Engineering the Future: Directed Evolution of XNA Polymerases for Advanced Synthetic Biology
The field of synthetic biology is being transformed by xenonucleic acids (XNAs), synthetic genetic polymers that offer nuclease resistance and novel chemical functionalities not found in nature.
Engineering the Future: Directed Evolution of XNA Polymerases for Advanced Synthetic Biology
Abstract
The field of synthetic biology is being transformed by xenonucleic acids (XNAs), synthetic genetic polymers that offer nuclease resistance and novel chemical functionalities not found in nature. This article explores the central role of directed evolution in engineering DNA polymerases capable of synthesizing and replicating these 'un-natural' genetic polymers. We cover the foundational principles of XNA polymerase engineering, detail advanced methodological pipelines for their evolution and application—including the synthesis of mixed XNA polymers like 2′-azide/2′-fluoro—and provide a comprehensive guide to troubleshooting and optimizing selection protocols. Furthermore, we examine the critical validation of these engineered enzymes through high-throughput sequencing and crystallographic analysis, highlighting their immense potential for creating secure therapeutic agents, durable biosensors, and catalytic XNA enzymes (XNAzymes) for biomedical and clinical research.
The XNA Frontier: Building Polymerases for Synthetic Genetics
The Critical Need for Engineered Polymerases in XNA Synthesis
Xeno Nucleic Acids (XNAs) are synthetic genetic polymers not found in nature, characterized by modified sugar-phosphate backbones or nucleobases that confer superior properties compared to natural DNA and RNA, such as enhanced nuclease resistance and novel functional capacities [1] [2]. These properties make XNAs invaluable for biomedical applications, including the development of advanced therapeutics, diagnostics, and aptamers [2]. However, a significant bottleneck impedes their widespread application: natural DNA polymerases are generally incapable of efficiently recognizing and incorporating XNA nucleotides [1]. This limitation creates a critical need for engineered DNA polymerases, developed through directed evolution and rational protein design, to enable the enzymatic synthesis, reverse transcription, and amplification of XNAs, thereby unlocking their full potential for synthetic biology and drug development [1] [3] [4].
Table 1: Key Challenges in XNA Synthesis and the Role of Engineered Polymerases
| Challenge | Impact on Research | Engineered Polymerase Solution |
|---|---|---|
| Poor Substrate Recognition | Natural polymerases fail to incorporate many XNA nucleotides [1]. | Directed evolution creates active site mutations to accept modified nucleotides [4]. |
| Low Fidelity | Error-prone synthesis and reverse transcription of XNA [1]. | Protein engineering of palm and finger domains to enhance accuracy [4]. |
| Limited Reverse Transcription | Inability to convert XNA back to DNA for analysis and evolution [3]. | Engineering of XNA-dependent DNA polymerases [3] [2]. |
| Strand Displacement | Hinders isothermal amplification methods like LAMP [4]. | Use of engineered Bst DNA polymerase large fragment [4]. |
Engineered Polymerases as Enabling Tools
Key Polymerase Families and Engineering Strategies
The engineering of XNA-compatible polymerases primarily focuses on thermostable enzymes from the A-family (e.g., the Stoffel fragment (SF) of Taq polymerase) and the B-family (e.g., polymerases from Pyrococcus and Thermococcus) [1] [2]. Thermostable polymerases are particularly amenable to engineering because their inherent structural stability allows them to tolerate mutations that would destabilize other proteins [2].
Engineering strategies involve:
- Directed Evolution: This involves creating vast mutant libraries and screening for variants with enhanced XNA synthesis activity. A key methodology is cross-chemistry selective enrichment by exponential amplification (X-SELEX), which enables the selection of functional XNA enzymes (XNAzymes) from diverse repertoires of synthetic genetic polymers [3].
- Rational Design: This approach leverages structural knowledge of the polymerase. Mutations are introduced into specific domains to improve interactions with the modified sugar or backbone of the XNA. For instance, mutations in the palm, thumb, and fingers domains can modulate fidelity, substrate specificity, and strand displacement efficiency [4].
- Fusion Proteins: Polymerases can be fused with other domains, such as DNA-binding motifs, to enhance properties like processivity, DNA affinity, and salt tolerance, which are crucial for diagnostic applications [4].
Quantitative Performance of Engineered Polymerases
The success of polymerase engineering is quantitatively demonstrated by the ability of these enzymes to synthesize and reverse transcribe various XNA chemistries with high efficiency and fidelity.
Table 2: Performance Metrics of Engineered Polymerases with Various XNAs
| XNA Chemistry | Engineered Polymerase | Key Application | Reported Fidelity / Efficiency |
|---|---|---|---|
| 2'-Azido (2'Az) / 2'-Fluoro (2'F) Mixed Polymers | Stoffel Fragment (SF) Mutants [1] | Synthesis of click-chemistry compatible, nuclease-resistant XNA [1] | Improved fidelity relative to previous systems; synthesis with high accuracy [1] |
| Arabino Nucleic Acids (ANA), 2'-F-ANA, HNA, CeNA | Engineered B-family polymerases [3] | XNAzyme development via X-SELEX [3] | Successful in vitro selection of fully substituted catalysts [3] |
| Phosphonate Nucleic Acid (phNA) | Engineered Polymerases [2] | In vitro evolution of aptamers with an uncharged backbone [2] | First demonstration of genetic function in an uncharged backbone [2] |
| General XNA Synthesis | Engineered Bst DNA Polymerase [4] | Reverse transcription of XNA for sequencing and analysis [4] | Recognizes and reverse transcribes templates with diverse chemical compositions [4] |
Application Notes & Experimental Protocols
This section provides detailed methodologies for the enzymatic synthesis and analysis of mixed XNA polymers, a key application for engineered polymerases.
Protocol: Enzymatic Synthesis of Mixed 2'Az/2'F XNA Polymers
This protocol describes the use of engineered XNA polymerases to synthesize mixed polymers containing 2'-Azide and 2'-Fluoro modifications, adapted from published research [1].
Research Reagent Solutions
Table 3: Essential Reagents for XNA Synthesis and Reverse Transcription
| Reagent / Material | Function / Role | Example / Source |
|---|---|---|
| Engineered XNA Polymerase | Catalyzes the incorporation of XNA nucleotides. | Stoffel Fragment (SF) mutants [1]. |
| 2'-Azide NTPs & 2'-Fluoro NTPs | Modified nucleotide substrates for XNA synthesis. | TriLink Biotechnologies [1]. |
| Primer & DNA Template | Provides the sequence information for directed synthesis. | Commercially synthesized (e.g., IDT) [1]. |
| Turbo DNase | Degrades the DNA template post-synthesis to isolate pure XNA. | Invitrogen [1]. |
| Phusion or Q5 DNA Polymerase | High-fidelity PCR amplification of the cDNA from reverse transcription. | New England Biolabs, Fisher Scientific [1]. |
Step-by-Step Procedure
Reaction Setup:
- Combine the following in a reaction tube to create a 2X mixture:
- 40 nM 5'-IRDye700-labeled primer.
- 80 nM 100mer DNA template.
- 1X SF buffer (50 mM Tris pH 8.5, 6.5 mM MgCl₂, 0.05 mg/mL Ac-BSA, 50 mM KCl).
- 20 nM engineered XNA polymerase (e.g., SF mutant).
- Prepare a separate 2X mixture of XNA nucleotide substrates. For 2'Az/2'F mixed polymers, use 25 µM 2'F-NTPs and 100 µM 2'Az-NTPs [1].
- Initiate the synthesis by mixing equal volumes of the two 2X mixtures.
- Incubate the reaction on a heat block at 50°C for 2 hours [1].
- Combine the following in a reaction tube to create a 2X mixture:
Product Analysis:
- Remove a 3 µL aliquot and quench it with 6 µL of Quenching Buffer (95% formamide, 12.5 mM EDTA, trace Orange G).
- Analyze the quenched samples on a 10% TBE-urea gel for 45 minutes at 120 V.
- Visualize the synthesized XNA products using an Odyssey CLx imager or similar system [1].
Protocol: One-Pot Reverse Transcription and Amplification of XNA
This protocol allows for the conversion of synthesized XNA back into DNA for sequencing and analysis, a process critical for in vitro selection [1].
XNA Purification:
- To the remaining 17 µL of synthesis reaction, add 2 µL of Turbo DNase (0.11 units/µL).
- Incubate at 37°C for 40 minutes to digest the DNA template.
- Purify the XNA product using a commercial oligonucleotide clean-up kit (e.g., Zymo Research Oligo Clean & Concentrator) according to the manufacturer's protocol [1].
Reverse Transcription and Amplification (RT/Amp):
- Set up a PCR mixture containing:
- 3 µL of the purified XNA.
- 1X manufacturer's buffer (e.g., Phusion GC buffer or Q5 reaction buffer).
- 0.5 µM barcoded reverse primer, 0.5 µM forward primer.
- 0.4 mM dNTPs.
- 1 U of a high-fidelity DNA polymerase (e.g., Phusion or Q5).
- Run the following thermocycling program:
- 98°C for 30 seconds.
- 3 cycles of: 98°C for 5 s, 50°C for 15 s, 72°C for 1 min.
- 12 cycles of: 98°C for 5 s, 67°C for 15 s, 72°C for 15 s.
- 72°C for 5 min.
- Hold at 4°C [1].
- Analyze the amplified cDNA products by agarose gel electrophoresis.
- Set up a PCR mixture containing:
Workflow Visualization
The following diagram illustrates the complete experimental workflow for the synthesis and analysis of XNA, from initial template-driven synthesis to final sequence analysis.
Discussion and Future Perspectives
The directed evolution of polymerases has been a transformative pursuit for synthetic genetics. Engineered enzymes now allow for the synthesis, reverse transcription, and evolution of numerous XNA chemistries, moving the field from fundamental proof-of-concept studies toward practical applications [3] [2]. The development of protocols like X-SELEX enables the discovery of XNAzymes (XNA-based enzymes) and high-affinity XNA aptamers, opening new avenues for therapeutic intervention [3].
Future directions in polymerase engineering will likely focus on:
- Expanding XNA Chemical Diversity: Engineering polymerases to handle an even wider array of backbone and nucleobase modifications, including completely neutral backbones like phosphonate nucleic acids (phNA) [2].
- Improving Fidelity and Processivity: Continuous optimization of polymerase domains to enhance the accuracy and length of XNA synthesis, which is critical for reliable data storage and functional molecule development [4].
- Integration with Diagnostics: Leveraging engineered polymerases like Bst DNA polymerase in point-of-care testing (POCT) devices for robust, isothermal detection of nucleic acids, including XNA-based biomarkers [4].
As these tools mature, XNAs are poised to become central components of next-generation therapeutics, diagnostics, and synthetic biological systems, fulfilling their promise as genetically encoded polymers with expanded chemical and functional capabilities.
Xeno-nucleic acids (XNAs) represent a class of synthetic genetic polymers characterized by modified sugar moieties or backbone structures, differing from the natural deoxyribose and ribose sugars of DNA and RNA. The enzymatic synthesis and replication of these molecules are cornerstone activities in the field of synthetic biology, enabling the in vitro evolution of XNAs for therapeutic and biotechnological applications. These processes rely on engineered polymerases capable of processing XNA substrates. This application note details the key properties of prominent XNAs, provides validated protocols for assessing critical enzymatic functions like fidelity, and discusses advanced methods to promote efficient XNA synthesis, thereby supplying researchers with the foundational tools for directed evolution of XNA polymerases.
Xeno-nucleic acids (XNAs) are artificially sugar-modified nucleic acids, with alterations to the sugar-phosphate backbone, that serve as alternative genetic polymers [5]. The development of XNAs is driven by their potential to address limitations of natural nucleic acids, particularly for therapeutic applications. Many XNAs exhibit increased biostability against nuclease digestion and enhanced thermodynamic properties for Watson-Crick base pairing compared to their natural counterparts [2]. These characteristics make them ideal candidates for the development of advanced aptamers, catalysts (XNAzymes), and genetic systems in synthetic biology [5] [2].
The ability to propagate genetic information using XNAs requires specialized laboratory-evolved polymerases. These enzymes must catalyze two fundamental steps: the synthesis of XNA from a DNA template (forward transcription) and the reverse transcription of XNA back into DNA. A functional comparison of such polymerases has revealed that the mutations enabling XNA synthesis often come with a trade-off, sometimes sacrificing protein-folding stability for greater substrate tolerance [6].
Table 1: Key Properties of Common Xeno-Nucleic Acids (XNAs)
| XNA Type | Full Name | Backbone/Sugar Modification | Key Properties | Notable Applications |
|---|---|---|---|---|
| FANA | 2'-Fluoroarabino nucleic acid | Arabinose sugar with 2'-fluorine | Capable of evolution; forms stable duplexes [6] [2] | In vitro selection of functional molecules [2] |
| HNA | 1,5-Anhydrohexitol nucleic acid | Hexitol sugar | Chemically and enzymatically stable; capable of evolution [7] [8] | Aptamer and XNAzyme development [5] [8] |
| ANA | Arabino nucleic acid | Arabinose sugar | Forms unstable duplexes with DNA at higher temperatures [5] | Model for studying synthesis constraints |
| TNA | Threose nucleic acid | Threose sugar | Capable of evolution; nuclease-resistant [9] [2] | In vitro selection of functional molecules [2] |
| phNA | Phosphonate nucleic acid | Uncharged phosphonate linkage | Genetic function in an uncharged backbone [2] | Demonstration of expanded chemical diversity for evolution [2] |
Experimental Protocols
Protocol: Measuring XNA Polymerase Fidelity Using Hydrogel Particles
Conventional methods for measuring the fidelity of XNA synthesis and reverse transcription involve large-scale reactions and cumbersome purification via denaturing polyacrylamide gel electrophoresis (PAGE). The following protocol utilizes a hydrogel particle-based system to drastically reduce the time, scale, and reagent consumption of this assay [9].
Principle: A DNA primer is covalently cross-linked within a polyacrylamide hydrogel matrix on magnetic particles. The XNA replication cycle (DNA→XNA→DNA) occurs entirely within this matrix, eliminating the need for intermediate physical purification steps [9].
Materials:
- DNA Primer: 5'-Acrydite-modified primer (cross-links to hydrogel).
- Template: DNA template of defined sequence.
- XNA Triphosphates (xNTPs): e.g., TNA, FANA, or HNA triphosphates.
- Engineered XNA Polymerases: For forward and reverse transcription.
- Hydrogel Particles: Dynabeads M-270 carboxylic acid.
- Chemicals: Acrylamide/Bis-acrylamide (19:1), ammonium persulfate (APS), TEMED, and appropriate reaction buffers.
Procedure:
- Functionalized Hydrogel Particle Preparation: [9]
- Resuspend magnetic beads in a degassed mixture of 6% acrylamide/bis-acrylamide, acrydite-primer, and APS.
- Add an oil mixture and TEMED, then emulsify by vigorous mixing.
- Incubate on ice for 2 hours to allow polymerization, forming primer-functionalized hydrogel particles.
- Wash particles thoroughly with breaking buffer (e.g., 10 mM Tris-HCl, pH 7.5, 1% SDS, 1% Triton X-100) to remove oil and unincorporated reagents.
XNA Synthesis on Hydrogels: [9]
- Anneal the hydrogel-bound primer to the DNA template.
- Perform the primer extension reaction by adding the appropriate engineered XNA polymerase and xNTPs. This step transcribes the DNA template into an XNA product that remains trapped within the hydrogel matrix.
- Remove the DNA template and reagents by washing the magnetic particles with a suitable buffer.
Reverse Transcription: [9]
- With the XNA product now serving as a template, perform a second primer extension reaction using a reverse transcriptase (engineered polymerase that synthesizes DNA from XNA) and dNTPs.
- This generates a complementary DNA (cDNA) strand, which is recovered from the hydrogel matrix for analysis.
Fidelity Analysis: [9]
- Amplify the recovered cDNA via PCR.
- Clone the PCR products and subject them to Sanger or next-generation sequencing.
- Compare the sequences of the initial DNA template and the final cDNA product to identify mutations. The error rate is calculated as the number of mutations per total nucleotides sequenced.
Protocol: Promoting XNA Synthesis with Polyamines
Thermophilic polymerase mutants are often used for XNA synthesis, but some XNAs that form unstable duplexes with DNA (e.g., ANA) dissociate at the high temperatures optimal for these enzymes. This protocol describes the use of polyamines to stabilize nascent duplexes and promote efficient synthesis [5].
Principle: Polyamines like spermine and spermidine bind to nucleic acids, stabilizing the duplex formed between the growing XNA strand and the DNA template. This stabilization prevents dissociation during synthesis, enabling fuller-length transcription, especially for challenging XNAs like ANA, 2'-OMe-RNA, and 2'-F-RNA mixtures [5].
Materials:
- DNA Template & Primer
- xNTPs (e.g., ANA triphosphates)
- Engineered Thermophilic Polymerase (e.g., B-strain polymerase variants)
- Polyamines: Spermine, spermidine, cadaverine, or putrescine.
- Standard PCR or Transcription Buffers
Procedure:
- Reaction Setup: [5]
- Prepare a standard XNA transcription reaction mixture containing the DNA template, primer, engineered polymerase, and xNTPs.
- Supplement the reaction with a polyamine (e.g., 1-6 mM spermine). A negative control without polyamines should be run in parallel.
Incubation:
- Incubate the reaction at the optimal temperature for the polymerase (typically 50-65°C) for a determined period (e.g., 1-2 hours).
Analysis:
- Analyze the products by denaturing PAGE or other appropriate methods.
