Techniques
| Method | Mechanism | Advantages | Limitations | Suitability for Combinatorial/Epistasis Studies |
|---|---|---|---|---|
| Error-Prone PCR | Low-fidelity polymerization introduces random mutations [33] | Low cost; technically simple; rapid implementation | Non-uniform mutation spectrum; biases toward specific nucleotide changes; difficult to achieve all amino acid substitutions [33] | Limited due to uncontrolled mutation distribution and inability to target specific residues |
| Oligo Pools with Degenerate Codons | Synthetic oligonucleotides containing NNN/NNK codons [33] | Customizable libraries; reduced bias compared to error-prone PCR; systematic amino acid coverage [33] | Higher cost; uneven amino acid distribution; includes stop codons [33] | Good for targeted single-site saturation mutagenesis |
| Trinucleotide Cassettes (T7 Trinuc) | Pre-synthesized trinucleotides encode specific amino acids [34] | Equiprobable amino acid distribution; eliminates stop codons [34] | Complex synthesis; specialized expertise required | Excellent for precise combinatorial library design |
| CRISPR-Mediated Mutagenesis | Cas9 cleavage with homology-directed repair using oligo donors [34] | Genomic integration; native expression context; barcoding capability [34] | Variable editing efficiency; PAM sequence dependence; potential indels [34] | Excellent for endogenous context studies with barcoded combinatorial variants |
Figure 1: Comprehensive DMS workflow for epistasis research, showing the integration of library generation, functional screening, and data analysis phases.
Functional Screening Modalities
Selection of appropriate phenotyping assays is crucial for capturing relevant functional consequences of combinatorial mutations. The choice of screening method dictates what types of epistatic interactions can be detected and quantified.
Growth-Based Assays
In growth-based selection, cell proliferation is linked to protein function through survival under selective pressure [37]. Variants that enhance function are enriched in the population over time, while deleterious variants are depleted [37] [36]. The functional score is derived from frequency changes measured via deep sequencing across multiple time points [37]. This approach is particularly valuable for studying essential genes and metabolic pathways where epistatic interactions might affect cell fitness.
Binding-Based Assays
Binding assays measure direct molecular interactions using techniques like phage display, yeast display, or mammalian surface display [36]. Variants with altered binding affinity are isolated through affinity selection methods, and their enrichment is quantified relative to the initial library [36]. This method is ideal for mapping epistatic interactions in antibody-antigen complexes or receptor-ligand systems.
Fluorescence-Based Assays
Fluorescence-activated cell sorting (FACS) enables high-throughput screening based on fluorescent reporters linked to protein function [36]. Variants are binned according to fluorescence intensity, and functional scores are calculated from the distribution shifts between pre- and post-selection populations [36]. This approach offers fine resolution for detecting subtle epistatic effects that cause intermediate phenotypic changes.
Analytical Frameworks for Variant Effect Scoring
Accurate quantification of variant effects from sequencing count data presents statistical challenges, especially with the small sample sizes typical of DMS experiments. The choice of analysis tool significantly impacts the reliability of epistasis detection.
Table 2: Comparison of DMS Data Analysis Tools
| Tool | Statistical Approach | Experimental Designs Supported | Key Features | Epistasis Analysis Capabilities |
|---|---|---|---|---|
| Enrich | Log-ratio of variant frequencies [35] [36] | Two-population (input/output) | First dedicated DMS tool; error correction via paired-end reads [35] [36] | Limited to basic variant effect quantification |
| dms_tools2 | Bayesian inference with Dirichlet priors [36] | Two-population | Estimates amino acid preferences per position; specialized for viral proteins [36] | Position-specific preferences enable some epistasis inference |
| Enrich2 | Random-effects model with Poisson variance assumption [37] [36] | Two-population, time-series | Graphical user interface; bin-based FACS data analysis [36] | Time-series data supports dynamic epistasis studies |
| DiMSum | Ratio-based method with overdispersion modeling [37] | One-round selection | Addresses overdispersion in count data; improved error control [37] | Improved single-mutant effect estimates for epistasis baselines |
| Rosace | Bayesian hierarchical model with positional shrinkage [37] | Growth-based, multiple time points | Incorporates positional information; shares information across variants [37] | Enhanced power for detecting positional epistasis |
Figure 2: Variant effect scoring workflow with emphasis on the Rosace framework, which incorporates positional information to enhance epistasis detection in combinatorial libraries.
Advanced Analytical Approaches for Epistasis
The statistical power of epistasis detection in combinatorial libraries depends heavily on the accuracy of individual variant effect estimates. Rosace introduces a Bayesian hierarchical model that leverages positional information to improve effect size estimation, addressing the small-sample-size problem inherent to DMS experiments [37]. By modeling variant-specific scores (βv) as partially constrained by position-level effects (φp(v)), Rosace achieves more robust effect estimates that reduce false discovery rates while maintaining sensitivity [37]. This approach is particularly valuable for combinatorial libraries where multiple mutations within the same protein domain may exhibit coordinated functional effects.
For combinatorial libraries with random multiple mutations, the position-unaware mode of Rosace can be employed when positional assignment is ambiguous [37]. This flexibility ensures that researchers can extract meaningful signals from diverse library designs, though positional information should be leveraged whenever possible to maximize statistical power.
Experimental Design for Epistasis Studies
Multi-Environment DMS
Recent advances in DMS methodology enable the exploration of genotype-phenotype relationships across diverse environmental conditions, revealing context-dependent epistasis. A multi-environment DMS study of a bacterial kinase demonstrated that temperature-sensitive variants distribute across both protein core and surface regions, challenging conventional stability-centric explanations for conditional phenotypes [38]. This approach identified variants with unchanged stability but altered enzymatic activity, highlighting how epistatic interactions can manifest differently under various conditions [38].
Structural Insights from DMS
The integration of DMS data with structural prediction algorithms has created new opportunities for understanding the structural basis of epistasis. DMS-Fold represents a significant innovation that uses residue burial information from single-mutant DMS to refine AlphaFold2 predictions [39]. By calculating burial scores from mutational stability effects (ΔΔG values), DMS-Fold embeds structural constraints that improve accuracy for 88% of protein targets compared to AlphaFold2 alone [39]. This integration is particularly valuable for interpreting epistatic interactions in combinatorial variants by providing structural context for non-additive effects.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Materials for DMS Experiments
| Reagent/Material | Function | Examples/Alternatives | Considerations for Combinatorial Libraries |
|---|---|---|---|
| Mutagenic Oligonucleotides | Introduce defined mutations into target gene [33] | Doped oligos; NNK/NNS codons; trinucleotide cassettes [33] [34] | Trinityucleotide cassettes reduce amino acid bias and stop codons in combinatorial libraries [34] |
| High-Fidelity DNA Polymerase | Amplify mutant libraries with minimal additional mutations | Pfu polymerase; commercial high-fidelity mixes | Critical for maintaining library integrity during amplification |
| Display System | Link genotype to phenotype for screening | Yeast display; phage display; mammalian surface display [36] | Choice affects post-translational modifications and physiological relevance |
| Selection Reagents | Apply selective pressure during screening | Antibiotics; fluorescent ligands; cytotoxic substrates [37] [34] | Stringency must be optimized to capture range of epistatic effects |
| Barcoded Vectors | Track individual variants in pooled screens | Commercial barcoding systems; custom designs | Essential for deconvoluting complex combinatorial libraries |
| Next-Generation Sequencing Platform | Quantify variant frequencies | Illumina; PacBio; Oxford Nanopore | Sufficient depth required for adequate combinatorial library coverage |
The experimental frameworks for phenotyping combinatorial variants through Deep Mutational Scanning have evolved into sophisticated pipelines that integrate diverse library generation methods, functional screening modalities, and statistical analysis tools. The selection of appropriate methods at each stage depends on the specific research goals, with particular considerations for epistasis studies including library comprehensiveness, screening context relevance, and analytical robustness. Emerging approaches such as multi-environment DMS and structure-guided analysis further enhance our ability to detect and interpret epistatic interactions in combinatorial libraries. As these technologies continue to mature, they promise to deliver deeper insights into protein structure-function relationships and enable more predictive modeling of genetic interactions in both basic research and therapeutic development contexts.
Protein engineering aims to tailor or create new molecular activities for research, industrial, and therapeutic applications. A significant hurdle in this endeavor is epistasis—a phenomenon where the functional effect of a combination of mutations differs from the sum of their individual effects [23] [40]. In protein active sites, which are densely packed with critical molecular interactions, epistasis is particularly pronounced [41] [23]. This non-additivity means that beneficial multipoint mutants often cannot be discovered through simple, stepwise mutagenesis, as many necessary intermediate variants may be non-functional [23]. Computational design methods must therefore account for these complex inter-residue interactions to successfully generate functional proteins. This guide evaluates two related computational methods, FuncLib and htFuncLib, which are explicitly developed to address the challenge of epistasis and enable the reliable design of functional multipoint mutants.
Methodological Comparison: FuncLib vs. htFuncLib
While both FuncLib and its successor, htFuncLib, leverage evolutionary information and atomistic modeling to design protein variants, they are built for distinct experimental scales and employ different strategies for managing combinatorial complexity.
FuncLib (Functional Library) is an automated method for designing and ranking epistatic multipoint mutants at enzyme active sites [42] [43]. It begins by identifying single-point mutations that are phylogenetically likely and energetically tolerated. It then exhaustively models all possible combinations of these pre-filtered mutations using the Rosetta biomolecular modeling suite, ranks them by calculated energy, and recommends the top 50-100 designs for experimental testing [42] [43]. Its exhaustive nature limits the scale of sequence space it can practically explore.
htFuncLib (high-throughput Functional Library) extends FuncLib for the design of much larger libraries suitable for high-throughput screening [42] [41]. Instead of exhaustively scoring every possible combination, htFuncLib aims to identify a set of mutually compatible point mutations. The key innovation is the use of a machine learning model, EpiNNet, which is trained to predict combinations of mutations that form low-energy, stable proteins [41]. This allows htFuncLib to generate a sequence space where mutations can be freely combined, creating libraries of hundreds to millions of variants that are computationally enriched for folded and functional proteins [42] [41].
The table below summarizes the core differences between the two approaches.
Table 1: A direct comparison of the FuncLib and htFuncLib methodologies.
| Feature | FuncLib | htFuncLib |
|---|---|---|
| Primary Goal | Design individual, highly optimized multipoint mutants [43] | Design large combinatorial libraries for experimental screening [42] [41] |
| Library Scale | Low- to medium-throughput (tens to hundreds of designs) [43] | Medium- to high-throughput (up to millions of variants) [42] |
| Design Strategy | Exhaustive combination and Rosetta energy ranking of pre-filtered mutations [43] | Machine learning (EpiNNet) to select compatible mutations for free combination [41] |
| Handling of Epistasis | Implicitly addressed by ranking full sequences based on total energy [42] | Explicitly addressed by filtering for mutations that form low-energy combinations [41] |
| Typical Output | A ranked list of specific protein sequences to synthesize [43] | A defined sequence space (set of mutations per position) for library synthesis [41] |
Experimental Workflow and Key Findings
The power of htFuncLib was demonstrated in a comprehensive study on Green Fluorescent Protein (GFP), which provided quantitative data on the method's performance [41]. The following diagram illustrates the integrated computational and experimental workflow of htFuncLib, from initial input to functional validation.
Key Experimental Protocol and Outcomes in GFP Optimization
The application of this workflow to the chromophore-binding pocket of a stabilized GFP (PROSS-eGFP) involved specific experimental steps and yielded measurable results [41].
Detailed Experimental Protocol:
- Library Design: The htFuncLib method was applied to 27 positions lining the GFP chromophore-binding pocket. The process resulted in two designed libraries: one excluding and one including positions that directly hydrogen-bond with the chromophore [41].
- Library Synthesis: The designed DNA libraries, comprising 11 million and 930,000 unique sequences, were synthesized and cloned using Golden-Gate assembly [41].
- Functional Screening: The variant libraries were expressed in E. coli, and functional fluorescent proteins were identified using Fluorescence-Activated Cell Sorting (FACS) [41].
- Deep Sequencing: The input and sorted libraries were analyzed by deep sequencing to identify the sequences of thousands of functional designs and quantify their enrichment [41].
- Characterization: Unique functional variants were characterized for properties including thermal stability (Tm), fluorescence lifetime, and quantum yield [41].
Quantitative Results: The htFuncLib approach proved highly successful, generating an unprecedented diversity of functional GFP variants from a single designed library. The following table summarizes the key experimental outcomes.
Table 2: Experimental outcomes from the application of htFuncLib to GFP design [41].
| Metric | Result | Implication |
|---|---|---|
| Unique Functional Designs Recovered | >16,000 | Demonstrates the ability to access a vast functional sequence space in one shot. |
| Maximum Number of Mutations per Design | Up to 8 | Confirms the method's capacity to efficiently design highly mutated active sites. |
| Thermal Stability Range (Tm) | Up to 96°C | Generated useful diversity, including variants with drastically improved stability. |
| Computational Energy Enrichment | >99% of nohbonds library designs had lower Rosetta energy than progenitor | Validates that the designed library is highly enriched for stable, folded proteins. |
The Scientist's Toolkit: Essential Research Reagents
The successful implementation of an htFuncLib project relies on a combination of specialized software, computational resources, and experimental reagents.
Table 3: Key research reagents and resources for implementing htFuncLib.
| Category | Item / Resource | Function and Description |
|---|---|---|
| Computational Tools | FuncLib/htFuncLib Web Server [42] | The primary online platform for running FuncLib and htFuncLib calculations. |
| Rosetta Software Suite [42] [41] | A comprehensive software for biomolecular structure prediction, design, and energy calculations. | |
| EpiNNet [41] | A machine learning model (neural network) used by htFuncLib to rank mutation compatibility. | |
| Experimental Reagents | Golden-Gate Assembly System [41] | A modular and efficient DNA assembly method used to construct the variant libraries. |
| Fluorescence-Activated Cell Sorter (FACS) [41] [44] | Enables high-throughput screening and isolation of functional fluorescent protein variants. | |
| Deep Sequencing Platform [41] [44] | Used to decode the identity and frequency of all variants in the input and selected libraries. |
FuncLib and htFuncLib represent a significant evolution in computational protein design, moving from the design of individual optimized sequences to the design of entire functional sequence spaces. By directly addressing the challenge of epistasis through a combination of evolutionary analysis, atomistic modeling, and machine learning, htFuncLib in particular enables a one-shot optimization strategy that can recover thousands of diverse, functional multipoint mutants [42] [41]. This approach has been experimentally validated in the design of GFP, demonstrating its power to generate variants with a wide range of improved properties, such as thermostability. For researchers in enzymology, antibody engineering, and therapeutic development, these methods offer a powerful and accessible platform to accelerate the discovery and optimization of protein function.
Leveraging Machine Learning and Zero-Shot Predictors for Navigating Epistatic Landscapes
Protein engineering, crucial for developing therapeutics, biocatalysts, and research tools, often relies on directed evolution (DE) to optimize protein fitness. This process empirically accumulates beneficial mutations through iterative cycles of mutagenesis and screening. However, the efficiency of directed evolution is significantly hampered by epistasis—the phenomenon where the functional effect of a mutation depends on the presence of other mutations within the same sequence [23]. In epistatic landscapes, the combined effect of mutations is not a simple sum of their individual effects, leading to non-additive interactions that can render beneficial single mutations deleterious when combined. This creates rugged fitness landscapes rich in local optima, where traditional greedy hill-climbing DE strategies often become trapped [26] [45].
The dense molecular packing and intricate interaction networks within protein active sites make these regions particularly prone to epistasis. Mutations that improve activity may undermine stability, requiring compensatory mutations that only confer benefits when introduced in specific combinations [23]. This complexity poses a substantial challenge for rational protein design and conventional directed evolution. Recently, machine learning (ML) has emerged as a powerful strategy to overcome these limitations. By learning the complex sequence-function relationships from experimental data, ML models can capture epistatic effects and guide exploration of the vast sequence space more efficiently. Particularly promising are approaches incorporating zero-shot predictors—models that leverage evolutionary, structural, or biophysical knowledge to estimate fitness without requiring experimental training data from the target protein [26] [46]. This guide provides a comparative analysis of current ML-assisted methods for navigating epistatic landscapes, evaluating their performance, experimental requirements, and optimal use cases to inform researchers' strategy selection.
Comparative Analysis of ML-Assisted Directed Evolution Strategies
Several ML frameworks have been developed to address epistasis in protein engineering. The table below compares four prominent strategies, their operating principles, and performance characteristics.
Table 1: Comparison of ML-Assisted Directed Evolution Strategies
| Method | Core Approach | Epistasis Handling | Key Advantages | Experimental Data Requirements | Reported Performance Gains |
|---|---|---|---|---|---|
| MLDE (Machine Learning-Assisted DE) | Single-round supervised model trained on initial variant screen | Captures non-additive effects for in-silico prediction | Reduces screening burden compared to DE; works with standard initial libraries | Requires initial combinatorial library data (~102-103 variants) | Outperforms DE across 16 diverse landscapes; advantage increases with landscape difficulty [26] |
| ALDE (Active Learning-Assisted DE) | Iterative batch Bayesian optimization with uncertainty quantification | Explores combinatorial space balancing exploitation/exploration | Prevents convergence to local optima; adaptively focuses screening | Multiple smaller rounds of screening (~50-500 variants per round) | Improved cyclopropanation yield from 12% to 93% in 3 rounds; superior to DE on epistatic landscapes [45] |
| ftMLDE (Focused Training MLDE) | Enriches training set using zero-shot predictors before model training | Leverages prior knowledge to avoid low-fitness variants | Enhances MLDE performance; reduces dependency on large initial screens | Can work with smaller initial datasets when combined with zero-shot | Consistently outperforms random sampling for binding and enzyme activity landscapes [26] |
| MODIFY (ML-optimized library design) | Co-optimizes predicted fitness and diversity using ensemble zero-shot models | Designs libraries to cover multiple fitness peaks | Addresses cold-start problem; no experimental fitness data required | No experimental fitness data needed for initial library design | Top Spearman correlation in 34/87 ProteinGym benchmarks; enables new-to-nature enzyme engineering [46] |
Performance Evaluation Across Diverse Landscapes
Recent large-scale benchmarking studies provide quantitative insights into how these methods perform across different types of epistatic landscapes. The following table summarizes experimental results from systematic analyses.
Table 2: Performance Comparison Across Protein Fitness Landscapes
| Method | Number of Landscapes Tested | Function Types | Key Performance Metrics | Optimal Use Cases |
|---|---|---|---|---|
| MLDE | 16 combinatorial landscapes [26] | Protein binding, enzyme activities | Greater advantage on landscapes challenging for DE (fewer active variants, more local optima) [26] | Landscapes with moderate epistasis; when initial screening capacity available |
| ALDE | 1 experimental + 2 computational landscapes [45] | Cyclopropanation activity, combinatorial fitness | 7.75x yield improvement in wet-lab; more efficient sequence space exploration in simulations [45] | Highly epistatic active sites; multiple experimental rounds feasible |
| ftMLDE with Zero-Shot | 16 combinatorial landscapes [26] | Protein binding, enzyme activities | Consistent outperformance over random sampling; combined with ALDE for maximum benefit [26] | Limited screening budget; availability of relevant zero-shot predictors |
| MODIFY | 87 DMS datasets + GB1 landscape [46] | Catalytic activity, binding, stability, growth | Best zero-shot predictor in 34/87 ProteinGym benchmarks; designs libraries with co-optimized fitness/diversity [46] | New-to-nature enzyme functions; cold-start problems without fitness data |
Critical Performance Insights
Landscape Navigability: MLDE provides greater advantages on landscapes that are more challenging for traditional DE, particularly those with fewer active variants and more local optima [26]. The ruggedness of epistatic landscapes that hinders DE actually creates opportunities for ML methods to demonstrate superior performance.
Zero-Shot Predictor Efficacy: Focused training using zero-shot predictors that leverage distinct evolutionary, structural, and stability knowledge sources consistently improves MLDE performance. The diversity of knowledge sources appears more important than any single predictor type [26].
Multi-Round Efficiency: For highly epistatic systems like enzyme active sites, ALDE significantly outperforms single-round MLDE by adaptively focusing experimental resources on promising regions of sequence space while maintaining diversity to escape local optima [45].
Experimental Protocols and Methodologies
Standard MLDE Implementation Protocol
Combinatorial Library Design: Select 3-5 target residues for simultaneous mutagenesis based on structural knowledge or previous experiments. For a 4-site library, this creates 160,000 (20^4) possible variants [26].
Initial Data Collection: Screen a randomly selected subset of the combinatorial library (typically 500-2000 variants) to generate sequence-fitness training data [26].
Model Training: Train supervised ML models (ensemble models often perform best) on the sequence-fitness data to learn the mapping from sequence to function, capturing epistatic interactions.
Prediction and Validation: Use the trained model to predict fitness across the entire combinatorial space. Select top-ranked predictions for experimental validation [26].
ALDE Workflow for Active Site Engineering
The following diagram illustrates the iterative ALDE workflow for optimizing epistatic active sites:
Diagram Title: ALDE Iterative Engineering Workflow
Critical Implementation Details:
Uncertainty Quantification: ALDE uses frequentist uncertainty estimation rather than Bayesian approaches, demonstrating more consistent performance in protein engineering contexts [45].
Acquisition Functions: Effective acquisition balances exploitation (high predicted fitness) and exploration (high uncertainty). Expected Improvement often performs well for protein engineering tasks [45].
Batch Selection: Each round selects a batch of variants (typically 50-500) to maintain diversity while focusing experimental resources.
MODIFY Library Design Protocol
Residue Selection: Input target residues for engineering without fitness data.
Zero-Shot Ensemble Prediction: Apply ensemble of protein language models (ESM-1v, ESM-2) and sequence density models (EVmutation, EVE) to predict fitness across combinatorial space [46].
Pareto Optimization: Solve the optimization problem: max(fitness + λ·diversity) to identify the Pareto frontier of optimal library designs balancing both objectives [46].
Stability Filtering: Filter designed variants using foldability and stability predictors to remove non-functional proteins.
Library Synthesis: Experimental construction of the optimized library for screening.
Zero-Shot Predictors for Epistatic Landscapes
Zero-shot predictors leverage various biological knowledge sources to estimate variant fitness without experimental training data. The table below compares prominent zero-shot approaches used in epistatic landscape navigation.
Table 3: Zero-Shot Predictors for Protein Fitness Prediction
| Predictor | Knowledge Source | Underlying Methodology | Strengths | Limitations |
|---|---|---|---|---|
| ESM-1v/ESM-2 [46] | Evolutionary information from protein sequences | Protein language models trained on UniRef | Strong performance across diverse proteins; no MSA required | Limited explicit structural constraints |
| EVmutation [26] [46] | Co-evolution patterns in multiple sequence alignments | Maximum entropy model from correlated mutations | Directly captures residue-residue dependencies | Requires sufficient MSA depth for accuracy |
| EVE (Evolutionary model of Variant Effect) [46] | Deep generative modeling of protein families | Variational autoencoder trained on MSAs | State-of-the-art for disease variant prediction | Computationally intensive; MSA depth dependent |
| MSA Transformer [46] | Joint evolutionary and sequence context | Transformer architecture with MSA inputs | Combines benefits of PLMs and co-evolution | High computational requirements for large MSAs |
| MODIFY Ensemble [46] | Multiple knowledge sources | Weighted combination of diverse predictors | Most robust performance across protein families | Increased complexity; requires implementation |
Ensemble Zero-Shot Strategies
The MODIFY framework demonstrates that ensemble approaches combining multiple zero-shot predictors achieve superior performance compared to individual methods. By leveraging complementary knowledge sources, ensemble models overcome limitations of individual predictors and provide more accurate fitness estimates across diverse protein families and functions [46]. This robustness is particularly valuable for engineering new-to-nature enzyme functions where evolutionary signals may be weak or non-existent.
Table 4: Key Research Reagents and Computational Tools
| Resource | Type | Function | Implementation Notes |
|---|---|---|---|
| ALDE Codebase [45] | Software package | Implements active learning with uncertainty quantification | Available at https://github.com/jsunn-y/ALDE |
| SSMuLA Dataset [26] | Benchmark data | 16 combinatorial landscapes for method evaluation | https://doi.org/10.5281/zenodo.13910505 |
| ProteinGym [46] | Benchmark suite | 87 DMS assays for zero-shot predictor evaluation | Comprehensive fitness prediction benchmark |
| ESM-1v/ESM-2 [46] | Protein language models | Zero-shot fitness prediction from evolutionary patterns | Available through HuggingFace Transformers |
| EVmutation [26] | Co-evolution model | Infers epistatic constraints from multiple sequence alignments | Python implementation available |
| MODIFY Algorithm [46] | Library design tool | Co-optimizes fitness and diversity in library design | Implements Pareto optimization for balanced libraries |
The comparative analysis reveals that optimal strategy selection depends on specific experimental constraints and landscape characteristics:
For well-characterized protein families with established assays, MLDE with focused training using diverse zero-shot predictors provides efficient optimization with moderate screening requirements [26].
For highly epistatic systems like enzyme active sites where local optima trap conventional approaches, ALDE offers superior performance through iterative exploration and uncertainty-guided sampling [45].
For new-to-nature functions or cold-start problems without existing fitness data, MODIFY's ensemble zero-shot approach with Pareto-optimized library design enables effective exploration of uncharted sequence space [46].
Hybrid approaches combining focused training with active learning consistently deliver robust performance across diverse landscape types, particularly when leveraging multiple, complementary zero-shot predictors [26].
The evolving toolkit of ML-assisted methods fundamentally transforms our approach to epistatic landscapes, moving from brute-force screening to intelligent navigation guided by computational prediction and strategic experimentation. As zero-shot predictors continue to improve and incorporate richer structural and biophysical information, their capacity to unravel complex epistatic interactions will further accelerate the design of novel proteins with tailored functions.
Navigating Challenges: Overcoming Bias, Noise, and Combinatorial Complexity
In the quest to understand complex biological systems, such as the effects of multiple genetic mutations, researchers frequently encounter the formidable barrier of combinatorial explosion. This phenomenon describes the rapid growth of complexity that occurs when the variables in a system combine, making it intractable to test all possible combinations [47]. For example, a library of combinatorial protein mutants can easily encompass more variants than can be practically synthesized or assayed [23]. This problem is particularly acute in research on epistatic effects, where the functional impact of one mutation depends on the presence of other mutations, creating a rugged fitness landscape that severely constrains evolutionary trajectories and experimental optimization [23].
The core of the issue lies in the mathematics of combination. The number of possible combinations grows at least exponentially with the number of variables, a problem that is computationally fundamental and often used to justify the intractability of certain problems [47] [48]. In protein engineering, this means that even armed with knowledge of all single-point mutations, one cannot reliably predict the function of higher-order combinations, rendering traditional stepwise optimization strategies ineffective [23]. This article objectively compares modern computational and experimental strategies designed to overcome this fundamental limitation, providing a clear analysis of their performance and the data-driven evidence supporting their use.
Computational & Algorithmic Strategies for Taming Explosion
Compression-Based Combinatorial Algorithms
Concept and Rationale: A cutting-edge approach involves finding and grouping similar combinations from a vast space to "compress" the problem. Instead of discarding data, this method represents the entire combinatorial space in a compact, database-like structure, allowing computations to be performed on the compressed representation. This leads to massive gains in efficiency without sacrificing accuracy [49].
Supporting Experimental Data:
- Application: Solving a tiling problem on an 8x8 grid.
- Performance Comparison: The compression-based algorithm reduced calculation time for 190 solutions from 16,475 seconds using existing methods to just 0.88 seconds, an improvement of over four orders of magnitude [49].
- Scalability: The performance advantage grows exponentially as the problem complexity increases (e.g., with more grid spaces or added conditions) [49].
Sparse Experimental Design for Linear Models (ED-S)
Concept and Rationale: When the relationship between variables (e.g., genomic features) and an outcome (e.g., disease progression) can be modeled with a sparse linear function, a budget-conscious Experimental Design (ED) strategy can be employed. The goal is to select a subset of experiments that maximizes the information gain for a given budget, ensuring the statistical model derived from the subset is as close as possible to the model from the full dataset [50].
Supporting Experimental Data:
- Application: Predicting future cognitive decline in Alzheimer's Disease (AD) using baseline neuroimaging data from approximately 1,000 subjects [50].