- Compare the yield and length of the XNA product in the polyamine-supplemented reaction versus the control. The promotion of synthesis is evidenced by a significant increase in full-length product.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for XNA Polymerase Research
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| Engineered Polymerases | Catalyze synthesis & reverse transcription of XNAs | Thermostable B-family polymerases (e.g., from Pyrococcus); variants for FANA, HNA, TNA [6] [2] |
| XNA Triphosphates (xNTPs) | Substrates for enzymatic XNA synthesis | Chemically synthesized TNA, FANA, HNA triphosphates [9] [8] |
| Polyamines | Stabilize DNA/XNA duplexes to promote synthesis | Spermine, spermidine; used in transcription/RT reactions for ANA, 2'-OMe-RNA [5] |
| ChromaTide Nucleotides | Fluorescent labeling for detection & tracking | Alexa Fluor-conjugated dUTP/UTP; incorporated enzymatically for FISH, microarrays [10] |
| Hydrogel Magnetic Particles | Solid-phase matrix for miniaturized fidelity assays | Polyacrylamide-encapsulated Dynabeads for DNA→XNA→DNA replication cycle [9] |
| Aminoallyl-dNTPs (aha-dNTPs) | Two-step labeling of synthesized nucleic acids | aha-dUTP/aha-dCTP enzymatically incorporated, then conjugated to amine-reactive dyes [10] |
Directed Evolution of XNA Polymerases
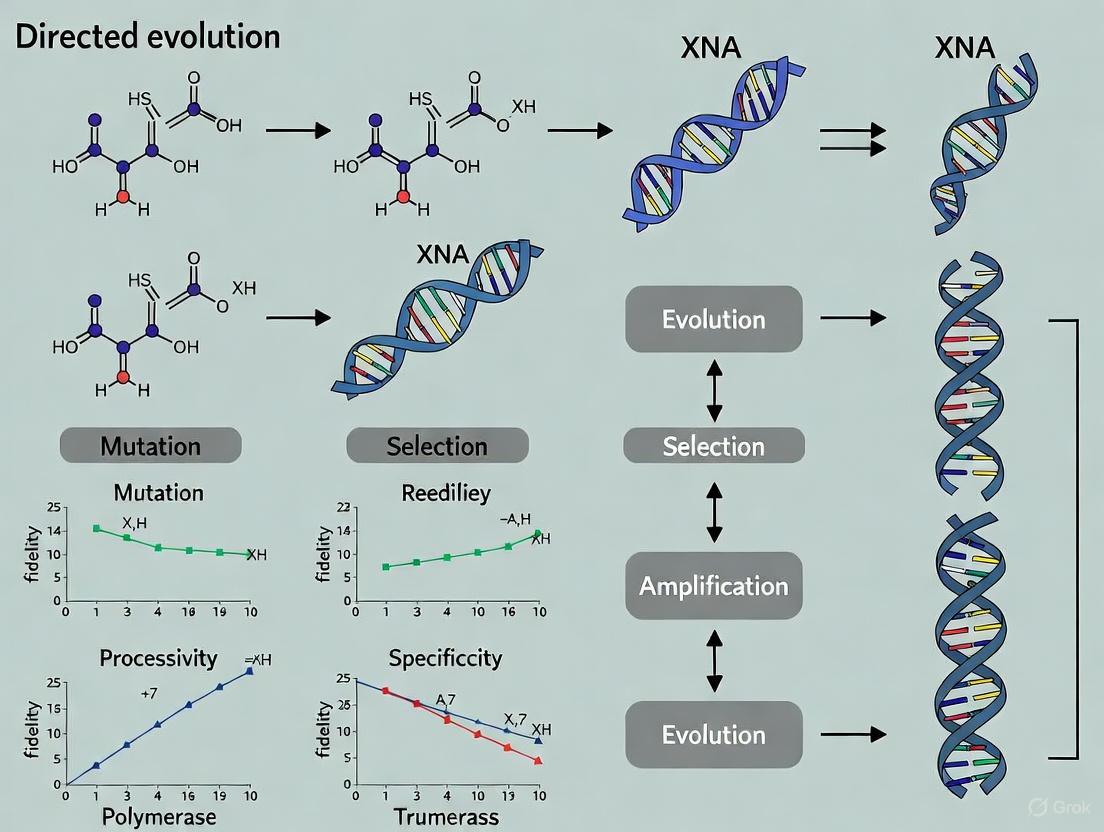
The discovery and optimization of polymerases that can handle XNA substrates is a cornerstone of the field. Directed evolution is the primary method for engineering these enzymes. This process involves creating vast libraries of polymerase mutants and applying stringent selection pressure to isolate variants capable of efficient XNA synthesis and reverse transcription.
Thermophilic B-family polymerases, particularly from Pyrococcus and Thermococcus species, have proven to be highly amenable starting points for engineering due to their high thermostability, which allows them to tolerate mutations that would be destabilizing in other enzymes [2]. Successfully evolved XNA polymerases have enabled the in vitro selection of functional XNA molecules, such as aptamers (binding molecules) and XNAzymes (catalysts), opening up new frontiers for therapeutic and diagnostic applications [5] [2].
The following diagram illustrates the core workflow for the directed evolution of XNA polymerases and their subsequent application in generating functional XNA molecules.
Diagram 1: Directed evolution of XNA polymerases for functional XNA discovery.
Advanced XNA Synthesis and Therapeutic Applications
The efficient enzymatic synthesis of diverse XNAs is critical for preparing high-quality libraries used in selecting XNA aptamers and XNAzymes. Expanding the chemical diversity of these libraries increases the potential for discovering molecules with high affinity and novel functions [5]. Recent advances have demonstrated that mixed backbone chemistries and the combination of multiple modifications can yield oligonucleotides with emergent properties, such as enhanced thermal stability or novel base-pairing rules [2].
The therapeutic potential of XNAs is immense. They form the basis of new modalities for therapeutic intervention, including antisense oligonucleotides (ASOs), siRNAs, and aptamers [2]. Several therapeutics based on modified nucleic acids have been approved, with many more under clinical evaluation. XNAs improve upon first-generation nucleic acid therapeutics by offering increased resistance to nuclease degradation and reduced off-target effects, thereby enhancing their potency and bioavailability [2]. Conjugating carbohydrates to oligonucleotides, as highlighted in recent research, is another powerful strategy for improving the therapeutic profile of these drugs [11].
The field of synthetic biology is advancing beyond the natural constraints of DNA and RNA toward the exploration of artificial genetic polymers known as xeno-nucleic acids (XNAs). These synthetic genetic systems feature structurally distinct sugar-phosphate backbones while retaining the capacity for hereditary information storage and enzymatic function [12]. However, a fundamental challenge persists: natural DNA polymerases exhibit stringent substrate specificity, making them inefficient at synthesizing and reverse-transcribing XNA polymers [2]. This application note examines the directed evolution of XNA polymerases, focusing on methodologies to overcome substrate specificity barriers for applications in therapeutic development and biotechnology. The ability to efficiently replicate XNAs enables their development as biostable therapeutics, functional catalysts, and diagnostic tools resistant to nuclease degradation [13] [2].
The Substrate Specificity Challenge
Natural DNA polymerases have evolved exquisite specificity for their native substrates—deoxyribonucleotide triphosphates (dNTPs). Their catalytic efficiency drops dramatically when confronted with XNA nucleotides (xNTPs), which feature modified sugars or backbone structures [2]. This specificity arises from structural constraints within the polymerase active site that sterically and chemically exclude non-native substrates.
Key structural barriers include:
- Sugar-pocket constraints: The active site pocket accommodating the nucleotide sugar is precisely sized for natural ribose or deoxyribose rings, excluding larger or differently shaped sugar modifications [13].
- Geometric incompatibility: The spatial orientation of functional groups in XNA substrates often differs from natural nucleotides, disrupting the precise positioning required for phosphodiester bond formation [2].
- Metal ion coordination: Natural polymerases utilize metal ions to facilitate catalysis, and modifications to the triphosphate or sugar can disrupt essential coordination chemistry [2].
These limitations manifest in dramatically reduced catalytic performance. Quantitative studies reveal that wild-type polymerases exhibit substrate specificities of approximately 0.1-5-fold for xNTPs versus dNTPs, with synthesis rates as slow as 1-80 nucleotides per minute for XNA synthesis [14].
Table 1: Key XNA Polymerases and Their Properties
| Polymerase Name | Origin | XNA Substrates | Catalytic Rate (nt/s) | Fidelity | Key Applications |
|---|---|---|---|---|---|
| 10-92 (evolved variant) | Directed evolution | TNA | ~1 | >99% [13] | TNA aptamer selection, information storage [13] |
| TgoT 6G12 | Thermococcus gorgonarius | HNA | Not specified | Not specified | HNA synthesis [12] |
| Kod-RI | Archaeal | TNA | Not specified | Not specified | TNA synthesis [12] |
| SF mutants | Thermus aquaticus (Stoffel fragment) | 2′-azido (2′Az), 2′-fluoro (2′F) XNA | Not specified | High accuracy for mixed polymers [1] | Synthesis of click-chemistry compatible XNA [1] |
| Therminator DNA polymerase | Engineered variant | TNA | Not specified | Not specified | TNA synthesis [12] |
Directed Evolution Strategies for Engineering XNA Polymerases
Directed evolution has emerged as a powerful approach to overcome the substrate specificity of natural polymerases. These strategies mimic natural selection in the laboratory to identify polymerase variants with enhanced XNA synthesis capabilities.
Library Creation through Homologous Recombination
Recent advances utilize homologous recombination libraries to create diverse starting populations. This approach allows amino acids to recombine independently, generating novel protein sequences with increased evolutionary potential [13]. Initial libraries created through DNA shuffling of polymerase genes from diverse species provide a rich source of structural variation from which functional XNA polymerases can emerge [13].
Compartmentalized Screening Methodologies
A critical innovation in polymerase evolution is the use of hydrogel particle display for fidelity measurements. This approach encapsulates primer-template complexes within polyacrylamide hydrogel matrices covalently linked to magnetic beads [9]. The hydrogel environment enables solution-like enzyme kinetics while physically linking the genetic template to the synthesized XNA product throughout replication cycles, eliminating the need for laborious purification steps between transcription and reverse transcription [9].
Polymerase Kinetic Profiling (PKPro)
The PKPro method enables high-throughput quantification of XNA polymerase activity using standard qPCR instrumentation. This approach monitors XNA synthesis in real-time through fluorescent dye intercalation as the template strand is copied into XNA [14]. The method allows parallel analysis of up to 288 different reaction conditions, dramatically accelerating the screening process [14].
Diagram 1: Directed Evolution Workflow for XNA Polymerase Engineering. The process involves iterative cycles of library creation, screening, and selection to evolve polymerases with enhanced XNA synthesis capabilities.
Experimental Protocols for XNA Polymerase Evaluation
Polymerase Kinetic Profiling (PKPro) for Activity Assessment
Purpose: To quantitatively measure the average rate and substrate specificity of XNA polymerases using standard qPCR instrumentation [14].
Materials:
- Purified XNA polymerase variant
- Self-priming DNA template with defined sequence
- xNTP substrates (chemically synthesized)
- HRM fluorescent dye (e.g., SYBR Green, EvaGreen)
- qPCR instrument with melting curve analysis capability
- Chemically synthesized XNA standards for calibration
Procedure:
- Prepare reaction mixture containing:
- 40 nM self-priming template
- 1× polymerase reaction buffer
- 6.5 mM MgCl₂
- HRM fluorescent dye at manufacturer's recommended concentration
- Variable xNTP concentrations (typically 25-100 μM each)
Initiate reaction by adding polymerase (final concentration 20 nM)
Monitor fluorescence continuously in qPCR instrument at appropriate temperature (typically 50-60°C)
Calculate synthesis rate by comparing fluorescence increase to calibration curve generated with chemically synthesized XNA standards
Determine substrate specificity by comparing rates with xNTPs versus dNTPs under identical conditions
Data Analysis:
- Plot fluorescence versus time to determine initial velocity
- Calculate catalytic rate in nucleotides per second
- Determine substrate specificity as the ratio of catalytic efficiency (kcat/KM) for xNTP versus dNTP
Hydrogel-Based Fidelity Measurement
Purpose: To rapidly determine error rates of XNA polymerases without denaturing PAGE purification [9].
Materials:
- Acrydite-modified DNA primer
- Magnetic Dynabeads M-270 carboxylic acid
- Acrylamide/bisacrylamide (19:1) solution
- Ammonium persulfate and TEMED
- DNA template with defined sequence for fidelity assessment
- XNA triphosphates
- XNA polymerase and reverse transcriptase
- PCR reagents for amplification (Phusion or Q5 polymerase)
- TOPO-TA cloning kit
Procedure: Hydrogel Particle Preparation:
- Resuspend magnetic beads in 6% acrylamide/bisacrylamide solution containing acrydite-modified primer
- Add 0.6% ammonium persulfate and mix with oil phase containing emulsifiers
- Initiate polymerization by adding TEMED and mix vigorously to form particles
- Incubate 2 hours on ice, then wash with breaking buffer to remove oil
XNA Synthesis on Hydrogels:
- Anneal template to primer-functionalized hydrogel particles
- Perform XNA synthesis reaction with polymerase and xNTPs
- Remove template by denaturation and washing
- Reverse transcribe XNA back to cDNA using engineered reverse transcriptase
Fidelity Analysis:
- Amplify cDNA by PCR using barcoded primers
- Clone products using TOPO-TA kit
- Sequence multiple clones and compare to original template sequence
- Calculate error rate as (total mutations)/(total nucleotides sequenced)
Advantages: Reduces assay time from 1 week to 1-2 days; decreases xNTP consumption 10-fold; eliminates tedious gel purification steps [9].
Mixed XNA Polymer Synthesis
Purpose: To assess polymerase capability to incorporate multiple modified nucleotides simultaneously, creating mixed XNA polymers [1].
Materials:
- Engineered XNA polymerase (e.g., SF mutant)
- 5′-IRDye700-labeled DNA primer
- DNA template (100mer)
- 2′-azido (2′Az) NTPs
- 2′-fluoro (2′F) NTPs
- Reaction buffer: 50 mM Tris (pH 8.5), 6.5 mM MgCl₂, 0.05 mg/mL BSA, 50 mM KCl
Procedure:
- Prepare reaction mixture with:
- 40 nM fluorescently labeled primer
- 80 nM template
- 1× reaction buffer
- 20 nM XNA polymerase
- Mixed xNTP substrates (e.g., 25 μM F-NTPs, 100 μM Az-NTPs)
Incubate at 50°C for 2 hours
Quench with 2 volumes of formamide/EDTA loading buffer
Separate products by 10% TBE-urea PAGE
Visualize using infrared imaging system
For reverse transcription assessment:
- Treat synthesized XNA with Turbo DNase to remove template
- Purify using Oligo Clean & Concentrator Kit
- Perform reverse transcription and amplification with barcoded primers
- Analyze by high-throughput sequencing
Table 2: Research Reagent Solutions for XNA Polymerase Engineering
| Reagent Category | Specific Examples | Function & Application | Commercial Sources |
|---|---|---|---|
| XNA Triphosphates | 2′-azido (2′Az) NTPs, 2′-fluoro (2′F) NTPs, TNA triphosphates, HNA triphosphates | Substrates for XNA synthesis; 2′Az enables click chemistry conjugation | TriLink Biotechnologies, Metkinen, Chemical synthesis [1] [9] |
| Engineered Polymerases | SF mutants, TgoT 6G12, Kod-RI, Therminator variants | Catalyze XNA synthesis and reverse transcription; specialized for different XNA backbones | Laboratory evolution, commercial suppliers [1] [12] |
| Specialty Oligonucleotides | Acrydite-modified primers, IRDye700-labeled primers, DNA templates | Enable immobilization and detection in various assay formats | Integrated DNA Technologies [1] [9] |
| Hydrogel Matrix Components | Acrylamide/bisacrylamide, TEMED, ammonium persulfate, magnetic Dynabeads | Create compartmentalized environments for screening and fidelity assays | Bio-Rad, Thermo Fisher Scientific, Sigma-Aldrich [9] |
| Analysis Kits | TOPO-TA cloning kit, Oligo Clean & Concentrator Kit, DNA Clean & Concentrator | Purify and analyze XNA synthesis products | Thermo Fisher Scientific, Zymo Research [9] |
Applications in Synthetic Biology and Therapeutics
The ability to synthesize and replicate XNAs enables diverse applications leveraging their enhanced biostability and functional capacity:
- Biostable Aptamers: TNA and FANA aptamers have been evolved to bind disease targets like PD-1/PD-L1 for cancer immunotherapy, exhibiting superior serum stability compared to DNA/RNA counterparts [13].
- Catalytic XNAs (XNAzymes): TNA and FANA enzymes capable of RNA cleavage and ligation have been developed, demonstrating that catalysis is not unique to natural nucleic acids [15].
- Genetic Information Storage: TNA polymers have been used for digital information storage and retrieval, creating biochemically stable archives resistant to nuclease degradation [13].
- Mixed Polymer Therapeutics: XNAs containing 2′-azido modifications enable precise conjugation of therapeutic payloads via click chemistry, creating targeted delivery systems [1].
Diagram 2: Application Pipeline for Engineered XNA Polymerases. Engineered polymerases enable synthesis of functional XNAs with applications in therapeutics, catalysis, and information storage.
Directed evolution has transformed our ability to engineer polymerase enzymes capable of synthesizing and reverse transcribing XNA polymers. Through methods like homologous recombination library generation, hydrogel-based fidelity screening, and polymerase kinetic profiling, researchers can now generate specialized polymerases that overcome the innate substrate specificity of natural enzymes. These engineered polymerases serve as enabling tools for synthetic biology, facilitating the development of XNAs with applications as stable therapeutics, functional catalysts, and robust information storage systems. As evolution strategies continue to advance, the functional gap between natural and synthetic genetic polymers will narrow, potentially ushering in a new era of genetic engineering based on XNA systems with tailor-made chemical properties.
The directed evolution of Xeno Nucleic Acid (XNA) polymerases represents a pivotal advancement in synthetic biology, enabling the replication and functional exploitation of synthetic genetic polymers. These engineered enzymes facilitate the synthesis, reverse transcription, and evolution of XNAs, which possess novel chemical properties such as enhanced nuclease resistance and increased thermodynamic stability. This application note details the use of evolved XNA polymerases in three key domains: the selection of high-affinity XNA aptamers, the discovery of catalytic XNAzymes, and the development of robust DNA data storage systems. Supported by structured experimental data and detailed protocols, this document provides a framework for researchers to integrate these tools into their synthetic biology and therapeutic development pipelines.
Natural DNA polymerases exhibit high substrate specificity, which historically precluded the enzymatic synthesis and replication of nucleic acids with non-natural backbones or sugars, known as Xeno Nucleic Acids (XNAs) [16]. Directed evolution has been used to engineer novel polymerase variants capable of processing these synthetic genetic polymers, thereby expanding the central dogma of molecular biology [17] [2]. These engineered polymerases are foundational to a new field of "synthetic genetics," which applies principles of heredity and evolution to artificial genetic systems [15].
XNAs are characterized by modifications to the sugar-phosphate backbone or nucleobases, conferring advantageous properties including superior biostability against nucleases and enhanced affinity for their targets compared to natural DNA and RNA [18] [2] [19]. The ability to enzymatically synthesize and reverse transcribe XNAs is a prerequisite for their functional exploration through in vitro evolution. This technical note outlines practical applications and methodologies for utilizing these evolved polymerases in aptamer, catalyst, and data storage development.
Application Note 1: XNA Aptamers for Therapeutics and Diagnostics
Rationale and Background
Aptamers are structured oligonucleotides that bind to specific molecular targets with high affinity and specificity. Conventional DNA and RNA aptamers are limited by their rapid degradation in biological environments. Fully modified XNA aptamers address this limitation by combining the evolvability of nucleic acids with the biostability and novel chemistries of XNAs [2]. Selections from XNA libraries (X-SELEX) have been enabled by engineered polymerases that can synthesize and reverse transcribe XNA, facilitating the isolation of aptamers against therapeutic targets [15] [19].
Key Experimental Data and Reagents
The following table summarizes key XNA chemistries and their properties relevant to aptamer development:
Table 1: XNA Modifications for Advanced Aptamer Development
| XNA Type | Key Modification | Relevant Properties | Example Polymerase |
|---|---|---|---|
| 2'-Fluoro (2'F) | 2'-F ribose | Nuclease resistance, improved duplex stability [1] | Engineered SF mutant [1] |
| Hexitol (HNA) | 1,5-anhydrohexitol | Nuclease resistance, stable duplex formation [18] | KOD-H4, Tgo-H4 [18] |
| Threose (TNA) | α-L-threofuranosyl sugar | Nuclease resistance, stable against degradation [20] | Engineered B-family polymerase [20] |
| 2'-Azide (2'Az) | 2'-N₃ ribose | Biorthogonal handle for click chemistry conjugation [1] | Engineered SF mutant [1] |
Experimental Protocol: X-SELEX for XNA Aptamer Selection
Objective: To isolate target-specific aptamers from a fully modified XNA library. Key Reagents:
- Engineered XNA polymerase (e.g., Tgo-H4 for HNA [18])
- XNA nucleoside triphosphate mix (e.g., 2'F-, 2'Az-NTPs [1])
- Synthetic DNA template library (randomized region flanked by constant sequences)
- Purified target protein (e.g., VEGF-165, PCSK9 [17] [2])
Procedure:
- Library Synthesis: Incubate the single-stranded DNA (ssDNA) template library (100 nM) with the engineered XNA polymerase (20-100 nM) and XNA-NTPs (25-100 µM each) in the appropriate reaction buffer (e.g., 1x ThermoPol buffer) for 1-2 hours at 50-55°C [1] [18].
- Target Selection: Incubate the synthesized XNA library with the immobilized target. Wash thoroughly to remove unbound and weakly bound sequences.
- Elution: Recover the target-bound XNA sequences.
- Reverse Transcription: Use the same or a specialized engineered polymerase (e.g., KOD-H4 for HNA [18]) to reverse transcribe the eluted XNA into complementary DNA (cDNA). This is a critical step that requires a polymerase with efficient XNA → DNA activity.
- Amplification: Amplify the cDNA by PCR using standard DNA polymerases.
- Transcription (Optional): For subsequent selection rounds, use the PCR product as a template to generate a new DNA library for the next round of XNA synthesis. Alternatively, if the polymerase can directly use a DNA template for XNA synthesis, the PCR product can be used directly.
- Iteration: Repeat steps 1-6 for 5-15 rounds to enrich high-affinity binders. The resulting pools can be characterized by high-throughput sequencing and individual clones tested for binding.
Application Note 2: XNAzymes as Catalytic Biomolecules
Rationale and Background
XNAzymes are catalytic XNAs that can mediate biochemical reactions, such as transphosphorylation and RNA cleavage [15]. They combine the programmability and evolvability of nucleic acid enzymes with the enhanced stability of XNA backbones, making them promising candidates as gene therapeutic agents and diagnostic sensors. The discovery of XNAzymes demonstrates that catalytic function is not exclusive to natural biopolymers and can be realized in synthetic genetic polymers with backbones structurally distinct from RNA [15] [17].
Key Experimental Data
Table 2: Characteristics of Representative XNAzymes
| XNAzyme Type | Catalytic Activity | Turnover Capability | Key Feature |
|---|---|---|---|
| FANAzyme | RNA cleavage, Transphosphorylation | Multiple turnover [15] | Function in human serum [2] |
| HNAzyme | RNA cleavage | Multiple turnover [15] | Stable folded structure |
| TNAzyme | RNA cleavage | Multiple turnover [15] | Nuclease resistant backbone |
Experimental Protocol: In vitro Evolution of XNAzymes
Objective: To evolve a catalytic XNA (XNAzyme) from a random XNA library. Key Reagents:
- Engineered XNA polymerase (e.g., for FANA or HNA synthesis)
- XNA nucleoside triphosphate mix
- Substrate for the desired reaction (e.g., a chimeric DNA-RNA oligonucleotide for RNA cleavage)
- Partitioning system (e.g., biotin tagging and streptavidin beads)
Procedure:
- Library Generation: Synthesize a random XNA library (e.g., 10^14 unique sequences) from a DNA template using an engineered XNA polymerase, as described in the aptamer protocol.
- Positive Selection: Incubate the XNA library with the substrate. For a cleavage reaction, the substrate can be tagged with biotin and a fluorophore. Active catalysts will cleave the substrate.
- Partitioning: Use the tagging system to separate active from inactive sequences. In the cleavage example, cleaved products will lose the biotin tag. Apply the reaction mixture to a streptavidin column; cleaved (active) XNAzymes will flow through, while inactive sequences bound to the uncleaved substrate will be retained.
- Recovery and Amplification: Reverse transcribe the recovered active XNA populations into cDNA, amplify by PCR, and either transcribe back into XNA or use directly as a template for the next round.
- Iteration: Repeat the selection cycle under conditions of increasing stringency (e.g., shorter reaction time, lower divalent cation concentration) to evolve highly efficient catalysts.
- Characterization: Clone and sequence the final enriched pool. Chemically synthesize individual hit sequences for biochemical characterization of kinetics and specificity.
Application Note 3: XNAs for Molecular Data Storage
Rationale and Background
DNA is an emerging medium for ultra-dense, long-term digital data storage due to its high information density and longevity [21]. However, natural DNA is susceptible to hydrolytic and enzymatic degradation. XNAs offer a path to enhanced storage stability due to their inherent resistance to nucleases [17] [21]. Engineered polymerases are essential for writing (synthesizing) and reading (sequencing via reverse transcription) information stored in XNA formats.
Key Experimental Data
Table 3: Enzymatic Tools for DNA and XNA Data Storage
| Enzyme | Role in Data Storage | Advantage |
|---|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) | De novo DNA writing [21] | Scalable, green synthesis |
| Engineered XNA Polymerases | Writing data into XNA; Reading from XNA [21] | Enables use of nuclease-resistant media |
| DNA Ligases | Assembly of long DNA fragments [21] | Builds long sequences from short fragments |
| Standard DNA Polymerases | PCR-based random access & amplification [21] | Efficient data retrieval |
Experimental Protocol: Enzymatic Synthesis of Data-Encoding XNA
Objective: To store digital information in a nuclease-resistant XNA polymer. Key Reagents:
- Engineered XNA polymerase (e.g., SF mutant for 2'Az/2'F polymers [1])
- 2'Modified NTPs (e.g., 2'F-dCTP, 2'F-dGTP, 2'F-dTTP, 2'Az-dATP [1])
- DNA template encoding digital data (via A:00, C:01, G:10, T:11 mapping [21])
- Primer complementary to the template's binding site.
Procedure:
- Encoding and Template Design: Convert digital files from binary code into DNA sequences using error-correction algorithms. Design and chemically synthesize a corresponding DNA template.
- XNA Synthesis (Writing): Set up a synthesis reaction containing:
- Product Purification: Purify the synthesized XNA product using a commercial oligonucleotide clean-up kit. Treat with DNase to remove the DNA template [1].
- Storage: Store the purified XNA in stable conditions (e.g., encapsulated in silica nanoparticles [21]).
- Data Retrieval (Reading): To recover the data, reverse transcribe the stored XNA back to DNA using an engineered reverse transcriptase (e.g., the same polymerase used for synthesis or a specialized variant). Amplify the resulting cDNA by PCR and sequence it using standard platforms (e.g., Illumina, Nanopore). Decode the sequence to recover the original digital information.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs key reagents that form the foundation of research in XNA polymerase applications.
Table 4: Essential Reagents for XNA Polymerase Research
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| Engineered Polymerases (Tgo, KOD mutants) | Synthesize and reverse transcribe various XNAs [18] [2] | HNA synthesis & RT (KOD-H4) [18] |
| 2'-Modified NTPs (2'F, 2'OMe, 2'Az) | Building blocks for XNA synthesis [1] [2] | Creating nuclease-resistant aptamers & data polymers [1] |
| Unnatural Base Pairs (e.g., TPT3:NaM) | Expand genetic alphabet for increased information density [20] | Incorporating novel functional groups into aptamers [20] |
| XNA-Compatible DNA Ligases | Enzymatic ligation of XNA oligonucleotides [2] | Assembly of XNA nanostructures or encoded libraries |
| Solid-Phase Synthesis Reagents | Chemical synthesis of defined XNA oligos [2] | Production of substrates, primers, and defined catalysts |
The directed evolution of XNA polymerases has unlocked a new frontier in synthetic biology, enabling the practical application of synthetic genetic polymers. As outlined in these application notes, these engineered enzymes are crucial for developing stable therapeutics (XNA aptamers), novel catalysts (XNAzymes), and durable media for molecular-scale information storage. The continued refinement of polymerase fidelity, efficiency, and substrate range will further accelerate the exploration of the vast chemical space of XNAs, paving the way for groundbreaking applications in biomedicine and biotechnology.
Pipeline for Progress: Directed Evolution and Application of XNA Synthetases
Directed evolution has revolutionized synthetic biology by enabling the engineering of biomolecules with tailor-made properties. Within the specific context of evolving XNA polymerases—enzymes capable of synthesizing and replicating xenonucleic acids—selection platform choice is paramount. XNA polymerases are essential for advancing synthetic genetics, as they facilitate the replication, transcription, and functional exploration of synthetic genetic polymers with novel physicochemical properties [16] [2]. This application note details two powerful directed evolution platforms—emulsion-based compartmentalization and phage display—contrasting their methodologies and applications through structured protocols and quantitative comparisons. These platforms are instrumental for overcoming the central challenge in polymerase engineering: creating a genotype-phenotype link that allows for the selection of enzymes based on their ability to process non-natural substrates [22] [23].
Directed evolution requires a robust link between a gene (genotype) and the function it encodes (phenotype). The two platforms discussed herein establish this link through fundamentally different mechanisms.
Emulsion-based compartmentalization utilizes water-in-oil emulsions to create artificial cell-like compartments. Each aqueous droplet houses a single gene or library variant, along with the necessary components for transcription, translation, and, crucially, for polymerase evolution, the catalytic activity itself [24] [22]. The phenotype (e.g., polymerase activity) directly benefits the genotype within the same compartment, leading to its selective amplification.
Phage display, in contrast, is a display technology where the gene of interest is fused to a gene encoding a bacteriophage coat protein. The resulting fusion protein is displayed on the phage's surface, physically linking the protein (phenotype) to the encapsidated genetic material (genotype) [25]. Selections are typically based on affinity interactions (biopanning) to isolate variants that bind to a desired target.
Table 1: Core Characteristics of Directed Evolution Platforms
| Feature | Emulsion-Based Compartmentalization | Phage Display |
|---|---|---|
| Genotype-Phenotype Link | Spatial co-localization within a microdroplet [22] | Physical fusion to phage coat protein [25] |
| Primary Selection Principle | Function-based (e.g., catalysis, replication) [22] | Affinity-based (binding to an immobilized target) [25] |
| Typical Library Size | >10^7 - 10^8 variants [24] [22] | >10^8 - 10^10 variants [25] |
| Key Advantage | Selects for enzymatic activities beyond binding; conditions can be toxic to cells [22] | Well-established, high library diversity, direct physical link [25] |
| Key Limitation | Requires careful emulsion formation and stability [24] | Limited to proteins that can be displayed and secreted in functional form [26] |
| Ideal for Evolving | Catalytic activities (polymerases, ribozymes), biosynthetic pathways [24] [22] | Binding molecules (antibodies, peptides), protein-protein interactions [23] [25] |
For XNA polymerase engineering, emulsion-based methods are often preferred because the selection can be directly coupled to the polymerase's core function: the templated synthesis of nucleic acids. For instance, Compartmentalized Self-Replication (CSR) and its advanced derivative, Compartmentalized Partnered Replication (CPR), select for polymerases based on their ability to amplify their own coding sequence or a partner gene under selective pressure [22].
Application Notes for XNA Polymerase Evolution
Emulsion-Based Compartmentalization
The evolution of XNA polymerases demands selection pressures that mirror their desired function, such as the ability to utilize XNA as a template or to incorporate XNA nucleotides. Emulsion-based platforms are uniquely suited for this task.
Compartmentalized Partnered Replication (CPR) is a highly adaptable emulsion-based method. In CPR, the activity of a polymerase variant (or a partner protein that influences polymerase production) is linked to the expression of a thermostable DNA polymerase in vivo. Following expression, bacterial cells are emulsified, and the most active variants are enriched via emulsion PCR (ePCR) based on their superior ability to amplify their own coding sequence [22]. This system has been successfully used to evolve T7 RNA polymerase variants with orthogonal promoter recognition and to improve orthogonal tRNA-synthetase pairs, demonstrating its utility in complex genetic circuit engineering [22].
A key advantage of CPR and related methods like Compartmentalized Self-Replication (CSR) is the ability to apply selective pressures that are impossible in vivo, such as the presence of toxic XNA substrates or reaction conditions that would kill a host cell. Holliger and colleagues used CSR to evolve DNA polymerase variants with an ability to incorporate unnatural nucleotides, a foundational step towards XNA synthesis [22]. Furthermore, our group engineered a KOD DNA polymerase variant into a high-fidelity reverse transcriptase by selecting for its ability to use RNA as a template [22].
Microfluidic Emulsification offers superior control and uniformity for compartmentalization. A microfluidic device with a radial array of 110 aqueous nozzles intersecting a surrounding oil flow channel can generate highly uniform water-in-oil droplets (21.9 ± 0.8 μm radius) at high throughput (10^7–10^8 droplets per hour) [24]. This uniformity is critical for quantitative directed evolution, as it ensures that selective advantages arise from enzyme activity rather than random variation in compartment size. This platform was used to evolve RNA ligase enzymes resistant to neomycin inhibition, with each RNA molecule undergoing 10^8-fold selective amplification within its compartment [24].
Phage Display for Enzyme Engineering
While typically used for affinity selections, phage display can be adapted for enzyme evolution through systems based on conditional phage replication (CPR). In these platforms, the activity of the enzyme of interest is linked to the replication of the phage itself [23]. For example, the enzyme's function can be tied to the production of an essential phage coat protein or to a factor that suppresses a conditional defect in phage assembly. This approach bypasses a key limitation of conventional phage display by allowing for the selection of catalytic properties rather than just binding.
Phage-based systems function in both batch and continuous culture and have been applied to evolve a wide range of proteins, including transcription factors, polymerases, and proteases [23]. This makes them a versatile tool for the synthetic biologist's toolkit, particularly when the enzymatic function can be effectively coupled to phage propagation within a bacterial host.
Experimental Protocols
Protocol: Microfluidic Compartmentalized Directed Evolution
This protocol is adapted from a study that evolved neomycin-resistant RNA ligases and is applicable to the directed evolution of XNA polymerases [24].
1. Library and Solution Preparation: * Library Design: Generate a diverse library of XNA polymerase variants via error-prone PCR or other mutagenesis methods. * Aqueous Phase Preparation: Prepare an aqueous mixture containing: * Library DNA (or an in vitro transcription-translation system with library DNA) * Necessary reagents for polymerase activity (e.g., XNA triphosphates, natural dNTPs/NTPs, divalent cations like Mg²⁺) * A selection pressure (e.g., an XNA template, modified nucleotides) * Primers for PCR amplification of successful variants * Oil Phase Preparation: Prepare an oil-surfactant mixture. A stable formulation for biochemical reactions is 70% Ar20 silicone oil, 26% mineral oil, and 4% Abil EM90 emulsifier [24].
2. Microfluidic Emulsification: * Use a soft lithography-fabricated PDMS device containing a circular nozzle array [24]. * Load the aqueous phase and oil phase into their respective input reservoirs. * Drive the oil phase using a syringe pump at a flow rate of 70 μL/min and the aqueous phase at 5 μL/min. This generates droplets of ~44 pL volume at a rate of ~10^7 droplets per hour. * Collect the emulsion from the output reservoir.
3. Incubation and In-Droplet Amplification: * Incubate the emulsion under conditions that permit the polymerase reaction (e.g., thermal cycling for PCR). * Polymerase variants that successfully perform the desired catalytic function (e.g., XNA synthesis) will amplify their own genotype within the compartment.
4. Recovery and Analysis: * Break the emulsion by adding a destabilizing agent (e.g., perfluorooctanol) and centrifuging. * Recover the amplified DNA from the aqueous phase. * Purify the DNA and either sequence it directly or use it to seed the next round of selection.
The workflow for this protocol is as follows:
Protocol: Phage ESCape Selection
Phage ESCape is an emulsion-based method that combines the diversity of phage display with the compartmentalization and screening power of FACS [27].
1. Phage Library and Antigen Preparation: * Phage Library: Use a phage display library where the protein or enzyme variant is fused to a phage coat protein (e.g., pIII). * Antigen Labeling: Label the target antigen (or selection molecule) with a fluorescent probe (e.g., fluorescein).
2. Cell Infection and Emulsification: * Infect E. coli with the phage display library to allow for phage production and display of the variant proteins. * Prepare an emulsion containing the infected cells, the fluorescently labeled antigen, and reagents for phage secretion. The goal is to achieve compartments with no more than one cell.
3. Incubation and Secretion: * Incubate the emulsion to allow cells within the droplets to secrete phage particles. The displayed variant proteins on the phage surface can bind to the co-compartmentalized fluorescent antigen.
4. Fluorescence-Activated Cell Sorting (FACS): * Break the emulsion. * Use FACS to isolate fluorescent phage particles (or phage-bound complexes) that have bound the antigen. * The FACS machine acts as a high-throughput "colony picker."
5. Phage Recovery and Amplification: * Infect fresh E. coli with the sorted phage to amplify the selected variants. * The resulting phage can be subjected to additional rounds of selection to further enrich for high-binders or catalysts.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Emulsion-Based Directed Evolution
| Reagent / Material | Function / Application | Example Formulation / Notes |
|---|---|---|
| Abil EM90 | Non-ionic silicone-based emulsifier for stabilizing water-in-oil emulsions [24]. | Used at 4% in combination with silicone and mineral oil [24]. |
| Polydimethylsiloxane (PDMS) | Material for fabricating microfluidic devices via soft lithography [24]. | Allows for creation of custom droplet generator circuits. |
| Silicone Oil (e.g., Ar20) | Component of the continuous oil phase in emulsions [24]. | Provides stability; used at 70% with mineral oil and emulsifier [24]. |
| Thermostable DNA Polymerase (e.g., Taq) | Core component for in-compartment PCR amplification of selected genotypes [22]. | In CPR, its expression is linked to the activity of the gene circuit being evolved [22]. |
| XNA Nucleotide Triphosphates | Substrates for XNA polymerase enzymes; applied as a selective pressure [2]. | Required for selecting polymerases that can synthesize or replicate XNA. |
The directed evolution of XNA polymerases is a cornerstone of synthetic genetics, enabling the replication and functional exploration of synthetic genetic polymers. Emulsion-based compartmentalization and phage display offer complementary pathways to this goal. Emulsion methods, particularly microfluidic CPR and CSR, excel at directly selecting for the catalytic activity of polymerases under highly customizable and even toxic conditions, making them the premier choice for engineering XNA-synthesizing capability. Phage display, especially next-generation systems like conditional phage replication and Phage ESCape, provides a powerful alternative, particularly when enzyme activity can be coupled to phage viability or when high-affinity binders are also desired. The choice of platform, therefore, depends on the specific catalytic trait targeted for improvement, with both offering robust and scalable solutions for creating the next generation of biocatalysts for synthetic biology.