- Protocol: The ED-S (Spectral Experimental Design) algorithm selects a subset of subjects for costly longitudinal follow-up (to obtain the response variable
y) by optimizing thelog detof the subject covariance matrix, a criterion known as D-optimality [50]. - Outcome: The proposed models (geometric and algebraic formulations) were shown to be robust and effective, yielding consistent results with each other and with the "full" model that used all data, thereby validating the subset selection approach for high-dimensional data [50].
Scaling Laws and Pairwise Interaction Models
Concept and Rationale: For specific biological processes like RNA splicing, quantitative laws can help predict the effects of mutations. Research has revealed that the effects of mutations on splicing scale non-monotonically with the inclusion level of an exon, with each mutation having its maximum effect at a predictable intermediate inclusion level [51].
Supporting Experimental Data:
- Application: Generating a combinatorially complete genotype-to-phenotype map for the evolution of a human exon [51].
- Protocol: Quantifying the effects of all possible combinations of exonic mutations accumulated during exon emergence. Mathematical modeling indicated that competition between alternative splice sites is the key driver of this non-linearity [51].
- Outcome: Combining this global scaling law with specific pairwise epistatic interactions allowed the researchers to accurately predict the effects of complex genotype changes involving more than ten mutations [51]. This approach directly addresses epistasis to make accurate high-order predictions.
Comparative Analysis of Strategic Performance
The table below summarizes the key performance characteristics of the strategies discussed, based on experimental data.
Table 1: Objective Comparison of Strategies Against Combinatorial Explosion
| Strategy | Core Mechanism | Reported Performance Gain | Key Application Context | Addresses Epistasis? |
|---|---|---|---|---|
| Compression Algorithms [49] | Groups similar combinations; performs computation on compressed data. | Time reduced from 16,475s to 0.88s (>18,000x faster) for a tiling problem. | General combinatorial problems (e.g., network design, tiling). | Indirectly, by enabling exhaustive search. |
| Sparse Experimental Design (ED-S) [50] | Selects an optimal subset of data points that maximize information under a budget. | Produced model estimators consistent with the full-model estimator in a neuroimaging study (n ≈ 1000). | High-dimensional data with costly experiments (e.g., longitudinal studies). | No, focuses on efficient data acquisition. |
| Scaling Law & Pairwise Models [51] | Uses a global scaling law and specific pairwise interactions to predict complex effects. | Accurate prediction of phenotypic effects for combinations of >10 mutations. | Genotype-to-phenotype mapping (e.g., RNA splicing). | Yes, explicitly. |
Experimental Protocols for Key Methodologies
Protocol: Sparse Experimental Design for Predictive Modeling
This protocol is adapted from budget-constrained experimental design for sparse linear models [50].
- Problem Formulation: Define the linear model
yi = xi^T β + ε, wherexiare the feature vectors (e.g., baseline neuroimaging data),yiis the response variable (e.g., cognitive decline), andβis the sparse regressor to be estimated. - Objective Setting: The goal is to select a binary vector
μthat maximizes the D-optimality criterionf(Σ μ_i x_i x_i^T + εI) = log det(Σ μ_i x_i x_i^T + εI)subject to the budget constraint1^T μ ≤ B. - Algorithm Execution:
- Geometric Motivation (ED-S): The selection is motivated by choosing data points that optimize the spectral properties (eigenvalues) of the resulting covariance matrix.
- Algebraic Manipulation: An alternative formulation involves algebraic manipulation to achieve a similar optimal subset selection.
- Model Estimation: Once the optimal subset
Sis selected, solve the LASSO regression problemβ* = argmin_β (1/2 ||X_S β - y_S||_2^2 + ε ||β||_1)to obtain a sparse and interpretable model. - Validation: Compare the model
β*derived from the subsetSwith the model estimated from the full dataset to ensure consistency and validate the design.
Protocol: Mapping Epistatic Interactions in Splicing
This protocol is derived from research that quantified the effects of combinatorial mutations on RNA splicing [51].
- Library Construction: Synthesize a combinatorially complete set of DNA sequences encompassing all possible combinations of exonic mutations of interest that have accumulated during the evolution of an alternatively spliced exon.
- Phenotype Quantification: For each genotype variant in the library, quantify the exon inclusion level (phenotype) using a high-throughput splicing assay, such as RNA-seq or a fluorescent reporter system.
- Identify Scaling Behavior: Analyze the data to identify the non-monotonic scaling law, where the effect of a given mutation is smallest at low and high inclusion levels and greatest at an intermediate level. Model this using a mathematical framework based on splice-site competition.
- Detect Pairwise Epistasis: Statistically identify significant pairwise genetic interactions that deviate from the expected additive effects based on the global scaling law.
- Predictive Model Building: Integrate the global scaling law with the identified pairwise epistatic coefficients into a unified model. This model can then be used to predict the splicing phenotype of any higher-order genetic combination within the defined sequence space.
Visualizing Strategic Workflows
Sparse Experimental Design Workflow
Epistasis Mapping & Prediction Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Combinatorial Genetics Research
| Reagent / Material | Function in Research |
|---|---|
| Combinatorial DNA Synthesis Library | A pool of DNA sequences containing all defined combinations of mutations, serving as the starting genetic material for high-throughput functional assays [51]. |
| High-Throughput Phenotyping Assay | A scalable method (e.g., deep mutational scanning, mass spectrometry, fluorescent reporter) to quantitatively measure the function or fitness of thousands of variants in parallel [23] [51]. |
| Sparse Linear Model Solver (LASSO) | A computational software package (e.g., in R or Python) that implements L1-regularized regression to estimate a sparse parameter vector β, identifying the most predictive features from high-dimensional data [50]. |
| Covariance Matrix Optimization Tool | Software capable of solving the D-optimal (or other) experimental design problem by optimizing the selection of data points from a large covariate matrix [50]. |
| Decision Graph / Compression Algorithm | A specialized algorithmic tool that can compress combinatorial spaces by grouping equivalent or similar states, enabling efficient computation on the compressed representation [49]. |
In genetics, epistasis describes the phenomenon where the phenotypic effect of one mutation depends on the genetic background in which it occurs [9] [52]. Disentangling the sources of epistasis is fundamental to understanding fitness landscapes, yet represents a significant statistical challenge. Epistasis generally arises from two distinct sources: specific epistasis (SE), resulting from direct physical interactions between residues (e.g., amino acids in close proximity in a protein structure), and global epistasis (GE), which arises from nonlinearities in the genotype-to-phenotype map itself [9]. Traditional methods for detecting epistasis often rely on strong assumptions about the form of these nonlinearities, leading to potential model misspecification that can over- or underestimate specific interactions [9]. In response, researchers have developed rank-based and semiparametric methods that require fewer assumptions, offering more robust frameworks for distinguishing these entangled effects in combinatorial mutagenesis experiments [9] [52].
Rank-Based Methods: Core Principles and Applications
Theoretical Foundation of Rank-Based Approaches
Rank-based methods leverage the observation that global epistasis, under the assumption of monotonicity, imposes strong constraints on the rank statistics of combinatorial mutagenesis data [9]. Specifically, if the genotype-to-phenotype map involves a monotonic nonlinear transformation (global epistasis) without specific interactions, then the rank-order of mutational effects should remain preserved across different genetic backgrounds [9]. The core principle is that rank statistics are invariant under monotonic transformations; thus, systematic violations of rank-order preservation provide evidence of specific epistasis arising from direct interactions [9]. This foundational insight allows researchers to detect SE without explicitly modeling the form of the nonlinearity, making these methods particularly valuable when the underlying biological mechanisms are complex or poorly understood.
Rank-order statistical methods are ideally suited for biological data analysis in several key scenarios: when the primary data naturally occur in rank form; when sample sizes are too small to verify distributional assumptions for parametric tests; or when the sampling distribution of continuous data is skewed or otherwise violates assumptions of continuous-based methods [53]. By reducing sensitivity to extreme outliers and distributional shape, rank methods provide a safer, more robust alternative to traditional parametric approaches for analyzing epistasis in high-throughput mutagenesis datasets [53].
Key Rank-Based Methods and Workflows
Resample and Reorder (R&R) Method
The Resample and Reorder (R&R) method is a semiparametric approach specifically designed to detect specific epistasis in the presence of global epistasis and measurement noise [9] [52]. This method operates on the principle that in the absence of SE, the rank-order of mutation effects should be consistent across genetic backgrounds, with any deviations attributable to either specific interactions or measurement noise [9]. The R&R procedure systematically accounts for heteroskedastic noise—a common feature in sequencing-based assays where measurement precision varies across the fitness range—by comparing observed rank variations against a null distribution generated through resampling [9].
The experimental workflow for implementing R&R involves specific steps for processing combinatorial mutagenesis data, particularly from deep mutational scanning (DMS) experiments:
RankCorr for Single-Cell RNA Sequencing
The RankCorr algorithm represents another application of rank-based methods in computational biology, designed for marker selection in high-throughput single-cell RNA sequencing (scRNA-seq) data [54]. While applied to a different domain, RankCorr shares foundational principles with epistasis detection methods: it operates by ranking mRNA counts data before performing linear separation, providing a non-parametric approach for analyzing count data with high variance and sparsity [54]. This method demonstrates the versatility of rank-based approaches across biological domains, particularly for handling large-scale datasets with characteristics common to modern high-throughput experiments.
Semiparametric Frameworks for Data Fusion
Semiparametric Efficiency in Data Integration
Semiparametric methods offer a powerful framework for integrating diverse data sources while maintaining robust statistical properties. Recent research has established semiparametric efficiency bounds for estimating general functionals when fusing individual data with external summary statistics [55]. This theoretical foundation demonstrates that properly integrated external summary statistics can improve estimation efficiency without introducing bias, resolving the "efficiency paradox" where naively incorporated external data sometimes reduces rather than improves precision [55]. The data-fused efficient estimator achieving this bound has a closed-form expression and inherits the Neyman orthogonality property, enabling the use of flexible machine learning methods for estimating nuisance parameters without compromising statistical validity [55].
Adaptive Fusion for Handling Untransportable Data
A significant challenge in data integration arises when external summary statistics are not fully transportable due to population heterogeneity or other biases. The adaptive fusion estimator addresses this by incorporating carefully designed weighting matrices that automatically downweight or exclude untransportable components [55]. This method maintains consistency and asymptotic normality even when some external summary statistics are biased, while remaining asymptotically equivalent to an oracle estimator that uses only transportable statistics [55]. For finite-sample applications, a re-bootstrap procedure helps mitigate undercoverage issues that can occur when distinguishing between transportable and untransportable components is challenging [55].
The conceptual relationship between different data types and integration methodologies follows a structured pathway:
Performance Comparison of Statistical Methods
Systematic Evaluation of Ranking Aggregation Methods
A comprehensive comparison of ranking aggregation methods relevant to meta-analysis of gene lists provides valuable insights into method performance under various conditions [56]. This systematic evaluation examined multiple algorithms under scenarios simulating real genomic data features, including heterogeneity of quality, noise level, and mixtures of unranked and ranked data with up to 20,000 entities [56]. The study implemented both existing methods and variations suitable for genomic data, assessing them on simulated datasets and real biological data from SARS-CoV-2, cancer (non-small cell lung cancer), and bacterial infection (macrophage apoptosis) research [56].
Table 1: Comparison of Ranking Aggregation Methods for Genomic Data
| Method Category | Example Methods | Handles Unranked Lists | Performance with High Noise | Computational Efficiency |
|---|---|---|---|---|
| Borda's Methods | MEAN, GEO, MED [56] | Yes (with modifications) [56] | Poor with significant noise [56] | High [56] |
| Complex Bayesian Methods | BiG, BARD [56] | Limited accommodation [56] | Varies | Lower for large datasets [56] |
| Specialized Genomic Methods | RRA, MAIC [56] | MAIC explicitly handles unranked [56] | Generally robust [56] | Generally high [56] |
Empirical Performance in Protein Fitness Landscapes
Machine learning-assisted directed evolution (MLDE) strategies provide practical evidence of epistasis management in protein engineering. Recent evaluation across 16 diverse combinatorial protein fitness landscapes revealed that MLDE consistently outperforms traditional directed evolution approaches, with advantages becoming more pronounced on landscapes challenging for conventional methods [26]. Landscapes with more local optima and fewer active variants—indicators of epistatic interactions—particularly benefited from ML approaches [26]. The study found that focused training using zero-shot predictors that leverage evolutionary, structural, and stability knowledge consistently improved performance for both binding interactions and enzyme activities [26].
Table 2: Machine Learning Performance Across Protein Fitness Landscape Types
| Landscape Characteristic | DE Performance | MLDE Performance | Key Advantage Factors |
|---|---|---|---|
| Smooth, additive landscapes | High | Moderate | Limited ML advantage |
| Rugged, epistatic landscapes | Low | High | ML captures non-additive effects [26] |
| Landscapes with few active variants | Low | High | Focused training effectiveness [26] |
| Landscapes with many local optima | Low | High | Broad sequence space exploration [26] |
Experimental Protocols and Methodologies
Protocol for Rank-Based Epistasis Detection
Implementing the Resample and Reorder (R&R) method for detecting specific epistasis requires careful attention to several key stages. First, data preparation involves processing fitness measurements from deep mutational scanning experiments, typically represented as a matrix where rows correspond to genetic variants and columns represent different genetic backgrounds or experimental conditions [9]. The next stage involves ranking mutations within each genetic background, transforming raw fitness measurements into rank orders that are invariant to monotonic transformations [9].
The core resampling procedure accounts for heteroskedastic noise by generating a null distribution through repeated sampling that respects the varying precision of measurements across different fitness ranges [9]. This is particularly crucial for sequencing-based fitness measurements where less fit variants typically have fewer read counts and higher measurement variance [9]. Finally, statistical testing compares observed rank correlations between genetic backgrounds against the null distribution, with significant deviations indicating specific epistasis [9]. This protocol requires minimal preprocessing of the data beyond generating variant read counts and remains agnostic to the form of the nonlinearity beyond monotonicity [9].
Protocol for Semiparametric Data Fusion
The implementation of semiparametric efficient estimation for fusing individual data and summary statistics follows a structured workflow. The initial data harmonization stage ensures internal individual data and external summary statistics are compatible, with careful attention to potential population heterogeneity [55]. The efficient influence function calculation comes next, deriving the specific form based on the target functional and available data sources [55].