The field of synthetic biology is increasingly leveraging artificial genetic polymers (XNAs) to develop new biotechnological and biomedical tools. XNAs, which are nucleic acids not found in nature, offer significant advantages over their natural counterparts, including increased nuclease stability and the capacity to incorporate non-natural functional groups [1]. Among various XNA chemistries, those featuring 2′-azide (2′Az) and 2′-fluoro (2′F) substitutions are particularly promising. The 2′Az modification enables participation in biorthogonal click chemistry, facilitating the conjugation of dyes, probes, and other molecules of interest, while 2′F modifications enhance nuclease resistance and duplex stability [1]. However, a significant challenge remains the enzymatic synthesis of these polymers, as natural DNA polymerases inefficiently incorporate XNA nucleotides. This application note explores the engineering and functional comparison of polymerases for synthesizing mixed XNA polymers containing 2′Az and 2′F modifications, framed within the broader context of directed polymerase evolution for synthetic biology applications.
Key Findings and Experimental Data
Recent research has demonstrated that laboratory-evolved XNA polymerases can successfully synthesize fully substituted mixed polymers containing both 2′Az and 2′F modifications [1]. These engineered enzymes exhibit improved fidelity compared to previous systems, enabling accurate synthesis of complex XNA polymers. The fidelity of these polymerases is crucial for applications requiring high accuracy, such as the development of XNA aptamers and catalysts.
Table 1: Performance Metrics of Engineered XNA Polymerases
| Polymerase Type | XNA Substrate | Incorporation Efficiency | Fidelity (Error Rate) | Key Applications |
|---|---|---|---|---|
| Engineered A-family (SF mutants) | 2′Az/2′F mixed polymers | High, fully substituted synthesis | Improved accuracy over previous systems [1] | Biorthogonal conjugation, nuclease-resistant aptamers [1] |
| Bst DNA Polymerase | ANA, HNA templates | Variable reverse transcriptase activity | Weak for ANA, uncontrolled for HNA [6] | Reverse transcription of specific XNAs [6] |
| Engineered B-family (Pyrococcus, Thermococcus) | Various XNAs | High functional plasticity | Varies by engineering | Broad XNA synthesis [2] |
The functional comparison of various laboratory-evolved XNA polymerases reveals important trade-offs. While the mutations that enable XNA synthesis increase enzyme tolerance for sugar-modified substrates, this often comes with a sacrifice to protein-folding stability [6]. Furthermore, reverse transcriptase activity varies significantly among different polymerases and across XNA types. For instance, Bst DNA polymerase exhibits weak reverse transcriptase activity on arabino nucleic acid (ANA) templates but uncontrolled activity on hexitol nucleic acid (HNA), differing from its recognition of FANA and TNA templates [6].
Table 2: Comparison of XNA Polymerase Biochemical Properties
| Property | Polymerase A | Polymerase B | Polymerase C | Testing Method |
|---|---|---|---|---|
| Substrate Specificity | Broad XNA range | Narrow for 2′Az | Moderate for 2′F | Primer extension with various xNTPs [6] |
| Thermal Stability | Reduced vs. wild-type | Moderately reduced | Near wild-type | Thermal denaturation assays [6] |
| Reverse Transcriptase Activity | Efficient on FANA | Inefficient on ANA | Variable by XNA type | cDNA synthesis from XNA templates [6] |
| Processivity | High | Moderate | Low | Gel-based analysis of extension products [1] |
Experimental Protocols
Protocol 1: XNA Synthesis and Fidelity Assessment
This protocol describes the enzymatic synthesis of 2′Az/2′F mixed XNA polymers and the measurement of polymerase fidelity using a hydrogel particle-based method, which reduces xNTP consumption and simplifies the traditional workflow [1] [9].
Materials Required:
- 5′IRDye700-labeled DNA primer (e.g., 40mer K017, Integrated DNA Technologies)
- DNA template (e.g., 100mer K021, Integrated DNA Technologies)
- 2′Az-NTPs and 2′F-NTPs (TriLink Biotechnologies)
- Engineered XNA polymerase (e.g., SF mutant)
- 10% TBE-urea gel (Bio-Rad)
- Hydrogel-coated magnetic particles (for fidelity assay)
- Heparin affinity column (for enzyme purification)
Procedure:
XNA Synthesis Reaction:
- Prepare a 2× reaction mixture containing:
- 40 nM 5′IRDye700-labeled primer
- 80 nM DNA template
- 1× SF buffer (50 mM Tris pH 8.5, 6.5 mM MgCl₂, 0.05 mg/mL Ac-BSA, 50 mM KCl)
- 20 nM engineered XNA polymerase
- Add an equal volume of 2× NTP mixture containing 25 μM F-NTPs and 100 μM Az-NTPs [1].
- Incubate at 50°C for 2 hours.
- Quench an aliquot with 2 volumes of Quenching Buffer Orange (95% formamide, 12.5 mM EDTA, trace Orange G).
- Analyze by 10% TBE-urea gel electrophoresis at 120V for 45 minutes and image.
- Prepare a 2× reaction mixture containing:
Hydrogel-Based Fidelity Measurement (Alternative to Gel Purification):
- Functionalize hydrogel particles with acrydite-modified DNA primer covalently distributed throughout the gel matrix [9].
- Anneal the template to the primer on hydrogels.
- Extend primer with XNA using engineered polymerase.
- Remove DNA template by washing particles.
- Reverse transcribe XNA product back to cDNA within the hydrogel matrix.
- Recover cDNA, amplify via PCR, clone, and sequence to quantify errors [9].
XNA Fidelity Assay Workflow
Protocol 2: One-Pot Reverse Transcription and Amplification of XNA
This protocol enables the conversion of 2′Az/2′F mixed polymers back to DNA and their subsequent amplification using commercially available enzymes, facilitating sequence analysis and downstream applications [1].
Materials Required:
- Synthesized and purified 2′Az/2′F mixed XNA polymer
- Turbo DNase (Invitrogen)
- Oligo Clean & Concentrator Kit (Zymo Research)
- Phusion or Q5 DNA Polymerase with corresponding buffer
- dNTPs
- Barcoded primers for multiplexing
Procedure:
XNA Purification:
- Add Turbo DNase (0.11 units/μL) to XNA synthesis reaction.
- Incubate at 37°C for 40 minutes to digest DNA template.
- Purify using Oligo Clean & Concentrator Kit per manufacturer's instructions.
One-Pot Reverse Transcription and Amplification:
- Prepare PCR mixture containing:
- 3 μL purified XNA
- 1× manufacturer's buffer (SF, Phusion GC, or Q5 with GC enhancer)
- 0.5 μM barcoded reverse primer
- 0.5 μM forward primer
- 0.4 mM dNTPs
- 1 U Phusion or Q5 DNA Polymerase
- Use the following cycling conditions:
- 98°C for 30 seconds
- 3 cycles: 98°C for 5 seconds, 50°C for 15 seconds, 72°C for 1 minute
- 12 cycles: 98°C for 5 seconds, 67°C for 15 seconds, 72°C for 15 seconds
- 72°C for 5 minutes
- Hold at 4°C
- Analyze amplified products by 2% agarose gel electrophoresis.
- Prepare PCR mixture containing:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for XNA Polymerase Engineering and Application
| Reagent/Category | Specific Examples | Function/Application | Commercial Sources |
|---|---|---|---|
| XNA Nucleotide Triphosphates | 2′-azide NTP (2′Az-NTP), 2′-fluoro NTP (2′F-NTP) | Substrates for enzymatic XNA synthesis; 2′Az enables click chemistry conjugation [1]. | TriLink Biotechnologies, Metkinen [1] |
| Engineered XNA Polymerases | SF mutants (A-family), B-st polymerases | Templated synthesis of XNA from DNA templates; specialized for specific XNA backbones [1] [6]. | Laboratory-evolved, commercially available variants |
| High-Fidelity DNA Polymerases | Phusion, Q5 | PCR amplification of cDNA from reverse-transcribed XNA; high fidelity reduces errors in analysis [1]. | New England Biolabs, Fisher Scientific [1] |
| Specialized Oligonucleotides | IRDye700-labeled primers, acrydite-modified primers | Detection and immobilization for synthesis and fidelity assays; acrydite enables hydrogel coupling [9]. | Integrated DNA Technologies [1] [9] |
| Purification & Processing Enzymes | Turbo DNase | Degrades DNA templates post-XNA synthesis, enabling XNA purification [1]. | Invitrogen [1] |
Discussion and Future Perspectives
The ability to enzymatically synthesize mixed 2′Az/2′F XNA polymers represents a significant advancement in XNA technology. These mixed backbone chemistries can display novel emergent properties not present in homopolymers [2]. The 2′Az modification provides a handle for post-synthetic functionalization via click chemistry, while the 2′F modification enhances nuclease resistance, creating multifunctional XNAs with applications in diagnostics, therapeutics, and materials science [1].
Future directions in polymerase engineering will likely focus on expanding substrate diversity and improving fidelity and efficiency. The discovery that thermophilic B-family polymerases from Pyrococcus and Thermococcus genera are particularly amenable to engineering suggests these enzymes offer a robust platform for further development [2]. Additionally, the application of high-throughput screening methods and advanced computational design will accelerate the evolution of next-generation XNA polymerases with enhanced capabilities.
As the field progresses, the integration of XNA polymerase engineering with other synthetic biology tools, such as CRISPR systems and cell-free expression platforms, will unlock new applications in molecular medicine, biosensing, and data storage. The ongoing development of efficient and accurate XNA synthesis and reverse transcription methods will be crucial for realizing the full potential of artificial genetic polymers in synthetic biology.
The field of synthetic biology is increasingly leveraging Xeno Nucleic Acids (XNAs)—nucleic acid analogs not found in nature—to develop advanced biotechnological and biomedical tools. XNAs offer significant advantages over their natural counterparts, including greatly increased nuclease stability and the capacity to incorporate non-natural functional groups for novel applications [1] [2]. Among the diverse array of XNA chemistries, those containing 2'-azide (2'Az) substitutions are particularly valuable due to the azide group's ability to participate in biorthogonal click chemistry reactions, enabling specific conjugation with molecules, dyes, or probes without interfering with native biological processes [1].
While laboratory-evolved XNA polymerases have demonstrated the ability to incorporate 2'Az nucleotides during DNA synthesis, their capacity to incorporate these modifications during XNA synthesis has remained unclear. This application note addresses this gap by demonstrating that engineered XNA polymerases can successfully synthesize fully substituted mixed polymers containing both 2'Az and 2'-fluoro (2'F) modifications [1]. We present a comprehensive toolkit for the accurate and efficient synthesis, reverse transcription, and amplification of these mixed XNA polymers, enabling researchers to harness their unique properties for synthetic biology applications, including the development of next-generation therapeutics and diagnostics.
Background & Significance
XNA Polymerases in Synthetic Genetics
The emergence of XNA technology has been propelled by advances in polymerase engineering, enabling the enzymatic synthesis and replication of artificial genetic polymers. Natural DNA polymerases typically cannot utilize XNA substrates efficiently, necessitating the engineering of specialized polymerases. Thermophilic B-family polymerases, especially those from Pyrococcus and Thermococcus genera, have proven particularly amenable to engineering for XNA substrates due to their high thermostability and functional plasticity [2]. Additionally, A-family DNA polymerases, such as the Stoffel fragment (SF) of Thermus aquaticus DNA polymerase I, have been successfully engineered to synthesize XNAs bearing various 2' modifications [1].
These engineered polymerases now enable the synthesis of XNAs with diverse backbone chemistries, including 2'-fluoroarabino nucleic acid (FANA), 1,5-anhydrohexitol nucleic acid (HNA), threose nucleic acid (TNA), and various 2'-modified RNA analogs [9] [2]. This expanding polymerase toolkit forms the foundation for exploring the functional potential of XNAs in synthetic biology.
Advantages of Mixed XNA Polymers
Mixed XNA polymers incorporating multiple modifications offer unique advantages over uniformly modified XNAs:
- Enhanced nuclease resistance: Combining different 2' modifications can provide synergistic protection against enzymatic degradation [1].
- Tunable physicochemical properties: Mixed polymers allow fine-tuning of duplex stability, hybridization characteristics, and structural properties [2].
- Orthogonal functionality: Incorporation of 2'Az modifications provides specific handles for biorthogonal conjugation without interfering with other functional groups [1].
- Emergent properties: Certain combinations of modifications can yield novel properties not present in singly-modified XNAs, expanding their application potential [2].
The ability to site-specifically incorporate 2'Az modifications within mixed XNA polymers creates powerful opportunities for constructing sophisticated nucleic acid-based materials and therapeutics with precision functionality.
Experimental Protocols
Enzymatic Synthesis of 2'Az/2'F Mixed XNA Polymers
Materials and Reagents
Table 1: Key Reagents for XNA Synthesis
| Reagent | Specification | Source | Function |
|---|---|---|---|
| 2'-azide NTPs | 2'Az-ATP, 2'Az-CTP, 2'Az-GTP, 2'Az-UTP | TriLink Biotechnologies | Azide-modified nucleotide substrates |
| 2'-fluoro NTPs | 2'F-ATP, 2'F-CTP, 2'F-GTP, 2'F-UTP | TriLink Biotechnologies | Fluoro-modified nucleotide substrates |
| XNA Polymerases | Engineered SF mutants | Laboratory expression & purification | Catalyze XNA synthesis from DNA template |
| Primer K017 | 5'IRDye700-labeled 40mer | Integrated DNA Technologies | Fluorescently labeled synthesis primer |
| Template K021 | 100mer | Integrated DNA Technologies | DNA template for XNA synthesis |
| SF Buffer | 0.05 M Tris (pH 8.5), 0.05 M KCl | Research Products International | Reaction buffer |
Synthesis Procedure
Reaction Setup: Prepare a 2× reaction mixture containing:
- 40 nM 5'IRDye700-labeled primer K017
- 80 nM template K021
- 1× SF buffer (0.05 M Tris, pH 8.5)
- 6.5 mM MgCl₂
- 0.05 mg/mL Ac-BSA
- 0.05 M KCl
- 20 nM XNA polymerase
NTP Addition: Add an equal volume of 2× NTP mixture containing:
- 25 μM 2'F-NTPs (each)
- 100 μM 2'Az-NTPs (each)
- For control reactions: 100 μM dNTPs (each)
Incubation: Incubate the reaction mixture on a 50°C heat block for 2 hours.
Analysis: Remove a 3 μL aliquot and quench with 6 μL Quenching Buffer Orange (95% formamide, 12.5 mM EDTA, trace Orange G). Separate products on a 10% TBE-urea gel at 120 V for 45 minutes and image using an Odyssey CLx imager [1].
One-Pot Reverse Transcription and Amplification
Materials and Reagents
Table 2: Reagents for Reverse Transcription and Amplification
| Reagent | Specification | Source | Function |
|---|---|---|---|
| Turbo DNase | 0.11 units/μL | Invitrogen | Degrades DNA template |
| Oligo Clean & Concentrator Kit | - | Zymo Research | Purifies synthesized XNA |
| Phusion DNA Polymerase | - | Fisher Scientific | High-fidelity PCR amplification |
| Q5 DNA Polymerase | - | New England Biolabs | High-fidelity PCR amplification |
| dNTPs | 0.4 mM in reaction | New England Biolabs | Nucleotide substrates for cDNA synthesis |
| Barcoded Primers | Sequence-specific | Integrated DNA Technologies | Enable multiplexing for HT-Seq |
Procedure
Template Degradation: Add 2 μL Turbo DNase (0.11 units/μL) to 17 μL of XNA synthesis reaction. Incubate at 37°C for 40 minutes.
XNA Purification: Purify the XNA using the Oligo Clean & Concentrator Kit according to the manufacturer's protocol.
RT/Amp Reaction Setup: Prepare a reaction mixture containing:
- 3 μL purified XNA
- 1× manufacturer's buffer (SF buffer, Phusion GC buffer, or Q5 reaction buffer with GC enhancer)
- 0.5 μM barcoded reverse primer
- 0.5 μM forward primer
- 0.4 mM dNTPs
- 1 U DNA polymerase (Phusion or Q5)
- Milli-Q water to volume
Thermal Cycling: Perform amplification with the following conditions:
- 98°C for 30 seconds
- 3 cycles of: 98°C for 5 seconds, 50°C for 15 seconds, 72°C for 1 minute
- 12 cycles of: 98°C for 5 seconds, 67°C for 15 seconds, 72°C for 15 seconds
- 72°C for 5 minutes
- Hold at 4°C [1]
Analysis: Separate amplified products on a 2% agarose gel with GelRed and visualize using a UV transilluminator.
High-Throughput Sequencing for Fidelity Analysis
Purification: Purify amplified PCR products using the DNA Clean and Concentrator Kit.
Quantification: Quantify DNA concentration using a Qubit 3 Fluorometer and dsDNA High Sensitivity Assay Kit.
Sequencing: Perform sequencing using the AmpliconEZ protocol (GeneWiz/Azenta).
Data Analysis: Analyze resulting data using a custom Python script that:
- Separates samples by barcodes
- Compares sequences to reference
- Counts errors in template regions that were formerly XNA
- Removes sequences deviating from predicted length by >15%
- Omits sequences without identical matched reads to account for HTS process errors [1]
The script is available at: https://github.com/Leconte-Group/Thompsonetal2020
Results and Data Analysis
Polymerase Performance and Fidelity
Table 3: Quantitative Analysis of XNA Polymerase Performance
| Parameter | Value | Conditions | Significance |
|---|---|---|---|
| 2'Az-NTP Concentration | 100 μM | Optimal for synthesis | Higher than 2'F-NTPs required |
| 2'F-NTP Concentration | 25 μM | Optimal for synthesis | Lower concentration sufficient |
| Polymerase Concentration | 20 nM | Standard reaction | Sufficient for complete synthesis |
| Reaction Temperature | 50°C | Optimal for SF mutants | Balance of enzyme activity and stability |
| Incubation Time | 2 hours | Complete synthesis | Ensures full-length product |
| Fidelity | Improved over previous systems | Error rate analysis | High accuracy for applications |
The synthesized 2'Az/2'F mixed polymers demonstrate complete resistance to DNase treatment, a critical advantage for applications in complex biological environments where nuclease degradation would limit the utility of natural nucleic acids [1]. The improved fidelity of the engineered XNA polymerases represents a significant advancement over previous systems, enabling the synthesis of mixed XNA polymers with accuracy sufficient for demanding applications in diagnostics and therapeutics.