For the estimation step, researchers can employ the data-fused efficient estimator when transportability assumptions are satisfied, or the adaptive fusion estimator when dealing with potentially untransportable components [55]. The final inference stage utilizes the re-bootstrap procedure to ensure proper coverage rates, particularly important when distinguishing between transportable and untransportable components is challenging in finite samples [55]. Throughout this process, the Neyman orthogonality property allows incorporation of machine learning methods for nuisance parameter estimation without compromising the asymptotic properties of the final estimator [55].
Computational Tools and Software Implementations
Table 3: Essential Computational Tools for Epistasis Research
| Tool/Resource | Function | Access |
|---|---|---|
| R&R Method Implementation | Detects specific epistasis in presence of global epistasis [9] | Custom code based on publication [9] |
| RankCorr | Marker selection for scRNA-seq data using rank-based approach [54] | https://github.com/ahsv/RankCorr [54] |
| MAIC Algorithm | Ranking aggregation for meta-analysis of gene lists [56] | https://github.com/baillielab/maic [56] |
| Comparison of RA Methods | Code for simulated data generation and ranking aggregation methods [56] | https://github.com/baillielab/comparisonofRA_methods [56] |
Experimental Design Considerations
When planning experiments for epistasis analysis, several key considerations emerge from methodological research. For rank-based methods, researchers should ensure sufficient replication across genetic backgrounds to reliably estimate rank correlations, with particular attention to statistical power for detecting specific interactions [9]. The sample size requirements depend on the expected effect sizes of specific epistatic interactions and the noise characteristics of the measurement system [9].
For data fusion approaches, careful assessment of transportability between internal and external datasets is crucial before integration [55]. Experimental designs should prioritize collecting high-quality internal data with appropriate negative and positive controls, as this provides the foundation upon which external information is added [55]. For protein engineering applications, considering landscape navigability attributes—including the number of active variants, fitness distribution properties, and ruggedness—can inform the selection of appropriate MLDE strategies [26].
Addressing Experimental Noise and Thermodynamic Bias in Library Generation
In the field of protein engineering, the generation of combinatorial mutant libraries is a fundamental strategy for discovering novel proteins with enhanced functions. However, two significant challenges complicate this process: experimental noise, which can obscure true functional signals, and thermodynamic bias, which can skew library representation towards stable but not necessarily functional variants. These issues are particularly acute when studying epistasis—the phenomenon where the effect of one mutation depends on the presence of other mutations—which dramatically shapes evolutionary trajectories and functional outcomes in proteins [23].
The presence of epistasis means that the functional landscape of combinatorial mutations is often rugged, with many potentially beneficial multi-mutant combinations being inaccessible because their constituent single mutations are deleterious when introduced alone [23]. Accurately navigating this landscape requires computational and experimental methods that can distinguish true epistatic effects from artifacts introduced by noise and thermodynamic bias. This guide evaluates contemporary computational platforms based on their capabilities to address these challenges, providing researchers with a framework for selecting appropriate tools for library generation and analysis.
Quantitative Comparison of Library Generation Platforms
The following table compares three advanced computational platforms used for designing and analyzing combinatorial mutant libraries, with a focus on their handling of experimental noise and thermodynamic constraints.
Table 1: Platform Comparison for Library Generation and Analysis
| Platform | Core Methodology | Noise Handling | Thermodynamic Bias Mitigation | Epistasis Modeling | Reported Performance |
|---|---|---|---|---|---|
| OpenProtein.AI | Sequence-to-function machine learning model trained on experimental data [57]. | Implicit via cross-validation and robust model training on large datasets (e.g., n=7,476 variants) [57]. | Not explicitly detailed; relies on data-driven constraints during design [57]. | Directly models non-additive effects by learning from combinatorial variant data [57]. | Spearman ρ = 0.69 between predicted and actual binding affinities [57]. |
| Chem3DLLM | Multimodal LLM for 3D molecular generation using reinforcement learning with scientific feedback (RLSF) [58]. | Robust training through systematic introduction of multisourced noise in spectral data [59]. | Explicitly addressed via RLSF using rewards based on energy minimization and stability [58]. | Considers 3D spatial packing and interaction networks within active sites [58]. | Vina score: -7.21 for structure-based drug design [58]. |
| Simulation-Trained Neural Networks (e.g., for 2DES spectra analysis) | Feed-forward neural networks trained on simulated spectral data with experimental "pollutants" [59]. | Systematically tests and establishes SNR thresholds for robust performance (e.g., SNR >12.4 for uncorrelated noise) [59]. | Not the primary focus; aimed at extracting electronic couplings from noisy data [59]. | Models are trained on Hamiltonian parameters that inherently include coupled interactions [59]. | ~84% to 96% accuracy in mapping noisy spectra to electronic couplings, depending on constraints [59]. |
Detailed Experimental Protocols
Protocol: Training a Sequence-to-Function Model with OpenProtein.AI
This protocol is used to create a predictive model for guiding the design of combinatorial libraries with optimized properties [57].
- Data Preparation and Upload: Compile a dataset of protein sequences and their corresponding measured properties (e.g., binding affinity, stability) into a structured table, typically a CSV file. The dataset should include combinatorial variants to capture epistatic effects. This file is then uploaded to the platform, which creates an
AssayMetadataobject containing sequence length, measurement names, and entry count [57]. - Model Training: Initiate a training job via the API (
session.train.create_training_job), specifying the dataset and the target measurement column (e.g.,log_kdnmfor binding affinity). The system trains a machine learning model to map sequence space to the functional property [57]. - Cross-Validation: Evaluate the trained model's performance and robustness through k-fold cross-validation. This step assesses how well the model generalizes to unseen data and provides an estimate of its predictive accuracy, reported as a Spearman correlation coefficient between predicted and actual values [57].
- Library Design and In-Silico Screening: Define design criteria using
ModelCriterionobjects (e.g., target affinity with a specific weight and direction). Optionally, apply constraints to restrict mutations to specific sites, such as active site residues. The platform's solver then searches the sequence space to identify variant sequences that Pareto-optimize the design criteria [57].
Protocol: Reinforcement Learning for 3D-Structure-Based Design with Chem3DLLM
This protocol leverages 3D structural information and physical priors to design valid molecular structures, mitigating thermodynamic bias [58].
- Reversible Molecular Tokenization: Convert the 3D molecular structure (e.g., from a Structure Data File - SDF) into a compact, reversible text sequence using the Reversible Compression of Molecular Tokenization (RCMT) method. This encodes 3D geometric and bond information into a format processable by a Large Language Model (LLM) [58].
- Multimodal Model Conditioning: Project the 3D spatial features of the target protein's binding pocket into the same semantic space as the tokenized molecule using a lightweight protein structure projector. This aligns the protein and ligand modalities within the unified LLM [58].
- Reinforcement Learning with Scientific Feedback (RLSF): The LLM generates candidate 3D molecular structures. A scientific critic module then evaluates these candidates based on physical and chemical priors (e.g., energy minimization, valency rules, steric feasibility). These evaluations are converted into differentiable reward signals that guide the LLM's policy via a reinforcement learning loop, iteratively refining the outputs toward thermodynamically stable and valid structures [58].
Workflow Visualization: Integrating Computational and Experimental Approaches
The diagram below illustrates a robust integrated workflow for library generation that embeds noise handling and bias correction at key stages.
Workflow for Robust Library Generation
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following table lists key computational tools and resources essential for implementing the described protocols.
Table 2: Key Research Reagent Solutions for Computational Library Generation
| Tool / Resource | Function / Application | Relevance to Noise/Bias |
|---|---|---|
| OpenProtein.AI Python Client [57] | Programmatic interface for training models and designing protein variant libraries. | Manages noise via statistical cross-validation; addresses epistasis by learning from combinatorial data. |
| Jupyter Notebook Environment [57] | Interactive computing platform for data analysis, visualization, and running computational protocols. | Essential for preprocessing data to identify outliers and for visualizing model performance to detect bias. |
| Structure Data File (SDF) [58] | Standard file format storing 3D molecular structures, atomic coordinates, and bond information. | Provides the ground-truth 3D structural data necessary for enforcing thermodynamic constraints. |
| Vibronic Exciton Hamiltonian Model [59] | A physical model used to simulate two-dimensional electronic spectroscopy (2DES) spectra. | Used to systematically study the impact of different noise types (additive, correlated, intensity-dependent) on ML performance. |
| Reinforcement Learning with Scientific Feedback (RLSF) [58] | A training paradigm that incorporates physical/chemical priors as rewards for an LLM. | Directly mitigates thermodynamic bias by rewarding chemically valid and stable conformations. |
The comparison of platforms reveals distinct strategic approaches to tackling the dual challenges of noise and bias. OpenProtein.AI employs a data-centric strategy, leveraging large-scale experimental datasets to implicitly learn the complex rules of protein function, including epistasis, while using statistical validation to ensure robustness [57]. In contrast, Chem3DLLM adopts a physics-informed approach, explicitly embedding thermodynamic principles through its reinforcement learning framework to actively steer the generative process away from implausible regions of chemical space [58].
The effectiveness of these tools is contextual. For projects with abundant, high-quality functional data, a powerful sequence-based predictor like OpenProtein.AI is highly effective. However, when designing for entirely new functions or when 3D structural interactions are paramount, the structure-based and physics-guided approach of Chem3DLLM provides a critical safeguard against thermodynamic bias. Furthermore, research into simulation-trained neural networks demonstrates a crucial general principle: systematically introducing and characterizing noise during training can make models remarkably robust to experimental imperfections, a strategy that can be adopted across platforms [59].
In conclusion, successfully addressing experimental noise and thermodynamic bias requires carefully matching the computational strategy to the biological question and available data. The emerging trend is the integration of these approaches—combining data-driven learning with physics-based reasoning—to create more powerful, generalizable, and reliable methods for designing functional protein variants in the face of pervasive epistasis.
Optimizing Training Sets for Machine Learning in Highly Epistatic Environments
In protein engineering, epistasis—a phenomenon where the functional effect of a mutation depends on the presence of other mutations—presents a fundamental challenge for predicting protein fitness and optimizing function [23]. This non-additive interaction creates rugged fitness landscapes where beneficial mutations may only confer advantages in specific combinations, rendering traditional greedy optimization approaches ineffective [60] [23]. In highly epistatic environments, such as protein active sites with densely packed amino acid constellations, the efficiency of machine learning-assisted directed evolution (MLDE) depends critically on the strategic design of training sets [60]. This guide compares the performance of MLDE against traditional directed evolution, providing experimental data and methodologies for researchers seeking to optimize protein functions in epistatic environments.
The molecular origins of epistasis stem from both direct physical interactions (electrostatics, van der Waals forces) and indirect conformational changes that can alter residue positioning and function [23]. When multiple mutations are required to improve activity, individual mutations may be deleterious when introduced alone, creating fitness valleys that block evolutionary trajectories [23]. This review contextualizes training set optimization within the broader thesis of evaluating epistatic effects in combinatorial mutant libraries, providing drug development professionals with practical frameworks for navigating complex fitness landscapes.
Performance Comparison: MLDE vs. Traditional Directed Evolution
Quantitative Performance Metrics
Table 1: Comparative performance of MLDE versus traditional directed evolution on epistatic fitness landscapes
| Method | Success Rate (%) at Global Maximum | Relative Efficiency (Fold Improvement) | Training Set Requirements | Optimal Training Set Composition |
|---|---|---|---|---|
| Traditional Directed Evolution | 1.2% | 1.0x (baseline) | Sequential screening of all variants | Single-step greedy walks |
| Standard MLDE | 42.7% | 35.6x | 200-500 variants | Random sampling of combinatorial space |
| Optimized MLDE with Informed Training Sets | 97.1% | 81.0x | 200-500 variants | Zero-shot prediction to minimize "holes" |
Key Performance Advantages
Machine learning-assisted directed evolution demonstrates substantial advantages over traditional methods when applied to epistatic fitness landscapes. In a comprehensive study evaluating a four-site combinatorial fitness landscape characterized by significant epistasis and "holes" (variants with zero or extremely low fitness), optimized MLDE achieved the global fitness maximum 81-fold more frequently than single-step greedy optimization [60]. This remarkable efficiency gain stems from MLDE's ability to screen full combinatorial libraries in silico after training on a subset of experimentally characterized variants, bypassing the path dependency that plagues traditional approaches [60].
The critical differentiator between standard and optimized MLDE performance lies in training set design. Research demonstrates that reducing the inclusion of minimally informative "holes" (protein variants with zero or extremely low fitness) in training data significantly enhances MLDE effectiveness [60]. Implementation of zero-shot prediction strategies enables construction of more informative training sets that sample the fitness landscape more strategically, dramatically improving the identification of high-fitness variants in epistatic regions [60].
Experimental Protocols and Methodologies
MLDE with Informed Training Set Design
Protocol Objective: To efficiently identify high-fitness protein variants in epistatic environments through machine learning-assisted directed evolution with optimized training sets.
Materials Required:
- Parent protein sequence for mutagenesis
- Library design platform (e.g., htFuncLib web server)
- High-throughput screening capability
- MLDE computational framework
Experimental Workflow:
Figure 1: MLDE with informed training set design workflow for epistatic environments.
Step-by-Step Procedure:
Library Design: Using computational tools like htFuncLib, design combinatorial mutant libraries focused on active-site regions. This server enables scalable library design by generating compatible sets of mutations likely to yield functional multipoint mutants [42].
Zero-Shot Prediction: Apply computational models (e.g., evolutionary statistical potentials, protein language models) to rank all possible variants without experimental data. Use these predictions to minimize selection of "holes" (variants with predicted zero fitness) in training sets [60].
Training Set Selection: Strategically select 200-500 variants that maximize coverage of sequence space while minimizing inclusion of predicted low-fitness variants. This represents approximately 5-15% of a typical 4-6 site combinatorial library [60].
Experimental Characterization: Express and purify selected training set variants. Measure fitness parameters relevant to the engineering goal (e.g., enzymatic activity, binding affinity, fluorescence intensity) using high-throughput assays.
Model Training: Train machine learning models (typically gradient boosting trees or neural networks) using protein sequence features as input and experimental fitness measurements as output. Standard encodings include one-hot, biophysical, and evolutionary representations [60].
In Silico Screening: Use trained models to predict fitness of all possible variants in the combinatorial space (typically thousands to millions of variants).
Validation and Iteration: experimentally test top-ranked predictions. For further optimization, incorporate newly characterized variants into expanded training sets for iterative model refinement.
Traditional Directed Evolution
Protocol Objective: To improve protein function through sequential rounds of mutagenesis and screening without computational guidance.
Materials Required:
- Parent protein sequence
- Mutagenesis method (error-prone PCR, site-saturation mutagenesis)
- Medium-throughput screening capability
Experimental Workflow:
Figure 2: Traditional directed evolution workflow with sequential optimization.
Step-by-Step Procedure:
Initial Mutagenesis: Generate diversity through random mutagenesis or focused mutagenesis at positions believed important for function.
Screening: Screen library for improved variants (typically 100-1000 clones) using functional assays.
Variant Selection: Identify the single best variant from the screening process.
Iteration: Use the selected variant as the new parent for subsequent rounds of mutagenesis and screening.
Termination: Continue process until fitness improvements plateau, typically requiring 5-10 rounds for modest improvements.
Key Limitation: This approach is path-dependent and often fails to identify optimal combinations of mutations in epistatic environments, as beneficial mutations may not be tolerated in intermediate steps [23].
Table 2: Key research reagent solutions for MLDE in epistatic environments
| Resource Category | Specific Tools | Function | Key Features |
|---|---|---|---|
| Library Design | htFuncLib Web Server [42] | Designs multipoint mutant libraries with compatible mutations | Integrates evolutionary and structure-based metrics; scalable design |
| Library Design | Rosetta Computational Suite [42] | Provides energy-based scoring for variant stability and function | Atomistic modeling of epistatic interactions |
| Machine Learning | MLDE Framework [60] | Implements training set optimization and model training | Multiple protein encodings; zero-shot prediction capabilities |
| Experimental Screening | Deep Mutational Scanning [23] | Enables high-throughput fitness characterization | Couples genotype to phenotype; large variant coverage |
| Epistasis Analysis | Epistatic Interaction Mapping [23] | Identifies and quantifies non-additive mutation effects | Reveals functional constraints and evolutionary paths |
Discussion: Strategic Implementation for Research and Development
The comparative data clearly demonstrates that informed training set design is the pivotal factor determining success in highly epistatic environments. Whereas traditional directed evolution succeeds in only ~1% of attempts at finding global fitness maxima in rugged landscapes, optimized MLDE approaches achieve success rates exceeding 97% [60]. This performance advantage translates to an 81-fold improvement in efficiency, dramatically accelerating protein engineering timelines.
For research and development teams, the strategic implication is clear: investment in computational infrastructure and expertise for training set optimization yields substantial returns in experimental efficiency. This is particularly valuable in drug development contexts where engineering therapeutic proteins with novel functions or specificities requires navigating complex epistatic landscapes [23] [42]. The integration of zero-shot prediction methods with experimental validation creates a virtuous cycle of improvement, with each round of data enhancing model accuracy for subsequent designs.
The most successful implementations combine multiple protein engineering strategies—leveraging both sequence-based covariation analysis from natural homologs and structure-based atomistic calculations—to preemptively identify epistatic relationships before experimental testing [23] [42]. This hybrid approach has enabled the design of thousands of functional active-site variants, demonstrating that the space of possible functional sequences is dramatically larger than that explored by natural evolution alone.
Benchmarking and Translational Impact: Validating Predictions for Biomedical Applications
In the field of functional genomics and drug discovery, genetic interactions occur when the combined effect of two or more gene perturbations differs from the expected effect based on their individual impacts [61]. The most strategically valuable of these are synthetic lethal interactions, where simultaneous disruption of two genes results in cell death, while individual disruption does not [61]. These interactions represent promising therapeutic avenues, particularly in oncology, where targeting a gene that is synthetic lethal to a cancer-specific mutation can selectively kill tumor cells while sparing healthy tissues [61].
Advancing from computational predictions to experimentally confirmed interactions requires robust validation frameworks. These frameworks systematically integrate bioinformatic predictions with experimental assessments to distinguish true biological interactions from computational artifacts. The complexity of biological systems, combined with phenomena such as epistasis (where the effect of one mutation depends on the presence of other mutations), makes accurate prediction and validation particularly challenging [42]. This guide compares the leading methodologies and provides experimental data to help researchers select appropriate validation pathways for their specific research contexts.
Computational Prediction Methods
Computational methods form the essential first layer in identifying potential genetic interactions. These tools analyze diverse data types—from genomic sequences to evolutionary patterns—to prioritize gene pairs for experimental testing.
Table 1: Computational Methods for Predicting Genetic Interactions
| Method Name | Underlying Approach | Primary Application | Key Output |
|---|---|---|---|
| ISM (Informational Spectrum Method) | Transforms protein sequences into signals using electron-ion interaction potential (EIIP) and analyzes via Fourier transformation [62] | Predicting mutations affecting protein-receptor interactions, particularly in viral host tropism [62] | Frequencies corresponding to structural motifs with defined physico-chemical characteristics |
| SOCoM (Structure-based Optimization of Combinatorial Mutagenesis) | Uses cluster expansion (CE) to transform structure-based energy evaluations into efficient sequence-function relationships; optimizes libraries via integer linear programming [2] | Designing combinatorial mutagenesis libraries enriched in stable variants based on structural energies [2] | Library-averaged energy scores and optimized variant libraries |
| FuncLib/htFuncLib | Combines evolutionary conservation with Rosetta design calculations to create multipoint mutant libraries, emphasizing residues in active sites [42] | Enzyme and binder optimization through designed combinatorial mutation libraries [42] | Ranked list of multipoint mutant combinations |
| MutPred | Machine learning-based assessment of missense mutations' impact on protein structure and function [63] | Pathogenicity prediction for amino acid substitutions [63] | Pathogenicity score and potential molecular mechanism alterations |
| SynLethDB | Curated database of known synthetic lethal interactions from literature and experimental data [64] | Identification of previously reported synthetic lethal pairs for hypothesis generation [64] | Catalog of known genetic interactions with supporting evidence |
The Informational Spectrum Method (ISM) represents a distinctive approach that analyzes protein sequences without requiring alignment. By converting amino acid sequences into numerical signals based on their electron-ion interaction potentials and applying Fourier transformation, ISM identifies frequency-specific patterns associated with biological functions [62]. This method successfully predicted specific HA mutations (K153D, S223N, and G272S) in H5N1 influenza virus that enhance human receptor specificity, which were subsequently confirmed experimentally [62].
For protein engineering applications, SOCoM and htFuncLib employ structural information to design mutant libraries. SOCoM leverages cluster expansion to efficiently calculate structural energies across sequence space, enabling the optimization of combinatorial libraries containing millions of variants without explicitly modeling each member [2]. The htFuncLib server extends this capability specifically for active-site mutagenesis, generating compatible mutation sets that account for epistatic effects common in densely packed enzymatic centers [42].
Experimental Validation Platforms
Experimental validation provides the essential evidence to confirm computationally predicted genetic interactions. The choice of experimental platform depends on the organism, scale, and specific research questions being addressed.
Table 2: Experimental Platforms for Validating Genetic Interactions
| Platform | Organism/System | Throughput | Key Readout | Limitations |
|---|---|---|---|---|
| SGA (Synthetic Genetic Array) | Yeast | High (can systematically test ~5000 deletion mutants) [61] | Colony size/fitness measurements [61] | Limited to genetically tractable organisms |
| E-MAP (Epistatic Miniarray Profiles) | Yeast | Medium (focused on rationally chosen gene subsets) [61] | Quantitative genetic interaction scores for pathway analysis [61] | Requires pre-selection of gene sets |
| Combinatorial CRISPR | Human and other mammalian cells | Medium to high (test 100s-1000s of pairs) [64] | Fitness effects measured by guide depletion [64] | Delivery efficiency and interpretive complexity |
| shRNA screening | Human cells | Medium | Fitness effects measured by sequence depletion [61] | Higher false-positive rates than CRISPR |
| High-content imaging | Human cells | Low to medium | Multiparametric morphological profiling [61] | Complex data analysis requirements |
Combinatorial CRISPR Screening
Combinatorial CRISPR screening represents the most advanced platform for systematically testing genetic interactions in mammalian systems. This approach utilizes dual-guRNA vectors or paired-guide systems to simultaneously target two genes in the same cell [64]. A prominent study designed to identify synthetic lethal interactions among paralogous genes screened 1,191 gene pairs, including 645 paralogues, 447 predicted synthetic lethal pairs, and 95 literature-curated pairs [64].
Experimental Protocol:
- Library Design: Design 3-5 guides per gene paired both with each other and with non-targeting controls [64]
- Vector Construction: Utilize dual-promoter systems (e.g., human U6 and synthetic U6) assembled via Gibson cloning [64]
- Cell Line Engineering: Generate Cas9-expressing cell lines with ≥90% activity confirmed by reporter assays [64]
- Screen Execution: Transduce at 1000x library representation, maintain for 28 days, with sampling at day 14 and 28 [64]
- Fitness Assessment: Sequence genomic DNA to quantify guide abundance depletion [64]
Data Analysis Approach: The Bliss independence model is commonly applied to assess interaction significance [64]. This model compares the observed fitness effect of paired guides against the expected effect calculated from individual guide activities:
- Expected effect = (Effect of Guide A) + (Effect of Guide B) - (Effect of Guide A × Effect of Guide B)
- Significant interaction: Observed effect significantly exceeds expected effect
This approach identified 105 gene combinations whose co-disruption impaired cellular fitness, with 27 pairs affecting fitness across multiple cell lines [64]. Notable among these was the FAM50A/FAM50B paralogue pair, whose co-disruption not only reduced fitness but also promoted micronucleus formation and transcriptional dysregulation [64].
Protein Mutagenesis Library Validation
For studies focusing on protein engineering rather than genetic interactions, combinatorial mutagenesis libraries provide an alternative validation approach. The SOCoM methodology enables the design of structure-based combinatorial libraries that can be experimentally screened for improved properties [2].
Experimental Protocol:
- Library Design: Define mutable positions and possible amino acid substitutions based on structural constraints [2]
- Variant Construction: Use degenerate oligonucleotides or site-directed mutagenesis to create library members [2]
- Functional Screening: Implement high-throughput assays for target properties (fluorescence, enzymatic activity, binding) [2]
- Variant Characterization: Isolate and characterize individual hits for detailed functional assessment [2]
In application studies, SOCoM-designed libraries for green fluorescent protein, β-lactamase, and lipase A demonstrated improved energy scores compared to previous library design methods and random approaches [2]. The method successfully identified variants with improved stability while covering greater sequence diversity than focused designs [2].
Comparative Performance Assessment
Understanding the relative strengths and limitations of different validation approaches enables researchers to select optimal strategies for their specific needs.
Table 3: Performance Comparison of Validation Methods
| Method | True Positive Rate | Throughput | Cost Efficiency | Context Dependence |
|---|---|---|---|---|
| ISM Prediction + Experimental Confirmation | Successfully identified human-tropic mutations in H5N1 HA [62] | Medium (focused mutation testing) | High (targeted approach) | High (virus-host specific) |
| Combinatorial CRISPR | Identified 27 high-confidence SL pairs across multiple cell lines [64] | High (1000+ pairs) | Medium (requires sequencing) | Medium (varies by cell line) |
| MutPred Pathogenicity Prediction | 92.3% accuracy for ABCB4 variants compared to experimental results [63] | High (computational only) | Very high | Low (generalizable) |
| SOCoM Library Design | Variants with energies better than random library approaches [2] | Very high (millions of variants) | High (per variant cost low) | Medium (structure-dependent) |
The comparative accuracy of computational methods was rigorously assessed in a study of ABCB4 variants, where MutPred achieved 92.3% concordance with experimental results, outperforming Provean, Polyphen-2, and PhD-SNP in predicting pathogenic mutations [63]. Similarly, combinatorial CRISPR screening demonstrated strong validation rates, with approximately 26% of identified synthetic lethal interactions (27/105) confirmed across multiple cell lines [64].
The epistatic effects observed in protein engineering studies highlight the importance of combinatorial approaches. Single mutations often show minimal effects, while specific combinations can dramatically alter function—a phenomenon effectively captured by structure-based methods like SOCoM and htFuncLib [2] [42].
Integrated Validation Framework
Successful validation of genetic interactions requires coordinated application of computational and experimental approaches in an iterative framework.
Case Study: FAM50A/FAM50B Paralogous Pair
The discovery and validation of the FAM50A/FAM50B synthetic lethal interaction illustrates this framework in practice:
- Computational Selection: FAM50A/FAM50B were selected as candidate paralogues based on >20% DNA sequence homology and essentiality of their common orthologue in model organisms [64]
- Combinatorial Screening: Dual-guRNA CRISPR screening in multiple cell lines identified the pair as synthetic lethal [64]
- Phenotypic Characterization: Disruption of both genes led to reduced cellular fitness, apoptosis induction, micronucleus formation, and transcriptional dysregulation [64]
- Clinical Relevance: Approximately 4% of tumors in TCGA datasets show FAM50B silencing, suggesting therapeutic potential [64]
Case Study: Influenza HA Human Tropism
The ISM framework successfully predicted mutations enhancing H5N1 human receptor specificity:
- ISM Analysis: Identified K153D, S223N, and G272S as mutations likely to increase affinity for human receptors [62]
- Experimental Confirmation: Generated pseudotyped particles and recombinant viruses with these mutations [62]
- Functional Validation: Demonstrated enhanced entry and replication in cells expressing human influenza receptors [62]
- Natural Corroboration: Subsequent surveillance revealed increasing prevalence of these mutations in circulating strains [62]
Research Reagent Solutions
Selecting appropriate reagents is crucial for implementing robust validation workflows.
Table 4: Essential Research Reagents for Genetic Interaction Studies
| Reagent Category | Specific Examples | Function in Validation | Considerations |
|---|---|---|---|
| CRISPR Systems | Dual-guRNA vectors (U6/hU6 promoters), Cas9-expressing cell lines [64] | Simultaneous disruption of gene pairs | Promoter strength balance, delivery efficiency |
| Library Construction | Gibson assembly reagents, degenerate oligonucleotides [2] [64] | Generation of variant libraries | Coverage representation, synthesis quality |
| Cell Lines | A375, MeWo, RPE-1 (for CRISPR screening) [64]; MDCK-SIAT1 (for viral tropism) [62] | Provide biological context for functional assays | Genetic background, physiological relevance |
| Bioinformatic Tools | MAGeCK, BAGEL (for screen analysis) [64]; Rosetta (for protein design) [2] [42] | Computational analysis and design | Algorithm parameters, statistical thresholds |
| Detection Reagents | SNA lectin (for receptor detection) [62], viability assays, antibodies | Phenotypic characterization | Specificity, sensitivity, quantitative range |
The validation of genetic interactions has evolved from disconnected computational and experimental approaches to integrated frameworks that systematically connect in-silico predictions with experimental confirmation. Methods like combinatorial CRISPR screening and structure-based library design enable medium- to high-throughput assessment of genetic interactions and protein variants at unprecedented scales.