Hydrogel-Based Fidelity Assessment
Recent advances in fidelity assessment have led to the development of hydrogel particle-based methods that dramatically reduce the time and reagent consumption required for polymerase fidelity measurements [9]. This approach involves:
- Covalent distribution of DNA primers throughout a polyacrylamide-encapsulated magnetic particle gel matrix
- XNA transcription and reverse transcription within the hydrogel matrix
- Elimination of denaturing PAGE purification steps through simple washing procedures
- Recovery of DNA product for amplification, cloning, and sequencing
This method reduces the fidelity assessment timeline from approximately one week to 1-2 days and decreases xNTP consumption by 10-fold, making it particularly valuable for evaluating novel XNA polymerases where substrates are scarce and expensive [9].
Application in Biorthogonal Conjugation
The 2'Az modifications incorporated into mixed XNA polymers enable efficient bioorthogonal click chemistry for conjugation applications. The azide group specifically reacts with alkyne-modified molecules via copper-catalyzed or strain-promoted azide-alkyne cycloaddition, allowing for:
- Site-specific conjugation of fluorescent dyes for imaging probes
- Attachment of therapeutic payloads for targeted drug delivery
- Incorporation of hydrophobic groups to enhance aptamer binding properties [1]
- Cross-linking of nucleic acids to create dynamic hydrogels [1]
- Surface immobilization of functional XNAs for biosensing applications
The biorthogonal nature of these reactions ensures compatibility with biological systems, as the azide and alkyne functional groups are inert to most biological molecules, enabling specific conjugation even in complex biological environments.
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions for XNA Research
| Tool/Reagent | Function | Key Features | Applications |
|---|---|---|---|
| Engineered SF Mutants | XNA synthesis | A-family polymerases engineered for 2' modified XNAs | 2'Az/2'F mixed polymer synthesis |
| 2'-Azide NTPs | XNA substrates | Azide group for biorthogonal conjugation | Click chemistry functionalization |
| 2'-Fluoro NTPs | XNA substrates | Enhanced nuclease resistance & stability | Mixed polymer synthesis |
| Hydrogel Particles | Fidelity assessment | Encapsulated magnetic beads for XNA replication | Rapid polymerase screening |
| Barcoded Primers | Multiplexing | Unique sequences for sample identification | High-throughput sequencing |
| Custom Python Script | Error analysis | Automated sequence analysis | Fidelity quantification |
Workflow and Signaling Pathways
XNA Synthesis and Application Workflow: This diagram illustrates the complete process for synthesizing mixed 2'Az/2'F XNA polymers and their downstream applications. The process begins with DNA template and fluorescently labeled primer, combined with engineered XNA polymerase and modified NTP substrates. After synthesis, the mixed XNA polymer can undergo two main pathways: (1) fidelity assessment through reverse transcription, amplification, and high-throughput sequencing, or (2) functionalization via biorthogonal click chemistry conjugation for various applications.
Troubleshooting Guide
Table 5: Common Issues and Solutions in Mixed XNA Synthesis
| Problem | Potential Cause | Solution |
|---|---|---|
| Incomplete Synthesis | Insufficient NTP concentration | Increase 2'Az-NTPs to 100 µM and 2'F-NTPs to 25 µM |
| Low Fidelity | Suboptimal polymerase variant | Screen alternative engineered XNA polymerases |
| Poor Reverse Transcription | XNA secondary structure | Adjust thermal cycling conditions or use different DNA polymerase |
| Failed Conjugation | Azide group incompatibility | Ensure copper-free click conditions for sensitive applications |
| Template Degradation | Incomplete DNase treatment | Extend DNase incubation time or increase enzyme concentration |
The development of robust methods for synthesizing mixed XNA polymers containing 2'Az and 2'F modifications represents a significant advancement in the toolkit available for synthetic biology research. The protocols outlined herein enable researchers to efficiently create these novel nucleic acid polymers with improved fidelity and defined functionalization capacity. The integration of biorthogonal conjugation capability through 2'Az modifications opens new possibilities for creating advanced nucleic acid therapeutics, diagnostic probes, and functional materials with enhanced stability and programmable functionality.
As the field of synthetic genetics continues to evolve, these mixed XNA polymer systems provide a versatile platform for exploring the boundaries of genetic information storage and manipulation while offering practical solutions to current limitations in therapeutic nucleic acid applications. The continued refinement of XNA polymerase engineering and fidelity assessment methods will further expand the capabilities and applications of these innovative synthetic genetic polymers.
The replication of xeno nucleic acids (XNAs) is a cornerstone of synthetic genetics, an emerging field that applies genetic principles to artificial genetic polymers. A critical step in this process is the reverse transcription of information encoded in XNA back into DNA, enabling analysis and amplification using conventional molecular biology tools. While natural reverse transcriptases (RTs) are inefficient with these unnatural substrates, directed evolution has created engineered enzymes capable of processing a wide array of XNA chemistries. This application note details the methodologies—Compartmentalized Bead Labeling (CBL) and hydrogel-based fidelity measurement—that facilitate this process, providing researchers with protocols to implement these techniques within a directed evolution framework for synthetic biology and therapeutic development.
Key Experimental Platforms and Workflows
Compartmentalized Bead Labeling (CBL) for Evolving XNA Reverse Transcriptases
The CBL platform addresses the fundamental challenge of linking the genotype (RT-encoding gene) to the phenotype (cDNA product from XNA template) during directed evolution [28].
Table 1: Key Reagents for CBL Selection
| Research Reagent | Function in Protocol |
|---|---|
| E. coli cells expressing RT variants | Source of RT genes (genotype) and expressed polymerase enzymes. |
| Microbeads with conjugated XNA/RNA templates and plasmid capture probes | Solid support for reverse transcription reaction and genotype capture. |
| Water-in-oil (w/o) emulsion reagents | Creates aqueous compartments for single-cell and bead co-encapsulation. |
| Fluorescent antisense DNA probes and HCR reagents | Detects successful cDNA synthesis on beads via fluorescence. |
| Fluorescent Activated Cell Sorter (FACS) | Isolates fluorescent beads corresponding to active RT variants. |
Experimental Protocol: CBL for RT Evolution
- Bead Preparation: Covalently conjugate the target XNA template (e.g., 2'OMe-RNA, HNA) and DNA oligonucleotide capture probes to the surface of microbeads.
- Emulsion Formation: Co-encapsulate single E. coli cells (harboring the RT mutant library) and the prepared template beads within the aqueous compartments of a w/o emulsion. Heat-lyse the cells to release both the RT enzymes and the plasmid DNA encoding them.
- On-Bead Reverse Transcription: Within each compartment, the released RT enzyme reverse transcribes the bead-bound XNA template into cDNA.
- Genotype-Phenotype Linkage: The plasmid DNA encoding the RT is captured on the same bead via the conjugated DNA capture probes, creating a stable link.
- Detection and Sorting: Recover the beads after breaking the emulsion. Detect successful cDNA synthesis by hybridization with fluorescent antisense probes, with optional signal amplification via Hybridization Chain Reaction (HCR). Sort the fluorescently labeled beads using FACS.
- Variant Recovery: Isolate the plasmid DNA from sorted beads and transform into fresh E. coli to initiate the next round of selection or for analysis.
The following workflow diagram illustrates the CBL process:
Hydrogel Particle Fidelity Assay for XNA Polymerases
Conventional fidelity measurements for XNA synthesis are slow and require tedious gel purification. A hydrogel particle-based method streamlines this process, drastically reducing time and reagent consumption [9].
Experimental Protocol: Hydrogel-Based Fidelity Measurement
- Hydrogel Particle Preparation: Synthesize polyacrylamide hydrogel-coated magnetic beads. A DNA primer is covalently crosslinked throughout the gel matrix using a 5'-acrydite modification.
- Template Annealing: Anneal a defined-sequence DNA template to the gel-immobilized primer.
- XNA Synthesis ("Transcription"): Incubate the particles with an XNA polymerase and the corresponding xNTPs. The enzyme synthesizes an XNA strand within the gel matrix.
- Template Removal: Wash the particles extensively with buffer, then degrade the original DNA template using a DNase. This purification is performed without denaturing gel electrophoresis.
- Reverse Transcription ("cDNA Synthesis"): Inside the same gel matrix, reverse transcribe the XNA strand back into DNA using an appropriate RT.
- Analysis: Recover the cDNA, amplify it by PCR, clone, and sequence it. Compare the sequences to the original template to calculate the error rate of the XNA polymerase.
Key Research Findings and Data
Engineered Polymerases for Diverse XNA Chemistries
Directed evolution via CBL has yielded RTs with significantly enhanced performance across various XNA substrates [28].
Table 2: Performance of Evolved XNA Reverse Transcriptases
| XNA Template Chemistry | Evolved RT Variant | Key Mutations | Performance and Application |
|---|---|---|---|
| 2'-O-Methyl-RNA (2'OMe-RNA) | RT-C8 | RT521K backbone + I114T, S383K, N735K, F493V, Y496N, Y497L, Y499A, A500Q, K501H | ~12x more full-length product than RT521K; 3x more than commercial SSIII/RTx; works on complex N40 templates up to 80-90°C [28]. |
| Hexitol Nucleic Acid (HNA) | Evolved Tgo variants | Specific mutations not listed | Outperform previously described RTs for HNA [28]. |
| D-altritol Nucleic Acid (AtNA) | Evolved Tgo variants | Specific mutations not listed | De novo discovery of RTs for an "orphan" XNA chemistry with no prior known RTs [28]. |
| 2'-Methoxyethyl-RNA (MOE-RNA) | Evolved Tgo variants | Specific mutations not listed | De novo discovery of RTs for an "orphan" XNA chemistry with no prior known RTs [28]. |
| 2'-Fluoro-arabino NA (2'F-FANA) | SFM4-3, SFP1 (Taq-derived) | Not exhaustively listed | Capable of synthesizing 2'F XNA; error rates of ~5.6-6.9 errors per thousand base pairs [29]. |
Rational Engineering for Improved Fidelity
The accuracy of XNA synthesis is critical for applications. Rational design, incorporating mutations known to improve natural DNA synthesis fidelity, has successfully enhanced XNA polymerase performance.
Table 3: Fidelity Enhancement in Taq-Derived 2'F XNA Polymerases
| Parent XNA Polymerase | Fidelity (Errors per kbp) | Fidelity-Enhancing Mutations | Outcome and Significance |
|---|---|---|---|
| SFM4-3 | ~6.9 | Addition of mutations known to improve Taq's DNA fidelity (e.g., K659R, E708K, A759R) | Generated polymerases with significantly improved synthesis accuracy, demonstrating that fidelity determinants for DNA can be applied to XNA synthesis [29]. |
| SFM4-6 | ~19.1 | Same as above | Same as above [29]. |
| SFP1 | ~5.6 | Same as above | Same as above [29]. |
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions for XNA Replication
| Reagent / Material | Function and Importance |
|---|---|
| Engineered XNA Polymerases (e.g., RT-C8, SFM4-3) | Core enzymes capable of synthesizing and/or reverse transcribing various XNA chemistries with high efficiency [28] [29]. |
| XNA Nucleoside Triphosphates (xNTPs) | Activated monomers for enzymatic synthesis of XNA. Often require custom chemical synthesis [9]. |
| Compartmentalized Bead Labeling (CBL) Platform | A high-throughput selection system for evolving RT activity toward novel XNA templates by linking genotype to phenotype [28]. |
| Hydrogel-Coated Magnetic Beads | Solid support for miniaturized, purification-free fidelity assays and other XNA replication workflows, reducing xNTP consumption [9]. |
| Fluorescent Activated Cell Sorter (FACS) | Essential instrument for isolating active enzyme variants from large libraries based on fluorescence in CBL and other display methods [28]. |
The development of efficient reverse transcription and amplification methods for XNA is pivotal for advancing synthetic genetics. The CBL platform provides a powerful and generalizable strategy for evolving RTs with activity on diverse and novel XNA chemistries. Furthermore, the rational engineering of fidelity-enhancing mutations offers a pathway to achieving the high accuracy required for demanding biotechnological and therapeutic applications. These protocols and engineered enzymes collectively provide the foundational toolset for researchers to explore the information-coding potential of XNA polymers, enabling the development of stable aptamers, catalysts (XNAzymes), and functional molecules for drug development and diagnostic applications [15].
Optimizing the Selection: A Strategic Guide to Enhanced XNA Polymerase Activity
Systematic Pipeline for Screening and Benchmarking Selection Parameters
Directed evolution serves as a powerful methodology in synthetic biology for engineering proteins, such as xenobiotic nucleic acid (XNA) polymerases, without requiring complete understanding of sequence-function relationships [30]. By mimicking natural evolution in vitro, this approach enables researchers to isolate polymerase variants with desired activities, properties, and substrate specificities for processing unnatural genetic polymers [30] [31]. However, the success of directed evolution campaigns depends critically on establishing optimal selection conditions that maximize the recovery of desired variants while minimizing background noise and parasitic activities [30]. This application note details a systematic pipeline for screening and benchmarking selection parameters to efficiently engineer XNA polymerases, providing researchers with standardized protocols to accelerate progress in synthetic biology and therapeutic development.
Background
Directed Evolution of XNA Polymerases
Natural DNA polymerases generally lack the inherent ability to process XNA substrates, which feature alternative sugar-phosphate backbones or nucleobases compared to natural DNA and RNA [18] [32]. Directed evolution has enabled the engineering of polymerase variants capable of synthesizing, reverse transcribing, and replicating various XNAs, including 1,5-anhydrohexitol nucleic acid (HNA), 2'-fluoroarabino nucleic acid (FANA), and threose nucleic acid (TNA) [15] [18]. These engineered enzymes serve as foundational tools for developing XNA-based technologies with applications in therapeutics, diagnostics, and synthetic biology [31] [32].
Emulsion-based compartmentalization has emerged as a particularly effective selection platform, as it establishes a strong genotype-phenotype linkage by encapsulating individual cells expressing unique polymerase variants together with substrates and reaction products [30]. This approach minimizes cross-reactivity and enables partitioning of libraries based on enzyme function, including substrate recognition, product formation, and synthesis rate.
Key Challenges in Selection Optimization
A significant challenge in directed evolution involves optimizing selection parameters to favor variants with desired activities while suppressing "parasitic" pathways. For example, in compartments containing XNA triphosphates, parasitic activity might involve polymerases that utilize low concentrations of endogenous dNTPs instead of the provided XNA substrates [30]. The concentration of cofactors, such as Mg²⁺ and Mn²⁺, can profoundly influence polymerase activity and fidelity, potentially shifting the balance between polymerase and exonuclease activities [30]. Determining optimal selection parameters for polymerase libraries of unknown function remains non-trivial, particularly when engineering new-to-nature substrate specificities.
Systematic Pipeline Development
We propose a pipeline that incorporates Design of Experiments (DoE) methodology for screening and benchmarking selection parameters using small, focused protein libraries [30]. This approach enables efficient optimization of selection parameters and concentration ranges before committing to larger, more complex libraries. The strategy employs small libraries and cost-effective next-generation sequencing (NGS) to streamline selection processes while maintaining robust identification of significantly enriched mutants [30].
Table 1: Key Selection Parameters for Optimization in XNA Polymerase Engineering
| Parameter Category | Specific Factors | Impact on Selection | Typical Range Tested |
|---|---|---|---|
| Substrate Conditions | Nucleotide concentration, Nucleotide chemistry (dNTPs vs. xNTPs) | Influences enzyme kinetics, substrate specificity, and parasite suppression | Varies by XNA type [30] |
| Cofactor Requirements | Mg²⁺ concentration, Mn²⁺ concentration, Mg²⁺/Mn²⁺ ratios | Affects catalytic efficiency, fidelity, and polymerase/exonuclease balance [30] | 0.1-10 mM [30] |
| Reaction Conditions | Selection time, Temperature, pH | Impacts reaction completeness and stringency | Minutes to hours [30] |
| Additives | PCR enhancers, Stabilizers, Crowding agents | Modifies enzyme activity and stability | Variable [30] |
Library Design and Construction
Focused Library Generation:
- Design a small, focused library targeting active site residues. For example, a 2-point saturation mutagenesis library targeting a metal-coordinating residue (D404) and its neighboring residue (L403) in Thermococcus kodakarensis DNA polymerase (KOD DNAP) [30].
- For broader exploration, consider a 5-point saturation mutagenesis library targeting multiple adjacent residues (e.g., L403, D404, F405, L408, and Y409) [30].
- Generate libraries using inverse PCR (iPCR) with mutagenic primers on an appropriate plasmid backbone (e.g., pET23-KOD-Exo-) with a high-fidelity DNA polymerase for 28 cycles [30].
- Digest parental DNA with DpnI, purify amplified products, and blunt-end ligate using T4 DNA ligase and T4 Polynucleotide Kinase overnight at room temperature [30].
- Transform ligated libraries into high-efficiency competent E. coli cells (e.g., 10-beta competent Escherichia coli cells) using electroporation [30].
- Plate transformed cells on large LB plates with appropriate antibiotics, incubate overnight at 37°C, then scrape and resuspend cells for storage at -80°C in LB with glycerol [30].
Experimental Design for Parameter Screening
Design of Experiments (DoE) Approach:
- Implement a DoE framework to systematically evaluate multiple selection parameters simultaneously, reducing experimental effort while capturing potential interaction effects [30].
- Define key factors (independent variables) for testing: nucleotide concentration, nucleotide chemistry (dNTPs vs. 2'F-rNTPs), selection time, Mg²⁺ concentration, Mn²⁺ concentration, and common PCR additives [30].
- Establish response metrics (dependent variables): recovery yield, variant enrichment patterns, and variant fidelity [30].
- Execute selection experiments according to the DoE matrix using the constructed focused libraries.
Selection Protocol and Output Analysis
Emulsion-Based Compartmentalization:
- Perform compartmentalized selection using water-in-oil emulsions to encapsulate individual cells expressing polymerase variants together with substrates [30].
- Include appropriate controls, such as non-functional polymerase variants (e.g., KODΔ lacking amino acid residues A316 to R406), to assess background and parasitic activities [30].
- After incubation under selection conditions, recover active variants by breaking emulsions and extracting genetic material [30].
Output Analysis:
- Analyze selection outputs for recovery yield (total number of recovered variants or sequences) [30].