The most successful validation strategies share common elements: use of multiple orthogonal methods, application of appropriate statistical models (e.g., Bliss independence), and iterative refinement of computational predictions based on experimental results. As the field advances, increasing integration of high-throughput experimental data with sophisticated machine learning approaches promises to further enhance the accuracy and efficiency of genetic interaction validation.
For researchers designing validation studies, the key considerations include selecting methods matched to their throughput requirements, incorporating relevant biological contexts (e.g., cell lines, physiological conditions), and implementing rigorous statistical thresholds to distinguish true interactions from background noise. The frameworks and data presented here provide a foundation for developing optimized validation pipelines specific to particular research objectives and resource constraints.
Comparative Analysis of MLDE Strategies Across Diverse Protein Fitness Landscapes
Machine Learning-assisted Directed Evolution (MLDE) represents a paradigm shift in protein engineering, enabling researchers to navigate the vast sequence-function landscape more efficiently than traditional methods. The core challenge in this optimization process is epistasis—the phenomenon where the effect of a mutation depends on its genetic background [9]. Epistatic interactions can create rugged fitness landscapes with local optima that trap traditional directed evolution, making the protein's adaptive path difficult to predict [65]. This comparative analysis examines three predominant MLDE methodologies—Supervised Learning-based In Silico Optimization, Active Learning-assisted Directed Evolution (ALDE), and Bayesian Optimization (BO)—evaluating their performance across landscapes with varying epistatic complexity. Understanding how these methods handle genetic interactions is crucial for developing effective protein engineering strategies, with significant implications for therapeutic development, including approaches like protein degradation therapies [66].
MLDE Methodologies and Their Approach to Epistasis
Supervised Learning-based In Silico Optimization
This approach trains models on initial sequence-function data to predict fitness across the sequence space, which is then searched in silico to identify optimal variants. Models range from multi-layer perceptrons (MLPs) that learn nonlinear interactions to convolutional neural networks (CNNs) that capture local residue contacts and recurrent neural networks (RNNs) that process sequential information [67]. These models can infer epistatic relationships from the training data, but their performance is highly dependent on data quantity and quality. A significant advancement is the use of low-dimensional protein representations learned from unsupervised learning on large sequence databases (e.g., UniProt), which can distill structural and evolutionary constraints that contribute to epistasis, enabling more accurate prediction with limited experimental data [67].
Active Learning-assisted Directed Evolution (ALDE)
ALDE implements an iterative design-test-learn cycle where machine learning models actively select which sequences to test next based on existing data [45]. This strategy is particularly adept at handling epistasis because it can adaptively explore the fitness landscape, testing combinations of mutations that models are uncertain about or predict to be high-fitness. The acquisition function balances exploration of new regions with exploitation of known promising sequences, allowing ALDE to navigate around epistatic roadblocks [45]. A key strength is its use of frequentist uncertainty quantification, which helps identify sequences that could resolve complex genetic interactions and escape local optima [45].
Bayesian Optimization (BO)
BO employs probabilistic models, traditionally Gaussian processes, to model the sequence-function relationship and quantify prediction uncertainty [67]. By iteratively testing sequences that maximize an acquisition function (e.g., expected improvement), BO systematically reduces uncertainty about the fitness landscape while pursuing optimal variants. Recent advances use ensembles of deep learning models (CNNs, RNNs) for uncertainty estimation, enhancing their capacity to model complex epistatic networks compared to simpler Gaussian processes [67]. This approach is particularly valuable when experimental throughput is limited, as it aims to find optima with the fewest possible function evaluations.
Table 1: Key Characteristics of MLDE Methods
| Method | Core Mechanism | Epistasis Handling | Data Efficiency | Computational Complexity |
|---|---|---|---|---|
| Supervised Learning + In Silico Optimization | One-step training and prediction | Infers epistasis from data patterns; limited by model architecture and data | Lower (requires substantial initial data) | Moderate (depends on model architecture and search space) |
| Active Learning-assisted DE (ALDE) | Iterative cycles with active data selection | Actively probes uncertain epistatic interactions | High (leverages iterative learning) | High (requires repeated model retraining and evaluation) |
| Bayesian Optimization | Probabilistic modeling with uncertainty-directed sampling | Models uncertainty around epistatic interactions | Very High (optimizes for fewest experiments) | High (posterior updates can be computationally intensive) |
Performance Comparison Across Landscapes
Low-Epistasis Fujiyama Landscapes
On smooth, single-peaked landscapes where mutations have largely additive effects, all three MLDE methods significantly outperform traditional directed evolution by reducing experimental screening requirements [67]. Supervised learning approaches excel in this context, as the minimal epistasis allows accurate extrapolation from limited training data. For example, in engineering GB1 binding affinity, a hill-climbing optimization on neural network predictions designed a stable variant with 10 mutations that exhibited substantially increased binding affinity [67]. The relative simplicity of Fujiyama landscapes enables supervised models to identify optimal sequences in a single design step without iterative experimentation.
Rugged, High-Epistasis Landscapes
Landscapes with prevalent epistasis present substantial challenges, as genetic interactions create multiple local optima and complex fitness ridges. In these contexts, ALDE demonstrates particular strength due to its iterative, adaptive nature. In a challenging optimization of five epistatic residues in the active site of a protoglobin (ParPgb) for cyclopropanation activity, ALDE improved the product yield from 12% to 93% in just three rounds while exploring only ~0.01% of the design space [45]. This success occurred despite the failure of single-site saturation mutagenesis and simple recombination approaches, which became trapped by negative epistasis [45]. Bayesian Optimization also performs well on rugged landscapes, with one study engineering acyl-ACP reductases through ten design-test-learn cycles and fewer than 100 experimental measurements to achieve a two-fold increase in product yield [67].
Performance Metrics and Experimental Validation
Table 2: Quantitative Performance Comparison of MLDE Methods
| Method | Experimental Screening Burden | Maximum Fitness Achieved | Rounds to Convergence | Handling of Epistasis |
|---|---|---|---|---|
| Traditional DE | High (10^3-10^6 variants) | Often limited to local optima | 5-20+ | Limited (greedy hill-climbing) |
| Supervised Learning | Moderate (10^2-10^4 variants for training) | Variable (depends on landscape smoothness) | 1 (after initial data collection) | Moderate (requires sufficient training data) |
| ALDE | Low-Moderate (tens to hundreds per round) | High (escapes local optima) | 3-10 | High (actively probes interactions) |
| Bayesian Optimization | Low (as few as 100 total measurements) | High | 5-15 | High (explicit uncertainty modeling) |
The empirical superiority of ALDE for handling epistasis is further demonstrated in computational simulations on combinatorially complete fitness landscapes, where it consistently achieved higher fitness peaks than traditional directed evolution approaches [45]. Performance varied with model architecture, with ensembles providing more reliable uncertainty estimates for guiding exploration in epistatic regions [45].
Experimental Protocols for MLDE Implementation
ALDE Workflow for Epistatic Landscapes
Step 1: Define Combinatorial Space: Select k target residues (typically 4-6) based on structural proximity or known functional importance, creating a 20^k possible sequence space [45].
Step 2: Initial Library Construction: Use NNK degenerate codons or other mutagenesis methods to create an initial diverse library covering the chosen positions [45].
Step 3: High-Throughput Screening: Express and assay variants for target function (e.g., enzymatic activity, binding, stability) using appropriate phenotypic selection or screening assays.
Step 4: Machine Learning Model Training: Train supervised models (CNNs, RNNs, or transformers) on sequence-function data, using evolutionary or biophysical representations to enhance predictive power [67].
Step 5: Sequence Proposal and Iteration: Apply acquisition functions to propose new variant batches that balance exploration and exploitation, then return to Step 3 [45].
Diagram 1: ALDE iterative workflow for epistatic landscapes. The process alternates between wet-lab experimentation (yellow) and computational modeling (green) until an optimal variant is identified.
Bayesian Optimization Protocol
Step 1: Establish Baseline: Characterize starting sequence fitness and define search space boundaries.
Step 2: Initialize Model: Create prior distributions for sequence-fitness relationships using Gaussian processes or deep learning ensembles.
Step 3: Sequential Design: For each iteration: a. Identify sequence maximizing acquisition function (e.g., expected improvement) b. Experimentally characterize selected variant c. Update model with new data [67]
Step 4: Convergence Testing: Continue until fitness improvements plateau or resources are exhausted.
Essential Research Reagents and Tools
Successful implementation of MLDE requires integration of specialized experimental and computational resources.
Table 3: Key Research Reagent Solutions for MLDE
| Category | Specific Tools | Application in MLDE |
|---|---|---|
| Library Construction | NNK degenerate codons; Sequential PCR mutagenesis [45] | Creates diverse variant libraries targeting specific residues |
| High-Throughput Screening | Gas chromatography; Flow cytometry; Microplate assays [45] | Enables rapid functional characterization of thousands of variants |
| Sequence-Function Mapping | every variant Sequencing (evSeq); Long-read every variant Sequencing (LevSeq) [68] | Generates comprehensive training data by pairing genotype with phenotype |
| Protein Representations | eUniRep; Transformer models; Evolutionary coupling analysis [67] | Provides low-dimensional, information-rich sequence encodings |
| ML Frameworks | ALDE codebase (GitHub); Bayesian optimization packages; CNN/RNN architectures [45] [67] | Implements core machine learning algorithms for fitness prediction |
The comparative analysis of MLDE strategies reveals that the optimal choice depends critically on the epistatic complexity of the target protein's fitness landscape. For smooth, additive-dominant landscapes, supervised learning with in silico optimization provides an efficient one-step solution. For rugged, highly epistatic landscapes, ALDE emerges as the superior approach, consistently demonstrating an ability to navigate complex genetic interactions and escape local optima with manageable experimental screening. Bayesian Optimization offers a compelling alternative for resource-constrained environments where experimental throughput is severely limited. As protein engineering increasingly targets challenging therapeutic applications, including protein degraders for previously "undruggable" targets [66], the strategic selection and implementation of MLDE methods will be crucial for success. Future advances will likely come from improved protein representations, better uncertainty quantification, and the integration of structural and biophysical constraints into machine learning models.
The engineering of fluorescent proteins (FPs) for enhanced properties, such as brightness, photostability, or novel emission wavelengths, is a cornerstone of modern biological imaging. However, a central challenge in this endeavor is epistasis, a phenomenon where the functional effect of a mutation depends on the presence or absence of other mutations in the protein sequence [23]. This non-additive interaction creates a rugged fitness landscape, where beneficial mutations can appear deleterious in some genetic backgrounds and vice versa [23] [19]. Consequently, the stepwise accumulation of mutations, a common approach in protein engineering, often fails to reach optimal variants because potential trajectories are blocked by epistatic fitness valleys [23].
This case study is framed within the broader thesis that understanding and mapping epistatic networks is crucial for rational protein design. We will objectively compare two dominant strategies for navigating these complex landscapes: the computationally guided design of combinatorial libraries and the experimental construction and screening of comprehensive mutant libraries. By comparing the protocols, outputs, and practical applications of the htFuncLib web server and a combinatorial nicking mutagenesis method, this guide provides a framework for selecting the optimal strategy for mapping mutational trajectories in a fluorescent protein system, with the aim of mitigating the confounding effects of epistasis.
Experimental Protocols for Mapping Mutational Trajectories
Protocol A: Computational Library Design with htFuncLib
The htFuncLib web server provides a resource for designing focused combinatorial libraries for multipoint mutagenesis in protein active sites, which can be directly applied to the chromophore-containing regions of fluorescent proteins [42].
- Principle: The method leverages evolutionary information and a force-field based energy function to identify combinations of mutations that are predicted to be structurally and functionally compatible, thereby preemptively navigating epistatic constraints [42].
- Procedure: The user submits a protein structure, and the server identifies sensitive positions (e.g., the chromophore environment). It then uses the FuncLib algorithm to generate and rank sequences by calculating the energetic cost of mutations and their combinations. Finally, it outputs a list of designed multipoint mutants or a defined library of sequences for experimental testing [42].
- Key Strengths: This method is highly efficient, drastically reducing the experimental screening burden by focusing on a small, computationally vetted set of variants. It is particularly powerful for exploring profound changes in protein activity that require multiple mutations in core, densely packed positions [23] [42].
Protocol B: Experimental Library Generation via Combinatorial Nicking Mutagenesis
This laboratory protocol enables the rapid assembly of user-defined combinatorial mutagenesis libraries, ideal for systematically exploring epistasis between known beneficial sites in a fluorescent protein [29].
- Principle: This is a template-based mutagenic method where mutagenic oligonucleotides encoding defined degenerate codons are annealed to a single-stranded DNA plasmid template. Enzymatic synthesis, followed by selective degradation of the parental template, yields a dsDNA library encompassing all possible combinations of the targeted mutations [29].
- Procedure:
- Parental DNA Preparation: A plasmid containing the gene of interest and a unique
BbvCInicking restriction site is prepared [29]. - Oligonucleotide Design: Mutagenic primers are designed with degenerate codons (e.g., NNK) at each targeted position, allowing for the encoding of both the parental and mutant residues. Primers should have ~30 bp homology arms for efficient annealing [29].
- Library Synthesis: The mutagenic oligonucleotides are annealed to the ssDNA template in a 5:1 molar ratio. The complementary strand is synthesized using a DNA polymerase, and the original template is nicked and degraded enzymatically [29].
- Transformation and Screening: The resulting dsDNA library is transformed into E. coli, and clones are screened for desired fluorescent properties. The protocol can create libraries of up to 16,384 variants (14 mutated positions) with low parental carry-over in two days [29].
- Parental DNA Preparation: A plasmid containing the gene of interest and a unique
- Key Strengths: This method is highly scalable and can empirically test a vast number of combinatorial states without computational prediction, making it agnostic to the specific molecular mechanisms of epistasis.
Comparative Performance Analysis
The table below summarizes a direct comparison between the two featured methodologies for mapping trajectories in a fluorescent protein system.