- Assess variant enrichment patterns through next-generation sequencing (NGS) of selection outputs [30].
- Evaluate fidelity of enriched variants using functional assays. Recent advances include hydrogel-based fidelity measurements that dramatically reduce time and reagent consumption compared to traditional gel-based methods [9].
Table 2: Research Reagent Solutions for Directed Evolution of XNA Polymerases
| Reagent Category | Specific Examples | Function/Application | Notes |
|---|---|---|---|
| Polymerase Libraries | KOD D404/L403 saturation library, Tgo polymerase variants | Provides genetic diversity for selection | Focused libraries recommended for parameter optimization [30] |
| XNA Triphosphates | 2'F-rNTPs, HNA triphosphates, TNA triphosphates [9] | Substrates for XNA polymerase activity | Chemical synthesis required; valuable reagents [9] [31] |
| Specialty Primers | 5'-acrydite-modified primers [9] | Enables covalent attachment to hydrogel matrices | Essential for hydrogel-based fidelity assays [9] |
| Emulsion Components | Mineral oil, silicone emulsifiers (KF-6012, KF-6038) [9] | Creates water-in-oil compartments for genotype-phenotype linkage | Critical for compartmentalized self-replication [30] |
| Hydrogel Particles | Polyacrylamide-encapsulated magnetic Dynabeads M-270 [9] | Solid support for fidelity assays without purification steps | Enables miniaturized, purification-free fidelity measurements [9] |
Workflow Visualization
Systematic Pipeline for Selection Parameter Optimization
Implementation Protocols
Critical Protocol: DoE for Selection Parameter Screening
Step 1: Library Preparation
- Inoculate 50 mL of fresh LB media per library with an overnight culture of expression cells (e.g., BL21(DE3)) [30].
- Grow at 37°C until OD600 reaches 0.4-0.6, then induce with 1 mM IPTG for protein expression [30].
- Harvest cells and prepare for emulsion formation.
Step 2: Emulsion Setup and Selection
- Prepare aqueous phase containing induced cells, selection substrates (XNA triphosphates or nucleotide analogues), and selection buffers with varying parameters according to the DoE matrix [30].
- Create water-in-oil emulsions by mixing the aqueous phase with oil phase (e.g., mineral oil with silicone emulsifiers) [30].
- Incubate emulsions at appropriate temperatures (e.g., 55-65°C for thermophilic polymerases) for varied selection times as per experimental design [30].
Step 3: Output Recovery
- Break emulsions by centrifugation and remove the oil phase [30].
- Recover cells or genetic material from the aqueous phase for analysis [30].
- Extract plasmid DNA from recovered outputs for sequencing or subsequent selection rounds [30].
Advanced Protocol: Hydrogel-Based Fidelity Assessment
Hydrogel Particle Preparation [9]:
- Functionalize magnetic Dynabeads M-270 (∼2×10⁸ beads) with 5'-acrydite-modified DNA primers through polyacrylamide encapsulation.
- Resuspend beads in 6% acrylamide/bisacrylamide solution containing acrydite primer (100 μM) and 0.6% ammonium persulfate.
- Add TEMED initiator to catalyze polymerization and form hydrogel particles on ice for 2 hours.
- Wash particles with breaking buffer (10 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1% SDS, 1% Triton X-100) to remove oil and surfactants.
XNA Synthesis on Hydrogels [9]:
- Anneal DNA template to primer-functionalized hydrogel particles.
- Perform XNA transcription by incubating particles with XNA triphosphates and polymerase variant in appropriate buffer at optimal temperature (e.g., 60°C for thermophilic polymerases).
- Wash particles to remove DNA template and excess reagents.
Reverse Transcription and Fidelity Analysis [9]:
- Reverse transcribe XNA product back to DNA using polymerase variant with dNTPs.
- Recover cDNA product from hydrogel particles, amplify via PCR, clone, and sequence.
- Calculate fidelity by comparing sequences to original template to identify mutations.
Hydrogel-Based Fidelity Assessment Workflow
Data Analysis and Interpretation
Sequencing Coverage Requirements
- Minimum Coverage Threshold: Cost-effective and accurate identification of significantly enriched mutants can be achieved even at low NGS coverages, substantially below those required for genome assembly or other omics approaches [30].
- Variant Identification: Precisely identify enriched mutants from selection outputs by establishing appropriate sequencing depth based on library complexity and selection round.
Parameter Optimization Guidelines
- Balancing Activity and Fidelity: Selection conditions profoundly influence the trade-off between synthesis efficiency and fidelity, reflecting the polymerase/exonuclease equilibrium [30].
- Parasite Suppression: Optimize parameters to minimize recovery of variants utilizing undesired substrates (e.g., endogenous dNTPs instead of provided XNAs) [30].
- Condition Stringency: Adjust cofactor concentrations, substrate levels, and reaction times to control selection pressure and favor desired activities.
The systematic pipeline presented here provides a robust methodology for optimizing selection protocols in directed evolution of XNA polymerases. By implementing DoE with focused libraries, researchers can efficiently determine optimal selection parameters before proceeding to larger, more complex libraries. The integration of emulsion-based selection with advanced fidelity assessment methods, such as hydrogel particle displays, enables comprehensive evaluation of selection outputs. This approach significantly enhances the efficiency and effectiveness of directed evolution strategies for engineering XNA polymerases and other enzymes, accelerating progress in synthetic biology and expanding the toolbox for developing XNA-based therapeutics and applications.
Within the broader thesis on advancing synthetic biology through engineered xenobiotic nucleic acid (XNA) polymerases, the optimization of selection conditions is a critical determinant of success. Directed evolution campaigns aimed at generating polymerases capable of synthesizing artificial genetic polymers are highly sensitive to the biochemical environment [33] [34]. This application note provides a detailed protocol for systematically optimizing the key factors of divalent cation identity and concentration (Mg²⁺, Mn²⁺), nucleotide chemistry, and selection time. Proper management of these parameters directly influences the efficiency and outcome of the selection process by modulating polymerase activity, fidelity, and the enrichment of desired variants over parasitic pathways [33] [35]. The following sections provide a structured framework, including quantitative data and actionable protocols, to guide researchers in establishing robust and effective directed evolution experiments.
Systematic Analysis of Key Factors
A quantitative understanding of how cofactors and substrates influence polymerase behavior is essential for designing effective directed evolution experiments. The data presented below enable informed decision-making when setting selection conditions.
Table 1: Impact of Divalent Cations on Polymerase Kinetics and Fidelity Data derived from pre-steady-state kinetic analysis of BST DNA polymerase (A-family), illustrating general trends applicable to many engineered polymerases [35].
| Metal Cofactor | Relative Incorporation Efficiency (Correct dNTP) | Impact on Base Selectivity | Key Observed Effects |
|---|---|---|---|
| Mg²⁺ | 1.0 (Reference) | High | The physiological cofactor, providing the benchmark for fidelity and activity. |
| Mn²⁺ | Variable (often increased) | Dramatically Reduced | Impairs base discrimination; can enhance ground-state dNTP binding but promotes misincorporation. |
| Co²⁺ | ~6-fold increase | Improved vs. Mn²⁺ | Can enhance correct dNTP incorporation while decreasing incorrect incorporation; may help extension past mismatches. |
| Cd²⁺ | Supports catalysis | Moderate | Similar to Co²⁺ in supporting activity, but with distinct kinetic parameters. |
| Ni²⁺, Ca²⁺, Zn²⁺ | Little to no activity | N/A | Unable to effectively support nucleotidyl transfer for many polymerases. |
Table 2: Guide to Nucleotide Chemistry in XNA Synthesis Summary of common XNA substrates and their relevance to polymerase engineering campaigns [17] [2].
| Nucleotide Chemistry | Key Characteristics | Relevance to Directed Evolution |
|---|---|---|
| dNTPs (natural) | Natural substrates; high-fidelity incorporation. | Often a source of "parasite" activity; their concentration may be controlled to favor XNA synthesis. |
| 2′F-rNTPs | Common XNA substrate with a 2′-fluoro modification on arabinose. | A target chemistry for engineering; selection parameters are tuned to favor its use over dNTPs. |
| HNA TNA | 1,5-anhydrohexitol nucleic acid. α-l-threofuranosyl nucleic acid; nuclease resistant, acid-resistant. | Desired substrates for therapeutic and biotechnological applications; polymerases like 6G12 and 10-92 have been evolved for these XNAs. |
| Other XNAs (FANA, CeNA, LNA) | Various sugar and backbone modifications with altered stability and pairing properties. | Represent a broad class of targets for polymerase engineering; selection conditions must be optimized for each specific chemistry. |
Experimental Protocols for Parameter Optimization
Protocol: Screening Divalent Cations for XNA Synthesis
Objective: To determine the optimal type and concentration of divalent cations that maximize the activity and fidelity of an XNA polymerase library for a specific XNA chemistry.
Materials:
- Purified polymerase library (e.g., a focused library of KOD or Phi29 variants).
- DNA template/primer complex.
- Natural dNTPs and target xNTPs (e.g., 2′F-rNTPs, TNA triphosphates).
- 1 M Stock solutions of MgCl₂, MnCl₂, CoCl₂, CdSO₄.
- 10x Reaction buffer (e.g., 660 mM Tris-HCl, pH 7.3).
- Stopping solution (0.5 M EDTA).
- Denaturing Polyacrylamide Gel Electrophoresis (PAGE) equipment.
Procedure:
- Prepare Master Mix: Create a master mix containing 1x reaction buffer, DNA template/primer complex, and the polymerase library.
- Set Up Cation Reactions: Aliquot the master mix into separate tubes. To each tube, add a different divalent cation (Mg²⁺, Mn²⁺, Co²⁺, or Cd²⁺) from stock solutions to a final concentration of 10 mM. Include a control with no added metal.
- Initiate Reaction: Start the synthesis reaction by adding a mixture of dNTPs and xNTPs. A typical final concentration for each nucleotide is 500 µM.
- Time-Course Sampling: Incubate at the desired selection temperature (e.g., 55°C for thermophilic polymerases). Withdraw 5 µL aliquots at multiple time points (e.g., 10, 20, 40, 60, 120 seconds) and immediately quench with an equal volume of 0.5 M EDTA.
- Analyze Products: Resolve the extended products from the substrate primer using denaturing PAGE. Quantify the product bands using phosphorimaging or fluorescence scanning.
- Data Analysis: Plot the amount of product formed versus time for each condition. The condition yielding the highest amount of full-length XNA product with the least misincorporation (as judged by side-products) is optimal.
Protocol: Optimizing Selection Time via Compartmentalized Self-Replication (CSR)
Objective: To identify the selection time that maximizes the enrichment of highly active XNA synthetases while minimizing the recovery of parasites.
Materials:
- Expression strain (e.g., E. coli BL21(DE3)) transformed with the polymerase library.
- Emulsification oil phase (e.g., mineral oil with surfactants).
- Luria-Bertani (LB) medium with appropriate antibiotic.
- Inducer (e.g., IPTG).
- Substrates: dNTPs and target xNTPs.
- Lysis buffer.
Procedure:
- Emulsion Formation: Grow the expression strain to mid-log phase and induce polymerase expression. Formulate the aqueous phase containing the induced cells, selection buffer, Mg²⁺/Mn²⁺ (at concentrations determined in Protocol 3.1), and dNTP/xNTP substrates. Emulsify this aqueous phase into the oil phase to create water-in-oil microdroplets, compartmentalizing individual cells and selection reagents [33].
- Time-Course Selections: Incubate the emulsion to allow for in-compartment PCR or primer extension based on polymerase function. Prepare multiple identical emulsions and halt the reactions at different time points (e.g., 30, 60, 90, 120 minutes) by breaking the emulsion and cooling.
- Recovery and Analysis: Recover the plasmid DNA from each time-point sample. Transform a fresh expression host and plate for colonies. Count the number of colonies recovered at each time point.
- Determine Optimal Time: The optimal selection time is typically at the inflection point of the recovery curve, where the yield of active variants is high but before the exponential growth of parasites. This time point should be used for subsequent rounds of selection.
Protocol: Rapid Fidelity Assessment via Hydrogel Particle Display
Objective: To rapidly measure the fidelity of engineered XNA polymerases without the need for laborious gel purification, facilitating the screening of multiple variants under different cofactor conditions [9].
Materials:
- Hydrogel-coated magnetic particles (e.g., Dynabeads M-270) with covalently linked DNA primer.
- DNA template.
- Candidate XNA polymerase (wild-type and evolved variants).
- xNTPs and dNTPs.
- Lysis and wash buffers.
Procedure:
- Functionalize Particles: Anneal the DNA template to the primer that is cross-linked throughout the hydrogel matrix of the magnetic particles.
- XNA Synthesis: Incubate the particles with the candidate polymerase and the required xNTPs in optimized buffer/cofactor conditions to transcribe the template into XNA. Wash thoroughly to remove the DNA template and enzyme.
- Reverse Transcription: Inside the same hydrogel matrix, reverse transcribe the synthesized XNA strand back into DNA (cDNA) using a reverse transcriptase (which could be the same engineered polymerase if it has this activity).
- Recover and Sequence: Recover the cDNA, amplify it by PCR, clone the products, and sequence multiple clones.
- Calculate Error Rate: Compare the sequences of the cDNA clones to the original DNA template sequence. The error rate is calculated as (Total number of mutations) / (Total number of bases sequenced).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for XNA Polymerase Directed Evolution
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| Thermostable B-Family DNA Pol | Common starting scaffold for engineering XNA synthetases. | Polymerases from Pyrococcus and Thermococcus species (e.g., KOD, Tgo) are highly amenable to engineering [2]. |
| xNTPs (XNA triphosphates) | Activated monomers for the synthesis of XNA polymers. | Chemically synthesized (e.g., TNA triphosphates [9]); can be costly, motivating miniaturized assays. |
| Compartmentalized Self-Replication (CSR) | Ultra-high-throughput selection platform linking genotype to phenotype. | Emulsion-based system partitions individual cells/variants to select based on function [33]. |
| Homologous Recombination Library | A method for generating high-quality, diverse variant libraries. | Used to create the starting diversity for evolving a highly efficient TNA polymerase [36]. |
| Hydrogel-Coated Magnetic Particles | Solid support for miniaturized and purification-free fidelity assays. | Provides a solution-like environment for XNA synthesis and reverse transcription without gel purification [9]. |
| Next-Generation Sequencing (NGS) | Deep mutational scanning to identify enriched variants and analyze populations. | Critical for calculating enrichment scores and understanding selection outcomes [33] [34]. |
Workflow and Strategic Planning
The following diagram illustrates the integrated workflow for optimizing selection parameters and executing a directed evolution campaign for XNA polymerases.
Minimizing Parasites and False Positives in Emulsion-based Selections
Within the context of directed evolution for engineering xenobiotic nucleic acid (XNA) polymerases, emulsion-based compartmentalization is a powerful tool. It establishes a critical genotype-phenotype linkage by isolating individual enzyme variants into water-in-oil droplets, enabling the selection of variants based on desired activities like XNA synthesis or reverse transcription [30]. However, the success of this platform is often compromised by the enrichment of "parasites" and false positives. Selection parasites are variants that are recovered due to viable alternative but non-desired phenotypes, such as a polymerase variant that utilizes low cellular concentrations of dNTPs present in the emulsion instead of the provided XNA substrates [30]. False positives can also arise from random, non-specific processes or background noise [30]. This application note details optimized protocols and strategic considerations to minimize these artifacts, thereby streamlining the directed evolution of XNA polymerases for synthetic biology and therapeutic applications [2].
Core Principles and Strategic Optimization
The directed evolution of polymerases can be conceptualized as an adaptive walk on a fitness landscape. The goal is to guide this walk towards peaks representing high fitness for the desired activity (e.g., XNA synthesis) and away from local optima representing parasitic functions [30]. Key selection parameters, often screened using Design of Experiments (DoE) methodologies, profoundly influence this trajectory by altering the polymerase/exonuclease equilibrium and shaping the selective pressure on the library [30].
Table 1: Key Selection Parameters and Their Impact on Parasite Suppression
| Parameter | Biological Impact | Strategy for Minimizing Parasites |
|---|---|---|
| Substrate & Cofactor Concentration (XNA, dNTP, Mg²⁺, Mn²⁺) | Influences enzyme kinetics, specificity, and the polymerase/exonuclease fidelity balance [30]. | Optimize ratios to strongly favor the desired XNA-templated synthesis over DNA/DNA activity. Limit dNTP availability to starve DNA-polymerase parasites [30]. |
| Selection Time | Determines the window for enzyme activity. | Shorter times can select for faster, more efficient XNA synthetases before slow-growing parasites accumulate [30]. |
| PCR Additives | Can stabilize emulsions, enhance polymerase processivity, or alter fidelity. | Use additives that stabilize the emulsion and promote the desired activity without boosting background. |
| Emulsion Stability | Prevents coalescence of droplets, which is critical for maintaining genotype-phenotype linkage [37]. | Employ robust surfactants and optimized emulsification protocols to prevent cross-talk and catalysis between compartments [37]. |
Experimental Protocols
Protocol 1: Systematic Optimization of Selection Conditions Using DoE
This protocol uses a small, focused polymerase library to rapidly benchmark and identify optimal selection parameters before committing large, diverse libraries.
1. Library Design and Construction:
- Generate a focused saturation mutagenesis library targeting key metal-coordinating and adjacent residues (e.g., residues L403 and D404 in Thermococcus kodakarensis DNA polymerase) [30].
- Use inverse PCR (iPCR) with mutagenic primers and a high-fidelity DNA polymerase (e.g., Q5 from NEB) for 28 cycles [30].
- Digest template plasmid with DpnI, purify the product, blunt-end ligate, and transform into high-efficiency electrocompetent E. coli cells (e.g., 10-beta) [30].
2. Screening Selection Parameters:
- Factors/Variables: Systematically vary the concentrations of Mg²⁺, Mn²⁺, dNTPs, and XNA nucleotides (e.g., 2′F-rNTPs). Also test different selection times and common PCR additives [30].
- Setup: Perform compartmentalized self-replication (CSR) or similar emulsion-based reactions for each parameter combination.
- Responses/Analysis: Quantify the recovery yield, variant enrichment (via Next-Generation Sequencing, NGS), and assess the fidelity of the enriched population [30].
3. Data-Driven Optimization:
- Analyze the DoE results to identify selection conditions that maximize the recovery of variants with the desired XNA polymerase activity while minimizing the enrichment of parasites (e.g., variants with high DNA polymerase activity) [30].
Protocol 2: Emulsion-based Selection with micPCR for Absolute Quantification
This protocol adapts a highly accurate micelle-based PCR (micPCR) workflow to emulsion-based selections, minimizing amplification artifacts and enabling precise quantification.