Table 1: Comparison of Strategies for Mapping Epistatic Trajectories in Fluorescent Proteins
| Feature | Computational Design (htFuncLib) | Experimental Library (Combinatorial Nicking) |
|---|---|---|
| Underlying Principle | Force-field & evolutionary-based prediction of compatible mutations [42] | Empirical testing of all user-defined combinatorial states [29] |
| Typical Library Size | Hundreds to millions of designed variants [42] | Up to 16,384 variants (14 positions) demonstrated [29] |
| Coverage of Sequence Space | Focused; explores a specific, energetically favorable region [42] | Comprehensive for defined positions; can cover all possible combinations [29] |
| Primary Output | A ranked list of multipoint mutant sequences [42] | A physical library of DNA clones [29] |
| Handling of Epistasis | Attempts to pre-calculate and avoid negative epistasis [23] [42] | Empirically reveals epistatic interactions through functional screening [29] |
| Experimental Screening Burden | Low (library is pre-filtered) [42] | High (requires screening of a large library) [29] |
| Best Use Case | Introducing new activities or optimizing densely packed sites with strong epistasis [23] [42] | Comprehensive evaluation of epistasis between a known set of beneficial mutations [29] |
Data Interpretation and Visualization
Conceptualizing the Fitness Landscape
Epistasis shapes the protein fitness landscape from a smooth, mountaineering-like terrain into a rugged terrain with multiple peaks and valleys. The following diagram illustrates how epistasis affects evolutionary trajectories toward a high-fitness fluorescent protein.
This ruggedness means that the direct path from a starting variant to an improved one may be blocked by a non-functional intermediate (A→B). Successful engineering requires identifying alternative, viable trajectories, such as the neutral path (B→A) [23] [19].
Workflow for Mapping Trajectories
Integrating both computational and experimental approaches provides a powerful strategy for comprehensively mapping trajectories. The following workflow outlines this integrated process.
Successful mapping of epistatic trajectories requires a suite of computational and experimental tools. The table below lists key resources for such a project.
Table 2: Key Research Reagent Solutions for Epistasis Mapping
| Item/Tool Name | Function in Epistasis Mapping |
|---|---|
| htFuncLib Web Server | Computationally designs focused combinatorial libraries of multipoint mutants to pre-empt negative epistasis in active sites [42]. |
| Combinatorial Nicking Mutagenesis | Experimental protocol for generating physical DNA libraries containing all combinations of user-defined mutations at multiple positions [29]. |
| Deep Mutational Scanning | An experimental approach (not detailed here) that can provide the initial single-mutation data to identify candidate positions for combinatorial libraries [23]. |
| ImageJ Plugin CGE | A software tool for quantifying circadian gene expression in live-cell microscopy, exemplifying the type of analysis needed for dynamic, single-cell fluorescence tracking [69]. |
| MATtrack | An open-source MATLAB platform for analyzing protein trafficking from time-lapse microscopy, useful for processing fluorescent protein movies [70]. |
| PRO-Simat | A web-based tool for simulating and analyzing protein interaction networks, which can help model the systemic effects of mutations [71]. |
The choice between computational library design and empirical combinatorial library generation hinges on the specific goals and constraints of the fluorescent protein engineering project.
- Use
htFuncLibwhen the objective is to introduce a new function or make profound changes to a densely packed region like the chromophore environment. Its strength lies in leveraging physical models to propose novel, functional combinations that would be difficult to find by chance, effectively "smoothing" the fitness landscape through intelligent design [23] [42]. - Use Combinatorial Nicking Mutagenesis when you have a defined set of candidate mutations (e.g., from a deep mutational scan) and the goal is to understand the epistatic network between them comprehensively. It is the preferred tool for an unbiased, empirical mapping of all possible trajectories within a defined sequence space, directly revealing the ruggedness of the landscape [29].
For the most robust mapping of all possible trajectories, an integrated approach is optimal. Computational design can narrow the vast sequence space to promising regions, which are then explored comprehensively with empirical libraries. This synergistic strategy accelerates the discovery of high-fitness fluorescent proteins by directly confronting and leveraging the pervasive reality of epistasis.
The study of epistasis, or the context-dependence of mutational effects, has moved from a theoretical concept to a central consideration in protein engineering. The genetic architecture of a protein—the set of causal rules by which its sequence determines its specific functions—directly shapes its evolutionary potential and the functional impacts of mutations [72]. In practical terms, understanding epistasis is crucial for engineering enzymes with enhanced catalytic properties, developing stable therapeutic antibodies, and interpreting the pathological significance of disease-associated mutations. Historically, protein engineering often treated mutations as having additive effects, but contemporary research reveals that epistatic interactions are pervasive and fundamentally shape protein fitness landscapes [10] [72].
Recent methodological advances now enable researchers to move beyond studying single mutations and instead comprehensively analyze combinatorial mutant libraries. These approaches include deep mutational scanning, machine learning-guided engineering, and sophisticated computational modeling. This guide objectively compares how epistasis research is transforming different biotechnological domains by examining experimental data, protocols, and outcomes across enzyme engineering, antibody development, and disease mutation studies. We focus specifically on how epistatic effects in combinatorial mutant libraries create both constraints and opportunities for protein design, providing a framework for researchers to navigate the complex sequence-function relationships that define protein engineering challenges.
Comparative Analysis of Epistatic Effects Across Biological Systems
Table 1: Comparative Analysis of Epistatic Effects Across Protein Engineering Domains
| Domain | Primary Epistatic Pattern | Key Experimental Findings | Impact on Engineering Outcomes | Quantitative Evidence |
|---|---|---|---|---|
| Enzyme Engineering | Predominantly pairwise interactions enabling new functions | Machine learning models revealed epistatic mutations in PylRS tRNA-binding domain that improved catalytic efficiency 30.8-fold | Enables divergence of generalist enzymes into multiple specialists with optimized activities | 11-30.8x improvement in catalytic efficiency; 1.6-42x increased activity for amide synthetases [73] [74] |
| Antibody Development | Structural epistasis affecting stability and aggregation | Bispecific antibodies and scFv fragments show 2-3x lower stability and higher aggregation propensity than full-length IgGs | Constrains design of non-natural antibody formats, requiring stability-enhancing mutations | 90% purity for full-length mAbs vs. <90% for bispecifics; 2-3x more fragmentation in engineered formats [75] |
| Transcription Factor Evolution | Dense pairwise interactions determining DNA specificity | Global genetic architecture analysis of steroid hormone receptor revealed pairwise epistasis massively expands functional sequence space | Facilitates evolutionary transitions between DNA binding specificities rather than constraining them | 97% concordance in activation classification; thousands of functional variants identified [72] |
Table 2: Methodological Approaches for Studying Epistasis
| Methodology | Throughput | Epistatic Order Captured | Key Applications | Limitations |
|---|---|---|---|---|
| Combinatorial Deep Mutational Scanning | 160,000 variants for 4 sites | Up to 4th order interactions | Transcription factor specificity mapping; comprehensive genetic architecture dissection | Limited to focused site sets due to combinatorial explosion [72] |
| Machine Learning-Guided Cell-Free Engineering | 1,217 enzyme variants in 10,953 reactions | Pairwise and higher-order interactions | Enzyme substrate specificity engineering; fitness landscape mapping | Requires substantial initial dataset for model training [73] |
| Structure-Guided Consensus Design | Moderate (10-100 variants) | Primarily additive effects with structural constraints | Thermostability enhancement; ancestral sequence reconstruction | May miss long-range epistatic interactions [76] |
| 3DM Database Analysis | Family-wide (1,000+ sequences) | Correlated mutations indicating historical epistasis | Substrate range expansion; activity enhancement across protein families | Limited to naturally occurring variations [76] |
Experimental Protocols for Epistasis Research
Combinatorial Deep Mutational Scanning for Transcription Factor Specificity
Objective: To comprehensively map the genetic architecture of DNA recognition specificity in a steroid hormone receptor DNA-binding domain by characterizing all amino acid combinations at four critical recognition helix positions [72].
Materials and Reagents:
- Template plasmid containing ancestral steroid hormone receptor (AncSR1) DNA-binding domain
- Oligonucleotide library encoding all 20 amino acids at four targeted sites (160,000 total combinations)
- Yeast reporter strains with ERE-driven GFP and SRE-driven GFP
- FACS equipment for Sort-seq assay
- High-throughput sequencing platform
Procedure:
- Library Construction: Synthesize a combinatorial mutant library using degenerate codons to achieve full coverage of all 20 amino acids at four recognition helix positions (total library size: 160,000 variants).
- Transformation: Divide the library and transform into separate yeast reporter strains containing either ERE-GFP or SRE-GFP reporter constructs.
- Functional Assay: Induce transcription factor expression and sort cells based on GFP fluorescence intensity into three functional categories: null, weak, and strong activators for each response element.
- Sequencing and Analysis: Isolate plasmid DNA from sorted populations and perform high-throughput sequencing to quantify variant abundance in each functional category.
- Genetic Architecture Modeling: Apply logistic regression-based modeling to dissect main effects, pairwise interactions, and higher-order epistasis from the categorical functional data.
Key Considerations: This protocol requires careful normalization to controls and computational correction for measurement noise. The categorical functional classification (null/weak/strong) improves reproducibility beyond continuous fluorescence measurements, achieving >97% replicate concordance [72].
Machine Learning-Guided Enzyme Engineering in Cell-Free Systems
Objective: To engineer amide bond-forming enzymes with enhanced activity for specific pharmaceutical compounds by mapping sequence-function relationships and predicting epistatic interactions using machine learning [73].
Materials and Reagents:
- Wild-type McbA gene from Marinactinospora thermotolerans
- Cell-free protein synthesis system (E. coli extract-based)
- ATP regeneration system
- Substrate libraries (acids and amines)
- LC-MS equipment for reaction quantification
- Computational resources for machine learning (ridge regression models)
Procedure:
- Hot Spot Identification: Perform site-saturation mutagenesis at 64 active site residues (1,216 variants) using cell-free DNA assembly and expression.
- Substrate Profiling: Test wild-type McbA against 1,100 substrate combinations to identify potential engineering targets.
- Variant Screening: Express mutant libraries in cell-free system and assay activity against three target compounds (moclobemide, metoclopramide, cinchocaine) under industrially-relevant conditions.
- Model Training: Use single-mutant activity data to train augmented ridge regression machine learning models with evolutionary zero-shot fitness predictors.
- Prediction and Validation: Predict higher-order mutants with improved activity, synthesize top candidates, and experimentally validate performance.
Key Considerations: The cell-free approach enables rapid iteration without cellular transformation steps. The machine learning models specifically account for epistatic interactions when predicting higher-order mutants, requiring both positive and negative fitness data for robust training [73].
Figure 1: Machine Learning-Guided Enzyme Engineering Workflow. This diagram illustrates the integrated computational and experimental pipeline for engineering enzymes with improved functions, incorporating epistatic effects in predictive models.
Epistasis in Enzyme Engineering and Design
Engineering enzymes for industrial applications requires navigating complex fitness landscapes where epistatic interactions strongly influence outcomes. Rational enzyme engineering has evolved from simple additive models to approaches that explicitly account for epistatic interactions between mutations [76]. The growing recognition of epistasis has driven the development of sophisticated computational tools that can predict how mutations will interact in different sequence contexts.
Machine learning has emerged as a particularly powerful approach for modeling epistatic effects in enzyme engineering. Recent work on pyrrolysyl-tRNA synthetase (PylRS) demonstrates how machine learning can guide the engineering of enzymes with dramatically improved catalytic efficiency [74]. Researchers first applied FFT-PLSR machine learning models to explore pairwise combinations of 12 single mutations, generating a variant (Com1-IFRS) with an 11-fold increase in stop codon suppression efficiency. Subsequent rounds of engineering using deep learning models (ESM-1v, Mutcompute, and ProRefiner) identified additional mutation sites, resulting in a variant (Com2-IFRS) with a 30.8-fold improvement in catalytic efficiency. Importantly, these epistatic mutations in the tRNA-binding domain could be transplanted into seven other PylRS-derived synthetases, significantly improving yields of proteins containing six different noncanonical amino acids [74].
The practical implications of epistasis extend to enzyme stability and substrate specificity. Structure-guided consensus approaches have successfully enhanced thermostability by introducing ancestral or consensus mutations, but these efforts are complicated by epistatic interactions that can destabilize the protein or reduce activity [76]. For example, when engineering an α-amino ester hydrolase from Xanthomonas campestris, researchers combined consensus approach with B-factor iterative test method, resulting in a quadruple mutant (E143H/A275P/N186D/V622I) with 7°C improvement in thermostability and 1.3-fold higher activity compared to wild-type [76]. The success of such campaigns depends on identifying combinations of mutations that interact favorably to enhance stability without compromising catalytic function.
Antibody Engineering and Developability Challenges
Therapeutic antibody development faces unique challenges related to epistatic effects, particularly when engineering non-natural formats like bispecifics and antibody fragments. A systematic comparison of 64 antibody constructs targeting TNF revealed that overall developability is highest for the natural full-length antibody format, with more complex engineered formats exhibiting intermediate to poor developability properties [75]. This study measured 15 biophysical properties related to activity, manufacturing, and stability, demonstrating that epistatic interactions in engineered antibodies can lead to fragmentation and aggregation issues not observed in natural IgG structures.
Table 3: Antibody Format Developability Comparison Based on Biophysical Properties
| Antibody Format | Relative Developability | Key Stability Challenges | Manufacturing Concerns | Recommended Applications |
|---|---|---|---|---|
| Full-length IgG | High | Minimal fragmentation and aggregation | High purity (>95%) after standard purification | First-line therapeutic development |
| scFv-Fc | Intermediate | Moderate aggregation propensity | Acceptable purity with optimized processes | Extended half-life fragment applications |
| Bispecific mAb-scFv | Intermediate-low | Interface instability between domains | Lower purity, requires additional purification steps | Targets requiring dual specificity |
| Diabody/scFv-scFv | Low | High aggregation and fragmentation | Significant heterogeneity, low yield | Diagnostic and imaging applications |
The developability challenges observed in engineered antibody formats directly result from epistatic interactions between domains that did not co-evolve naturally. For instance, linking single-chain variable fragments (scFvs) in bispecific formats creates new molecular interfaces that can be destabilizing, leading to aggregation-prone regions not present in either parent antibody [75]. These epistatic effects necessitate extensive engineering efforts to introduce stabilizing mutations that counteract the destabilizing interactions, often through rational design or directed evolution approaches.