1. Emulsion Formulation:
- Prepare the water phase containing the PCR mixture: DNA library, primers, polymerase (e.g., LongAmp Taq 2x MasterMix for full-length amplicons), dNTPs/XNA substrates, and a single internal calibrator (IC) such as Synechococcus 16S rRNA gene copies [38].
- Formulate the oil phase with a biocompatible surfactant to stabilize the emulsion and prevent droplet coalescence during thermocycling [37].
2. Emulsification and Amplification:
- Generate a water-in-oil emulsion using a microfluidic device or vigorous mixing to create millions of monodisperse compartments.
- Perform the first round of micPCR with primers containing universal sequence tails. The emulsion state ensures clonal amplification within each micelle, preventing chimera formation and PCR competition [38].
- Break the emulsion and purify the amplicons.
3. Analysis and Quantification:
- Perform a second, standard PCR to barcode the amplicons for NGS.
- Use NGS to sequence the outputs. The internal calibrator allows for absolute quantification of template molecules, enabling the subtraction of background contaminating DNA and the accurate identification of significantly enriched mutants, even at low sequencing coverage [38].
The workflow below illustrates the critical steps in a robust, emulsion-based selection pipeline designed to minimize parasites.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Robust Emulsion-Based Selections
| Reagent / Material | Function / Rationale | Example / Specification |
|---|---|---|
| High-Fidelity DNA Polymerase | Reduces PCR errors during library construction. | Q5 High-Fidelity DNA Polymerase (NEB) [30]. |
| Thermostable B-Family Polymerase | Engineering scaffold with high plasticity for XNA synthesis. | KOD DNA Polymerase from Thermococcus kodakarensis [30] [2]. |
| Biocompatible Surfactants | Stabilizes water-in-oil emulsions to prevent droplet coalescence during thermocycling, maintaining genotype-phenotype linkage [37]. | Specific commercial surfactants for droplet-based assays (e.g., from BioRad). |
| Internal Calibrator (IC) | Allows absolute quantification and background subtraction in NGS data. | Synechococcus 16S rRNA gene copies [38]. |
| Xenobiotic Nucleic Acids (XNAs) | Substrates for positive selection pressure. | 2′-deoxy-2′-α-fluoro nucleoside triphosphate (2′F-rNTP) [30]. |
| Long-Amp PCR MasterMix | Efficiently amplifies full-length gene amplicons, crucial for full-length 16S sequencing or large gene variants. | LongAmp Taq 2x MasterMix [38]. |
Data Analysis and Validation
Post-selection analysis is critical for distinguishing true hits from artifacts.
1. Next-Generation Sequencing (NGS):
- A sequencing coverage of 50-100x per variant is often sufficient for the accurate and precise identification of significantly enriched mutants in directed evolution experiments, making the process cost-effective [30].
- Analyze the data to identify mutations that are significantly enriched above the background noise and parental sequence.
2. Fidelity Assessment:
- For polymerases, a key validation step is to assess the fidelity (error rate) of enriched variants. This provides a window into the polymerase/exonuclease equilibrium and can indicate whether the selection has enriched for specific functional properties [30].
- Parasites may exhibit altered fidelity profiles compared to variants with the desired XNA synthesis activity.
Table 3: Troubleshooting Common Issues in Emulsion-Based Selections
| Problem | Potential Cause | Solution |
|---|---|---|
| High Background / False Positives | Non-specific amplification; droplet coalescence; insufficient selection pressure. | Optimize emulsion stability with better surfactants; titrate substrate/cofactor concentrations to increase stringency [30] [37]. |
| Enrichment of Parasites | Selection conditions allow for alternative phenotypes (e.g., dNTP usage). | Use DoE to find conditions that disfavor the parasitic activity; employ substrate-limited conditions [30]. |
| Low Recovery of Desired Variants | Selection conditions are too stringent; desired activity is too weak. | Systematically relax stringency (e.g., increase time, substrate concentration) based on DoE results. |
| Poor Emulsion Stability | Inefficient emulsification; unsuitable surfactant. | Use cavitation-based emulsification devices for monodisperse droplets; optimize surfactant type and concentration [39]. |
In the field of synthetic biology, the directed evolution of Xeno-Nucleic Acid (XNA) polymerases represents a frontier for expanding the functional capabilities of genetic systems. These engineered enzymes must master a fundamental equilibrium: catalyzing efficient DNA and XNA synthesis while maintaining high fidelity through proofreading. Polymerase fidelity is quantified as the error frequency of misincorporation per nucleotide incorporated, a critical metric for genetic information transfer [40] [41]. For replicative DNA polymerases, this fidelity is governed by two major activities: the polymerase (pol) activity that adds nucleotides to the growing chain, and the 3'-5' exonuclease (exo) activity that excises misincorporated nucleotides in a process called proofreading [40] [42].
The kinetic competition between these two active sites determines the overall accuracy of DNA replication. Efficient nucleotide insertion governs polymerase fidelity, with high-fidelity polymerases demonstrating superior catalytic efficiency for correct nucleotide insertion over incorrect ones [41]. When this insertion fails and a mismatch occurs, the primer terminus must traverse a ~60-70 Å path from the polymerase site to the exonuclease site—a process involving DNA fraying, backtracking, and significant conformational changes in the polymerase structure itself [42]. For researchers engineering XNA polymerases, understanding and measuring this equilibrium is paramount, as these enzymes are tasked with replicating non-natural genetic polymers with accuracy sufficient for evolution and functional application [43].
Core Principles and Quantitative Fidelity Metrics
Determinants of Polymerase Fidelity
The intrinsic fidelity of a DNA polymerase arises from a multi-step mechanism that selectively incorporates correct nucleotides while rejecting incorrect ones:
- *Nucleotide Selection:* The polymerase active site preferentially incorporates correct nucleotides through large decreases in both nucleotide incorporation rate constants and ground-state binding affinities for incorrect versus correct nucleotides. This selectivity ranges from 10⁴ to 10⁷, driven by factors including base stacking, nucleotide desolvation, induced-fit conformational changes, and shape complementarity [40].
- *Proofreading Enhancement:* The 3'-5' exonuclease activity provides a critical secondary checkpoint by excising misincorporated nucleotides from the primer 3' terminus. This proofreading function enhances polymerization fidelity by an unprecedented 3.5 × 10² to 1.2 × 10⁴-fold in human DNA polymerase ε (hPolε) [40].
- *Mismatch Extension:* Mispaired primer termini significantly slow further DNA extension, altering the equilibrium toward primer release and transfer to the exonuclease site [42].
Quantitative Fidelity Across Polymerase Families
The table below summarizes fidelity measurements for natural and engineered polymerases, highlighting the critical balance between synthetic capability and accuracy:
Table 1: Quantitative Fidelity Measurements of Natural and Engineered Polymerases
| Polymerase Type | Polymerase Family | Error Rate (per nucleotide) | Exonuclease Enhancement | Key Functional Role |
|---|---|---|---|---|
| Human Polε (pol activity only) | B | 10⁻⁴ – 10⁻⁷ | - | Leading strand synthesis [40] |
| Human Polε (with proofreading) | B | 10⁻⁶ – 10⁻¹¹ | 350–12,000-fold | Overall genomic replication [40] |
| E. coli Pol III core | C | ~10⁻⁶ | Not specified | Bacterial replicative polymerase [42] |
| TgoT (HNA synthesis) | B (engineered) | Not specified | Exonuclease-deficient (D141A, E143A) | XNA synthesis [43] |
| CeNA Replication Cycle | N/A | 4.3 × 10⁻³ | Not specified | Full DNA→XNA→DNA cycle [43] |
| LNA Replication Cycle | N/A | 5.3 × 10⁻² | Not specified | Full DNA→XNA→DNA cycle [43] |
For XNA systems, the aggregate fidelity of a complete replication cycle (DNA→XNA→DNA) varies significantly between different XNA chemistries, with HNA, CeNA, ANA, and FANA demonstrating superior fidelity to LNA and TNA [43]. Engineered XNA polymerases often exhibit reduced fidelity compared to their natural counterparts, as polymerase structures are poorly adapted to detect mismatches in non-canonical XNA•DNA duplexes [43].
Experimental Protocols for Measuring Fidelity and Activity
This section provides detailed methodologies for key experiments that quantify the polymerase/exonuclease equilibrium, with particular consideration for applications in XNA polymerase engineering.
Pre-Steady-State Kinetic Analysis of Fidelity
This protocol determines the base substitution fidelity of a polymerase by measuring individual contributions of polymerase and 3'→5' exonuclease activities under single-turnover conditions [40].
Materials and Reagents
- Purified polymerase (wild-type and exonuclease-deficient mutant)
- DNA substrates: Primer-template duplexes with defined sequences (see Table 1 in [40] for examples)
- Nucleotides: dNTPs or xNTPs (e.g., hNTPs, ceNTPs for XNA synthesis)
- Reaction Buffer E: 50 mM Tris-OAc (pH 7.4 at 20°C), 8 mM Mg(OAc)₂, 1 mM DTT, 10% glycerol, 0.1 mg/ml BSA, 0.1 mM EDTA
- Radioisotope: [γ-³²P]ATP for 5' end-labeling of primers
- Enzymes: Optikinase for radiolabeling
- Separation Materials: Denaturing polyacrylamide gel (17% acrylamide, 8 M urea, 1× TBE)
Procedure
Prepare Radiola-beled DNA Substrate:
- 5'-end-label the primer strand using [γ-³²P]ATP and Optikinase
- Purify labeled primer from free [γ-³²P]ATP using a Bio-Spin 6 column
- Anneal to template strand using 1.15-fold excess of template by heating to 95°C for 5 minutes followed by slow cooling to room temperature
Perform Polymerization Single-Turnover Assay:
- Pre-incubate polymerase (260 nM) with radiolabeled DNA substrate (20 nM) in Buffer E
- Rapidly mix with Mg²⁺ (8 mM final) and varying concentrations of correct or incorrect dNTP/xNTP using a rapid chemical quench-flow apparatus
- Quench reactions at times ranging from milliseconds to seconds using 0.37 M EDTA
- Perform all reactions at 20°C to accurately measure fast incorporation rates [40]
Perform Exonuclease Assay:
- Pre-incubate exonuclease-proficient polymerase (200 nM) with radiolabeled DNA substrate (20 nM) containing a defined mismatch at the primer terminus
- Rapidly mix with Mg²⁺ (8 mM) in the absence of nucleotides to initiate excision
- Quench at appropriate time points with 0.37 M EDTA
Analyze Products:
- Separate reaction products by denaturing polyacrylamide gel electrophoresis
- Quantify product formation using a phosphorimager and appropriate software
- Fit polymerization data to the single-exponential equation:
Product = A[1 - exp(-kobs × t)]where A is the amplitude and kobs is the observed rate constant
Calculate Kinetic Parameters and Fidelity:
- Plot kobs versus nucleotide concentration for correct and incorrect nucleotides
- Determine catalytic efficiency (kpol/Kd) for each nucleotide
- Calculate fidelity as (kpol/Kd)correct / (kpol/Kd)incorrect for polymerase activity alone
- Determine overall fidelity including exonuclease contribution
Hydrogel Particle-Based Fidelity Assay for XNA Polymerases
This streamlined approach measures XNA polymerase fidelity while minimizing tedious purification steps and reducing consumption of valuable xNTPs [9].
Materials and Reagents
- DNA Primer-Functionalized Hydrogel Particles: Prepared from M-270 carboxylic acid Dynabeads with 5'-acrydite-modified primer crosslinked throughout polyacrylamide matrix
- XNA Triphosphates: Chemically synthesized (e.g., TNA, FANA, HNA triphosphates)
- Engineered XNA Polymerases: Expressed and purified from E. coli (e.g., TgoT variants)
- Transcription Buffer: ThermoPol buffer (or equivalent)
- Cloning and Sequencing Reagents: TOPO-TA cloning kit, plasmid prep kit, Sanger sequencing reagents
Procedure
XNA Synthesis on Hydrogels:
- Wash DNA primer-functionalized hydrogel particles with transcription buffer
- Anneal DNA template of defined sequence to the immobilized primer
- Perform XNA transcription by incubating with engineered XNA polymerase and xNTPs
- Wash thoroughly to remove template, enzymes, and excess nucleotides [9]
Reverse Transcription:
- Reverse transcribe the XNA product back to cDNA directly on the hydrogel particles using an appropriate engineered reverse transcriptase (e.g., RT521 for HNA, TNA, ANA, FANA; RT521K for CeNA and LNA) [43]
- Wash to remove enzymes and reagents
Recovery and Analysis:
- Recover cDNA product from hydrogel particles
- Amplify via PCR using primers flanking the variable region
- Clone amplified products using TOPO-TA cloning kit
- Sequence multiple clones (typically 50-100) by Sanger sequencing
- Calculate error rate by comparing sequences to the original template
Table 2: Key Engineered Polymerases for XNA Synthesis and Reverse Transcription
| Polymerase Variant | Base Polymerase | Key Mutations | XNA Activities |
|---|---|---|---|
| Pol6G12 | TgoT | V589A, E609K, I610M, K659Q, E664Q, Q665P, R668K, D669Q, K671H, K674R, T676R, A681S, L704P, E730G | HNA synthesis [43] |
| PolC7 | TgoT | E654Q, E658Q, K659Q, V661A, E664Q, Q665P, D669A, K671Q, T676K, R709K | CeNA, LNA synthesis [43] |
| PolD4K | TgoT | L403P, P657T, E658Q, K659H, Y663H, E664K, D669A, K671N, T676I | ANA, FANA synthesis [43] |
| RT521 | TgoT | E429G, I521L, K726R | HNA, TNA, ANA, FANA reverse transcription [43] |
| RT521K | RT521 | A385V, F445L, E664K | Enhanced CeNA RT, LNA RT [43] |
The Polymerase-Exonuclease Transition Mechanism
The transfer of the primer terminus from the polymerase to exonuclease active site represents a critical structural rearrangement with direct implications for fidelity. Recent structural and computational studies have illuminated this process:
Molecular Pathway of Primer Transfer
Molecular simulations and cryo-EM structures reveal a defined path for the pol-to-exo transition in E. coli Pol III [42]:
- Mismatch Recognition and Fraying: A misincorporated nucleotide at the primer terminus induces fraying of the primer-template junction
- Sequential Unpairing: Three nucleotides sequentially unpair at the primer-template junction to create a flexible single-stranded region
- DNA Backtracking and Rotation: The DNA duplex backtracks approximately 5.3 Å and rotates 32.9° within the cavity formed by the polymerase and β-clamp
- Subunit Repositioning: The ε exonuclease subunit tilts approximately 12° toward the α polymerase subunit, shortening the distance to the exo site by ~10 Å
- Thumb Domain Movement: The thumb domain moves outward to create space for the passing primer strand
This transition occurs on a timescale of tens of milliseconds, with the pathway stabilized by conserved positively charged residues along the fingers, thumb, and exonuclease domains that interact with the transitioning DNA backbone [42].
Diagram 1: The Polymerase-Exonuclease Switching Pathway. This diagram illustrates the sequence of molecular events that transfer a mismatched primer terminus from the polymerase active site to the exonuclease site for proofreading.
Experimental Monitoring of Active-Site Switching
The radiometric thin-layer chromatography (TLC) assay simultaneously measures polymerase and exonuclease activities during processive synthesis, providing direct insight into their coupling [44]:
- Assay Principle: Measure the ratio of nucleotide incorporation (polymerase activity) to excision (exonuclease activity) during strand displacement synthesis on a minicircle DNA substrate
- Key Insight: The polymerase-to-exonuclease activity ratio provides a quantitative measure of helicase-polymerase coupling, with uncoupling leading to excessive excision
- Application: This method is particularly valuable for assessing how replication hurdles (nucleotide depletion, DNA lesions) affect the polymerase/exonuclease equilibrium
Application Notes for XNA Polymerase Engineering
Strategic Considerations for Directed Evolution
When engineering XNA polymerases through directed evolution, specific strategies must address the unique challenges of synthetic genetic polymers:
- *Evolution Strategy:* Employ compartmentalized self-tagging (CST) for selecting XNA polymerase activity from large mutant libraries [43]. This links genotype to phenotype through in vitro transcription of a DNA barcode into XNA.
- *Mutation Targeting:* Focus mutations to regions >20 Å from the active site in the thumb domain to enhance XNA synthesis, and near catalytic aspartates for reverse transcription capability [43].
- *Fidelity-Speed Tradeoff:* Recognize that engineering for non-canonical substrate activity often comes at the cost of reduced fidelity, necessitating compensatory mutations or alternative strategies [43].
- *Functional Validation:* Beyond synthesis capability, validate evolved polymerases through functional aptamer selection from XNA libraries, demonstrating the capacity for Darwinian evolution in synthetic genetic systems [43].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Polymerase Fidelity and XNA Engineering Studies
| Reagent / Tool | Function / Application | Examples / Specifications |
|---|---|---|
| Engineered XNA Polymerases | Synthesis and reverse transcription of XNAs | TgoT variants (Pol6G12, PolC7, PolD4K, RT521) [43] |
| XNA Triphosphates (xNTPs) | Substrates for XNA synthesis | FANA, HNA, TNA triphosphates (chemical synthesis) [9] [43] |
| Rapid Chemical Quench-Flow | Pre-steady-state kinetic measurements | Millisecond-second timescale reactions at controlled temperature [40] |
| Defined DNA Templates | Fidelity measurement substrates | Specific sequences with primer-binding regions [40] |
| Hydrogel Magnetic Particles | Solid-phase XNA synthesis and analysis | Polyacrylamide-encapsulated M-270 Dynabeads with crosslinked primers [9] |
| Compartmentalized Self-Tagging (CST) | Directed evolution of polymerase activity | Links genotype to phenotype through XNA transcription [43] |
Diagram 2: XNA Polymerase Engineering and Characterization Workflow. This diagram outlines an iterative directed evolution pipeline for developing and characterizing XNA polymerases with optimized activity and fidelity.
The equilibrium between polymerase and exonuclease activities represents a fundamental design principle in DNA replication machinery that must be successfully engineered for synthetic biology applications. For XNA polymerase evolution, this requires balancing the competing demands of synthetic efficiency against informational fidelity. The experimental approaches detailed here—from pre-steady-state kinetics to modern hydrogel-based fidelity assays—provide robust methodologies for quantifying this balance.
Successful engineering strategies must account for the structural basis of the polymerase-exonuclease transition, the kinetic principles governing nucleotide selection, and the unique challenges posed by non-natural nucleic acid chemistries. As the field advances, the continued development of high-fidelity XNA polymerases will enable more sophisticated applications in synthetic genetics, including the development of XNA aptamers for therapeutic applications and the exploration of alternative genetic systems with expanded chemical functionality.