Antibody humanization represents another domain where epistatic interactions critically influence outcomes. The process of grafting complementarity-determining regions (CDRs) from non-human antibodies into human framework sequences often results in affinity loss due to epistatic interactions between CDR and framework residues [77] [78]. Restoration of binding typically requires back-mutation of specific framework residues to maintain the structural context necessary for proper CDR conformation, demonstrating how epistasis constrains the sequence space available for humanized variants with optimal properties.
Computational Tools for Analyzing Epistatic Effects
The growing recognition of epistasis in protein engineering has driven development of specialized computational tools for analyzing and predicting epistatic effects. These tools help researchers navigate complex fitness landscapes and identify beneficial mutations despite epistatic constraints [79].
Table 4: Computational Tools for Protein Engineering Categorized by Application
| Tool Category | Representative Tools | Primary Application | Epistatic Modeling Capability | Experimental Data Requirement |
|---|---|---|---|---|
| Molecular Docking | DOCK, GOLD, ICM, FlexX | Enzyme-substrate recognition, binding affinity prediction | Limited to structural constraints | Protein structure required |
| Machine Learning Models | FFT-PLSR, ESM-1v, Mutcompute, Ridge Regression | Fitness prediction, variant prioritization | Explicit modeling of pairwise and higher-order interactions | Large variant activity datasets |
| Sequence Analysis | 3DM, Consensus Design, SCHEMA | Thermostability enhancement, family-wide activity optimization | Correlated mutation analysis, historical epistasis | Multiple sequence alignments |
| Genetic Architecture Mapping | Logistic Regression (DMS analysis) | Comprehensive epistasis mapping, specificity determinants | Full dissection of main effects and interactions | Combinatorial library data |
Machine learning approaches have shown particular promise for modeling epistatic relationships in protein sequences. Supervised methods like ridge regression can capture epistatic effects when trained on large variant activity datasets, enabling prediction of higher-order mutants with improved properties [73]. Meanwhile, deep learning models like ESM-1v leverage evolutionary information to infer epistatic constraints from natural sequence variation, providing zero-shot predictions of mutation effects even without experimental data on the specific protein being engineered [74].
The FFT-PLSR (Fast Fourier Transform-Partial Least Squares Regression) model represents a specialized approach for engineering epistatic enzymes. This method transforms protein sequences into numerical representations using physicochemical properties from the AAindex database, then applies Fourier transformation to create protein "spectra" that capture both individual residue contributions and epistatic interactions between positions [74]. This approach has successfully engineered PylRS variants with substantially improved activity, demonstrating the practical value of explicitly modeling epistatic effects in enzyme engineering campaigns.
Figure 2: Computational Framework for Analyzing Epistatic Effects. This diagram illustrates how different computational approaches address epistasis across various protein engineering applications, leading to proteins with enhanced functions.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 5: Essential Research Reagents for Epistasis Studies in Protein Engineering
| Reagent/Resource | Function in Epistasis Research | Specific Application Examples | Key Providers/Platforms |
|---|---|---|---|
| Combinatorial Mutant Libraries | Comprehensive mapping of genetic interactions | Deep mutational scanning of transcription factor DNA recognition; enzyme active site saturation | Custom synthesized; Twist Bioscience, Integrated DNA Technologies |
| Cell-Free Protein Synthesis Systems | Rapid variant expression without cellular constraints | Machine learning-guided enzyme engineering; high-throughput protein stability screening | PURExpress (NEB), homemade E. coli extracts |
| Phage Display Libraries | In vitro selection of functional binders | Antibody affinity maturation; protein-protein interaction engineering | Human synthetic scFv libraries, immune libraries |
| 3DM Protein Super-Family Databases | Analysis of evolutionary constraints and correlations | Identifying functional residues; thermostability engineering | Bio-Prodict (commercial), custom-built databases |
| Machine Learning Platforms | Prediction of epistatic interactions and variant fitness | Ridge regression for enzyme engineering; deep learning for PylRS optimization | Python scikit-learn, PyTorch, TensorFlow |
| Surface Plasmon Resonance (SPR) | Quantitative binding affinity measurements | Antibody-antigen interaction kinetics; enzyme-substrate binding characterization | Biacore systems, Carterra LSA platform |
| Stable Cell Lines | Functional characterization in biological context | Antibody effector function assessment; therapeutic protein production | CHO, HEK293 expression systems |
The selection of appropriate research reagents critically influences the success of epistasis studies. Combinatorial mutant libraries with comprehensive coverage enable robust genetic architecture analysis, while cell-free expression systems accelerate the testing of variant libraries without the bottlenecks of cellular transformation and culture [73]. Specialized databases like 3DM platforms that integrate structural, sequence, and mutational data across protein families provide valuable insights into historical epistatic constraints that have shaped natural enzyme evolution [76].
Emerging technologies particularly machine learning platforms and deep mutational scanning methodologies are transforming epistasis research by enabling the systematic analysis of interaction networks that were previously intractable. These tools allow researchers to move beyond studying isolated mutations to comprehensively characterizing the complex interaction networks that define protein fitness landscapes. As these technologies mature, they promise to accelerate the engineering of enzymes and therapeutic proteins with customized functions and enhanced properties.
Conclusion
The systematic evaluation of epistasis in combinatorial libraries is fundamental to advancing protein engineering and understanding genetic disease. The integration of high-throughput experimentation, robust computational design, and sophisticated machine learning models is successfully overcoming the historical challenges posed by epistasis, particularly in critical regions like enzyme active sites. These approaches reveal that while epistasis creates rugged fitness landscapes that constrain evolutionary paths, it also provides the nonlinearity necessary for profound functional innovations. Future progress hinges on developing more predictive biophysical models, creating standardized databases of epistatic interactions, and further refining AI tools that can generalize across diverse protein systems. For biomedical research, this deeper understanding promises more effective strategies for engineering therapeutic proteins, interpreting the pathogenicity of genetic variants across individuals, and ultimately, harnessing the full complexity of the genotype-phenotype map for clinical benefit.
References
- 1. Epistasis in protein evolution - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4918427/]
- 2. Structure-based design of combinatorial mutagenesis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4420537/]
- 3. Epistasis facilitates functional evolution in an ancient ... [https://elifesciences.org/articles/88737]
- 4. Epistasis and evolution: recent advances and an outlook for ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-023-01585-3]
- 5. Epistasis facilitates functional evolution in an ancient ... [https://elifesciences.org/reviewed-preprints/88737]
- 6. Epistasis Is a Major Determinant of the Additive Genetic ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1005201]
- 7. The interplay of additivity, dominance, and epistasis on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8933436/]
- 8. A method for finding epistatic effects of maternal and fetal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11995191/]
- 9. Distinguishing direct interactions from global epistasis using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12501145/]
- 10. Review Epistatic drift in protein evolution [https://www.sciencedirect.com/science/article/pii/S0959437X25001042]
- 11. Fluidity and Predictability of Epistasis on an Intragenic ... [https://elifesciences.org/reviewed-preprints/104848]
- 12. Adaptation in protein fitness landscapes is facilitated by ... [https://elifesciences.org/articles/16965]
- 13. Epistasis in Allosteric Proteins: Can Biophysical Models ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283625003936]
- 14. Epistasis and the Structure of Fitness Landscapes [https://pmc.ncbi.nlm.nih.gov/articles/PMC4896198/]
- 15. Evolution of Epistatic Networks and the Genetic Basis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6925314/]
- 16. Rugged fitness landscapes minimize promiscuity in the ... [https://www.sciencedirect.com/science/article/pii/S2405471224000620]
- 17. Machine learning-coupled combinatorial mutagenesis ... [https://www.nature.com/articles/s41467-022-29874-5]
- 18. High-throughput analysis of epistasis in genome-wide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3400955/]
- 19. Considerations in the search for epistasis | Genome Biology [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03427-z]
- 20. Sparse modeling of interactions enables fast detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12461027/]
- 21. Epistasis facilitates functional evolution in an ancient ... [https://elifesciences.org/reviewed-preprints/88737v2]
- 22. FlyBase:Interaction Browser [https://wiki.flybase.org/wiki/FlyBase:Interaction_Browser]
- 23. Addressing epistasis in the design of protein function - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11348311/]
- 24. SBOL Visual: A Graphical Language for Genetic Designs - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4669170/]
- 25. Gene and Pathways Interactions Graph User's Guide [https://genome.ucsc.edu/goldenPath/help/hgGeneGraph.html]
- 26. Evaluation of machine learning-assisted directed evolution ... [https://www.sciencedirect.com/science/article/abs/pii/S2405471225002200]
- 27. Epistasis facilitates functional evolution in an ancient ... [https://elifesciences.org/reviewed-preprints/88737v2/reviews]
- 28. A Rapid, High-Throughput Method for the Construction of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12649946/]
- 29. Facile Assembly of Combinatorial Mutagenesis Libraries ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9730894/]
- 30. Random Mutagenesis: 8 Approaches for Success [https://bitesizebio.com/252/8-approaches-to-random-mutagenesis/]
- 31. An efficient one-step site-directed deletion, insertion, single ... [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/1472-6750-8-91]
- 32. Performance evaluation of commercial library construction kits ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-5583-7]
- 33. Deep mutational scanning: A versatile tool in systematically ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9878224/]
- 34. Deep Mutational Scanning in Immunology: Techniques ... [https://www.mdpi.com/2076-0817/14/10/1027]
- 35. Variant scoring tools for deep mutational scanning - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12494760/]
- 36. Variant scoring tools for deep mutational scanning [https://link.springer.com/article/10.1038/s44320-025-00137-x]
- 37. Rosace: a robust deep mutational scanning analysis ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03279-7]
- 38. Multi-environment deep mutational scanning reveals the ... [https://www.sciencedirect.com/science/article/pii/S2211124725012173]
- 39. Deep-learning structure elucidation from single-mutant ... [https://www.nature.com/articles/s41467-025-62261-4]
- 40. Statistical analysis of mutational epistasis to reveal ... [https://www.sciencedirect.com/science/article/abs/pii/S0076687920302871]
- 41. Designed active-site library reveals thousands of functional ... [https://www.nature.com/articles/s41467-023-38099-z]
- 42. htFuncLib: Designing Libraries of Active-site Multipoint ... [https://www.sciencedirect.com/science/article/pii/S0022283625000774]
- 43. About FuncLib - Ablift [http://ablift.weizmann.ac.il/about-funclib/]
- 44. Learning the pattern of epistasis linking genotype and ... [https://www.nature.com/articles/s41467-019-12130-8]
- 45. Active learning-assisted directed evolution [https://www.nature.com/articles/s41467-025-55987-8]
- 46. Machine learning-guided co-optimization of fitness and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11289365/]
- 47. Combinatorial explosion [https://en.wikipedia.org/wiki/Combinatorial_explosion]
- 48. complexity theory - What is combinatorial explosion? [https://cs.stackexchange.com/questions/19459/what-is-combinatorial-explosion]
- 49. Cutting-edge Algorithms to Overcome Combinatorial ... [https://www.rd.ntt/e/research/JN202501_31214.html]
- 50. Experimental Design on a Budget for Sparse Linear Models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5415092/]
- 51. Combinatorial Genetics Reveals a Scaling Law for the ... [https://www.sciencedirect.com/science/article/pii/S0092867418316246]
- 52. Distinguishing direct interactions from global epistasis using ... [https://pubmed.ncbi.nlm.nih.gov/40986352/]
- 53. Rank Method - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/mathematics/rank-method]
- 54. A rank-based marker selection method for high throughput ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03641-z]
- 55. Semiparametric Efficient Fusion of Individual Data and ... [https://arxiv.org/html/2210.00200v6]
- 56. Systematic comparison of ranking aggregation methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9620830/]
- 57. Library analysis, comparison, and cost efficiency calculation [https://docs.openprotein.ai/walkthroughs/quantitative-decision-making-library-design.html]
- 58. Chem3DLLM: 3D Multimodal Large Language Models for ... [https://arxiv.org/html/2508.10696v1]
- 59. Using machine learning to map simulated noisy and laser ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00125k]
- 60. Informed training set design enables efficient machine learning-assisted directed protein evolution - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S2405471221002866]
- 61. Prediction of Genetic Interactions Using Machine Learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4620407/]
- 62. In Silico Prediction and Experimental Confirmation of HA ... [https://www.nature.com/articles/srep11434]
- 63. Comparison of in silico prediction and experimental ... [https://www.sciencedirect.com/science/article/abs/pii/S1357272517301267]
- 64. Combinatorial CRISPR screen identifies fitness effects of ... [https://www.nature.com/articles/s41467-021-21478-9]
- 65. Exploring protein by directed evolution - PMC fitness landscapes [https://pmc.ncbi.nlm.nih.gov/articles/PMC2997618/]
- 66. and Sibylla Biotech announce strategic... | Newswise MD Anderson [https://www.newswise.com/articles/md-anderson-and-sibylla-biotech-announce-strategic-collaboration-to-discover-and-develop-small-molecule-protein-degraders]
- 67. Machine learning to navigate fitness landscapes for protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9177649/]
- 68. Methods in Protein Engineering - Arnold Group [http://fhalab.caltech.edu/?page_id=171]
- 69. Circadian Gene Expression (CGE) - Biomedical Imaging Group [https://bigwww.epfl.ch/sage/soft/circadian/]
- 70. MATtrack: A MATLAB-Based Quantitative Image Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4616565/]
- 71. PRO-Simat: Protein network simulation and design tool [https://www.sciencedirect.com/science/article/pii/S2001037023001757]
- 72. Epistasis facilitates functional evolution in an ancient ... [https://elifesciences.org/reviewed-preprints/88737v1/pdf]
- 73. Accelerated enzyme engineering by machine-learning ... [https://www.nature.com/articles/s41467-024-55399-0]
- 74. Machine learning-guided evolution of pyrrolysyl-tRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12274524/]
- 75. A comparative study of the developability of full-length ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11457596/]
- 76. Recent advances in rational approaches for enzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3962183/]
- 77. Development of therapeutic antibodies for the treatment of ... [https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-019-0592-z]
- 78. Technologies for Monoclonal Antibody Discovery and ... [https://www.mdpi.com/1422-0067/26/21/10470]
- 79. A Practical Guide to Computational Tools for Engineering ... [https://www.mdpi.com/1422-0067/26/3/980]