Proving Function: Validation and Structural Analysis of Engineered Polymerases
Within the field of synthetic biology, the directed evolution of XNA polymerases is a critical endeavor for advancing the development of functional xenonucleic acids. A paramount characteristic of any engineered polymerase is its fidelity—the accuracy with which it synthesizes or reverse-transcribes genetic polymers. High-throughput sequencing (HTS) technologies provide the means to quantify this fidelity with unprecedented depth and statistical power. This Application Note details protocols and methodologies for employing HTS to rigorously assess the error rates of XNA polymerases, providing a essential framework for research aimed at creating high-fidelity enzymes for next-generation synthetic genetics applications [2].
High-Throughput Sequencing Error Profiles
The first step in designing a fidelity assay is understanding the baseline error rates of the sequencing technology itself. Different HTS platforms exhibit distinct error profiles, which must be accounted for when calculating polymerase-dependent error rates. The table below summarizes the characteristic error rates of major sequencing platforms.
Table 1: Error Rates of Common High-Throughput Sequencing Platforms
| Sequencing Platform | Primary Error Type | Reported Error Rate | Key Characteristics |
|---|---|---|---|
| Illumina [45] | Substitution | ~0.1% - 1% (10-2 - 10-3) | Bridge amplification; all four fluorescently tagged dNTPs present during incorporation. |
| Roche/454 GS FLX [46] | Indel (in homopolymers) | ~0.49% - 1.07% | Pyrosequencing; signal intensity is proportional to homopolymer length, leading to insertion/deletion errors. |
| Ion Torrent [45] | Indel (in homopolymers) | Not explicitly quantified | Similar to 454; detects hydrogen ions from incorporation, struggles with homopolymer stretches. |
| Pacific Biosciences (PacBio) [45] | Random Insertions | Relatively high, but random | Single-Molecule Real-Time (SMRT) sequencing; random errors can be mitigated via Circular Consensus Sequencing (CCS). |
| Oxford Nanopore (ONT) [47] | Insertion, Deletion, Substitution | ~7.0% - 14.0% (pre-correction) | Direct electronic sensing of DNA/RNA; long reads but high raw error rate, correctable computationally (e.g., to ~1.1%). |
| Circle Sequencing [48] | All types (dramatically reduced) | As low as 7.6 × 10-6 | A specialized library prep method that enables computational error correction, putting HTS error on par with Sanger sequencing. |
Key Experimental Protocols for Fidelity Assessment
The following sections provide detailed protocols for assessing XNA polymerase fidelity using HTS. The core principle involves using the polymerase to copy a defined DNA template, sequencing the products, and comparing them to the original template to identify errors.
Hydrogel Particle-Based Fidelity Assay
This protocol, adapted from a recent study, streamlines the traditionally tedious process of XNA fidelity measurement by containing the replication cycle within hydrogel particles, eliminating the need for gel purification and enabling miniaturization [9].
1. Functionalization of Hydrogel Particles:
- Materials: Dynabeads M-270 carboxylic acid, 40% acrylamide/bisacrylamide (19:1), acrydite-modified DNA primer, ammonium persulfate (APS), TEMED.
- Procedure:
- Resuspend Dynabeads in a solution of 6% acrylamide/bisacrylamide, the acrydite-primer (100 µM), and 0.6% APS.
- Add an oil mixture (e.g., mineral oil with silicone emulsifiers) and degas with argon.
- Add TEMED to initiate polymerization and mix vigorously using a homogenizer (e.g., BeadBug at 2500 rpm) for 65 seconds. Incubate on ice for 2 hours.
- Wash the resulting primer-functionalized hydrogel particles extensively with a breaking buffer (e.g., 10 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1% SDS, 1% Triton X-100) to remove oil and unincorporated reagents [9].
2. XNA Synthesis and Reverse Transcription on Hydrogels:
- Materials: DNA template, XNA triphosphates (xNTPs), XNA polymerase, DNA reverse transcriptase, appropriate reaction buffers.
- Procedure:
- Annead the DNA template to the primer displayed on the hydrogel matrix.
- Perform the XNA transcription reaction by adding the XNA polymerase and the corresponding xNTPs. Wash the particles to remove the DNA template and excess reagents.
- Perform the reverse transcription reaction on the same particles to copy the XNA product back into DNA (cDNA) using a DNA polymerase.
- Recover the cDNA from the particles for downstream processing [9].
3. Downstream Analysis:
- Amplify the recovered cDNA using PCR.
- Clone the PCR products into a sequencing vector and transform into bacteria.
- Sequence individual clones via Sanger sequencing or prepare libraries for HTS.
- Align the resulting sequences to the original known template sequence to identify mutations (substitutions, insertions, deletions) introduced during the XNA synthesis and reverse transcription steps [9].
Diagram: Workflow for Hydrogel-Based XNA Polymerase Fidelity Assay
Circle Sequencing for Ultra-High-Fidelity Measurement
For applications requiring the utmost accuracy in distinguishing polymerase errors from sequencing errors, the circle sequencing method is recommended. This library preparation technique can reduce the effective HTS error rate by orders of magnitude [48].
1. Library Preparation:
- Materials: DNA template, circularizing ligase, rolling circle polymerase.
- Procedure:
- Circularize the DNA templates using a ligase.
- Use a rolling circle polymerase to generate multiple tandem copies of the original template in a single, long concatemeric DNA molecule. This physically links all copies derived from a single original molecule.
2. Sequencing and Computational Correction:
- Procedure:
- Sequence the concatemers on any HTS platform (e.g., Illumina MiSeq).
- Process the reads computationally to identify the tandem copies.
- For each original molecule, generate a consensus sequence from all its linked copies. Errors that are random (sequencing errors) will not be present in all copies, while errors introduced during the initial polymerization (polymerase errors) will be present in every copy and thus appear in the consensus.
- The final output is a set of consensus sequences with an error rate as low as ( 7.6 \times 10^{-6} ), enabling highly accurate fidelity measurement [48].
The Scientist's Toolkit: Essential Research Reagents
Successful execution of these protocols requires a suite of specialized reagents. The following table outlines key materials and their functions.
Table 2: Essential Research Reagents for XNA Polymerase Fidelity Assays
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| XNA Triphosphates (xNTPs) | Substrates for XNA polymerase during transcription. | Chemically synthesized TNA-, FANA-, or HNA-triphosphates [9] [15]. |
| Engineered XNA Polymerases | Enzymes capable of synthesizing XNA from a DNA template. | Thermostable polymerases from Pyrococcus or Thermococcus, engineered for XNA synthesis [2]. |
| Acrydite-Modified Primers | Covalent incorporation of DNA primers into polyacrylamide hydrogel matrix. | 5'-modified oligonucleotide; enables solid-phase biochemistry in hydrogel protocol [9]. |
| Functionalized Magnetic Beads | Solid support for separations and assays. | Dynabeads M-270 carboxylic acid; used as a core for hydrogel particle formation [9]. |
| Circle Sequencing Reagents | Library prep for ultra-low error rate sequencing. | Circularizing ligases and rolling circle polymerases [48]. |
| Computational Error Correction Tools | Post-sequencing analysis to reduce platform-specific errors. | isONcorrect: For correcting ONT transcriptome data [47]. PyroBayes: Base caller for 454/pyrosequencing data [46]. |
Data Analysis and Fidelity Calculation
After sequencing, the data must be processed to calculate the polymerase's error rate.
1. Primary Sequence Analysis:
- Quality Control: Use tools like FastQC to assess the overall quality of the raw sequencing data, including per-base sequence quality, adapter content, and sequence duplication levels [49] [50].
- Read Trimming/Filtration: Employ programs like Trimmomatics to remove low-quality bases and adapter sequences [50].
- Alignment: Map the processed reads to the reference template sequence using an appropriate aligner (e.g., minimap2 for long reads) [47].
2. Error Rate Calculation:
- Identify mismatches, insertions, and deletions (INDELs) in the aligned reads.
- The fidelity (error rate, ( R )) is calculated using the formula: ( R = E / (L \times N) ) where ( E ) is the total number of errors identified, ( L ) is the total number of bases sequenced and successfully aligned, and ( N ) is the number of molecules sequenced. This calculation should be performed separately for different error types (substitutions, INDELs) [46].
3. Advanced Visualization:
- For a comprehensive graphical representation of sequencing data, tools like SeqCode can be used to generate occupancy plots, density heatmaps, and other visualizations that can help illustrate the distribution and potential context of errors [51].
Diagram: Data Analysis Workflow for Fidelity Calculation
The accurate assessment of XNA polymerase fidelity is a cornerstone of their development for synthetic biology. The protocols outlined here, from the streamlined hydrogel particle method to the ultra-accurate circle sequencing approach, provide researchers with a versatile toolkit. By leveraging high-throughput sequencing and understanding the intrinsic error profiles of different platforms, scientists can obtain rigorous, quantitative fidelity metrics. This enables the iterative design and evolution of ever-more accurate XNA polymerases, which are vital for realizing the full potential of synthetic genetics in therapeutics and biotechnology.
The field of synthetic biology aims to extend the principles of genetics to artificial genetic polymers, known as xeno-nucleic acids (XNAs), to create biological systems with novel functionalities [2]. XNAs are synthetic genetic polymers in which the natural deoxyribose sugar found in DNA has been replaced with an alternative sugar or sugar-like moiety, conferring properties such as enhanced nuclease resistance and novel physicochemical characteristics [52] [2]. A critical bottleneck in working with XNAs has been the inability of natural polymerases to efficiently synthesize and reverse-transcribe these synthetic genetic polymers [16]. Through directed evolution, researchers have engineered specialized XNA polymerases capable of processing these unnatural substrates [16] [6].
Understanding the molecular basis of how these engineered enzymes recognize and catalyze XNA synthesis is essential for advancing synthetic biology applications. X-ray crystallography of polymerase-XNA post-catalytic complexes provides unprecedented structural insights into the mechanisms enabling this functionality [52] [53]. This Application Note details the experimental methodologies and key findings from crystallographic studies of engineered polymerases in complex with XNA substrates, providing a framework for researchers pursuing the structural characterization of nucleic acid-processing enzymes.
Experimental Protocols
Protein Expression and Purification
Objective: To obtain high-purity, crystallography-grade engineered polymerase proteins.
Detailed Protocol:
- Gene Cloning: Clone the gene for the laboratory-evolved polymerase (e.g., Kod-RI or Kod-RSGA) into the pET21a expression vector using NdeI and NotI restriction sites [52].
- Transformation: Transfect the constructed plasmid into E. coli expression cells such as Acella cells.
- Protein Expression:
- Grow cells aerobically at 37°C in LB media supplemented with 100 μg ml⁻¹ ampicillin to an OD₆₀₀ of 0.8.
- Induce protein expression with 1 mM isopropyl β-d-thiogalactoside (IPTG).
- Incubate cultures at 18°C for 20 hours to promote proper protein folding.
- Cell Harvesting: Harvest cells via centrifugation (e.g., 3,315 × g for 20 minutes at 4°C).
- Cell Lysis and Clarification:
- Resuspend the cell pellet in an appropriate lysis buffer.
- Lyse cells by sonication on ice.
- Clarify the lysate by centrifugation (e.g., 23,708 × g for 30 minutes at 4°C).
- Heat Treatment: Perform a heat treatment step (70°C for 20 minutes) to denature and precipitate heat-labile endogenous E. coli proteins. Centrifuge again to remove precipitates.
- Column Chromatography:
- Load the cleared supernatant onto a tandem column system, typically involving a 5-ml HiTrap Q HP column followed by a 5-ml heparin HP column.
- Wash with a low-salt buffer to remove weakly bound contaminants.
- Elute the target polymerase with a linear gradient of a high-salt buffer.
- Size Exclusion Chromatography (SEC): Further purify pooled fractions using SEC (e.g., Superdex 200 HiLoad 16/600 column) pre-equilibrated with a compatible storage buffer (e.g., Kod buffer).
- Concentration and Storage: Concentrate the purified protein to approximately 10 mg ml⁻¹ using a 30-kDa cutoff Amicon centrifugal filter. Flash-freeze in aliquots using liquid nitrogen and store at -80°C.
Primer Extension Assay for Functional Validation
Objective: To biochemically validate the activity of the purified polymerase with XNA nucleotide substrates.
Detailed Protocol:
- Reaction Setup: Prepare a 20 μl reaction mixture containing:
- 1× ThermoPol reaction buffer.
- 0.5 μM IRDye-labeled DNA primer.
- 0.5 μM DNA template.
- 100 μM of the appropriate XNA nucleoside triphosphate (e.g., pTNA, TNA, or 2'-azido-modified NTPs).
- Annealing: Anneal the primer and template by incubating at 95°C for 5 minutes, followed by gradual cooling on ice for 5 minutes.
- Enzyme Activation: Pre-incubate 0.5 μM of the engineered polymerase with 1 mM MnCl₂. MgCl₂ can be used for DNA synthesis controls, but MnCl₂ is often critical for efficient XNA incorporation [52] [1].
- Extension Reaction: Initiate the reaction by adding the activated polymerase to the primer-template-nucleotide mixture. Incubate at 55°C for a specified time (e.g., 2 hours) [1].
- Reaction Termination: Quench the reaction by adding an equal volume of stop buffer (e.g., containing 95% formamide and EDTA).
- Product Analysis:
- Denature samples at 95°C for 10 minutes.
- Resolve the products by 15% denaturing polyacrylamide gel electrophoresis (PAGE).
- Visualize the extended products using a fluorescence imaging system such as a LI-COR Odyssey CLx imager.
Crystallization of Post-Catalytic Complexes
Objective: To grow high-quality crystals of the engineered polymerase bound to a primer-template duplex extended with XNA nucleotides.
Detailed Protocol:
- Complex Formation:
- Annealing: Anneal the crystallization primer (Pc) and Cy5-labeled template (Tc) in crystallization buffer supplemented with 20 mM MgCl₂ by heating to 95°C for 5 minutes and cooling slowly [52].
- Binary Complex Formation: Incubate 1.5 molar equivalents of the annealed duplex with 5 mg ml⁻¹ of the purified polymerase at 37°C for 30 minutes.
- Post-Catalytic Complex Formation: Add 5 molar equivalents of the desired nucleotide (e.g., dATP for DNA control or ptADP for pTNA complex) to the binary complex and incubate further at 37°C for 30 minutes to form a stable post-catalytic complex [52].
- Crystallization Screening:
- Use a pipetting robot (e.g., Mosquito) to set up 0.5 μl hanging drops in 96-well format by mixing equal volumes of the protein complex and crystallization solution.
- Screen commercial crystallization kits (e.g., from Hampton Research and Qiagen) at various temperatures.
- Optimization: Optimize initial crystal hits manually in 24-well trays using 2 μl drops by varying pH and precipitant concentration ratios.
- Soaking (If Required): For certain complexes, obtaining clear electron density for the nucleotide may require a soaking step. For example, the closed ternary structure of Kod-RI with TNA required a 45-minute soak with 2 mM tATP and 20 mM MgCl₂ just prior to freezing [53].
- Harvesting: Flash-cool crystals in liquid nitrogen using a cryoprotectant solution matching the mother liquor.
Table 1: Key Reagents for Crystallization Complex Formation
| Reagent Type | Example | Function in Experiment | Source |
|---|---|---|---|
| Engineered Polymerase | Kod-RI, Kod-RSGA, TgoT-EPFLH | Catalyzes XNA incorporation; subject of structural study | Heterologous expression in E. coli [52] [53] |
| DNA Template | Tc (e.g., 5'-Cy5 labeled) | Provides sequence context for synthesis and phasing for crystallography | Commercial synthesis (IDT) [52] |
| DNA Primer | Pc (e.g., 5'-hexynyl) | Provides a starting point for 3' extension by the polymerase | Commercial synthesis (IDT) [52] |
| XNA Nucleotides | tATP, ptADP, 2'-Az NTPs | Substrates for polymerization; define the XNA chemistry studied | Chemical synthesis [52] [1] [53] |
| Divalent Metal Ions | MnCl₂, MgCl₂ | Essential cofactor for polymerase catalytic activity | Various suppliers [52] [1] |
Data Collection and Structure Determination
Objective: To collect X-ray diffraction data and solve the three-dimensional structure of the complex.
Detailed Protocol:
- Data Collection: Collect X-ray diffraction datasets at a synchrotron beamline. Maintain crystals at 100 K during data collection.
- Data Processing: Process the diffraction data using software packages like XDS or HKL-2000 to obtain a merged and scaled dataset of structure factor amplitudes.
- Molecular Replacement: Solve the phase problem by molecular replacement using a related apo or binary polymerase structure (e.g., PDB ID: 1WNS for Kod polymerase) as a search model [53].
- Model Building and Refinement: Iteratively build the atomic model into the electron density map using Coot and refine the model using phenix.refine or Refmac. The model includes the polymerase, the primer-template duplex, the incorporated XNA nucleotides, metal ions, and solvent molecules.
The following workflow diagram summarizes the key experimental stages from protein preparation to structure analysis:
Key Structural Findings and Data Analysis
Crystallographic studies of engineered polymerases like Kod-RI and Kod-RSGA in complex with XNA substrates have revealed critical details about molecular recognition and catalysis.
Active Site Architecture and Substrate Positioning
Structures of post-catalytic complexes show that the laboratory-evolved polymerases mediate Watson-Crick base pairing between the incoming XNA nucleotide and the DNA template strand, which is essential for faithful information transfer [52]. However, a key finding is that XNA substrates often bind in a sub-optimal geometry compared to natural nucleotides.
For instance, in the closed ternary structure of Kod-RI with TNA, the TNA triphosphate in the active site displays a sub-optimal binding geometry, which explains the significantly slower rate of TNA synthesis (~1 nucleotide per minute) compared to natural DNA synthesis [53]. Comparative analysis of pTNA complexes revealed that the sugar conformation and backbone positioning of pTNA are structurally more similar to TNA than to DNA, despite pTNA sharing the same six-atom backbone repeat length as DNA [52].
Comparative Analysis of XNA Polymerase Performance
The table below summarizes functional data for several engineered XNA polymerases, highlighting differences in their activity and fidelity with various XNA substrates.
Table 2: Performance Metrics of Laboratory-Evolved XNA Polymerases
| Polymerase | Parent Polymerase | XNA Substrate | Key Mutations | Efficiency | Fidelity (Error Rate) | Key Application |
|---|---|---|---|---|---|---|
| Kod-RI [53] | T. kodakarensis (Kod) | TNA | A485R, E664I | ~1 nt/min | ~4 errors / 1000 nt | TNA synthesis & evolution |
| Kod-RSGA [52] | Kod-RI | TNA, pTNA | Further evolution of Kod-RI | Improved over Kod-RI | Data not specified | Synthesis of orthogonal polymers (pTNA) |
| TgoT-EPFLH [52] | T. gorgonarius (Tgo) | pTNA | Not specified in results | Synthesizes up to 57-nt pTNA | Data not specified | Synthesis of orthogonal polymers (pTNA) |
| Engineered SF Mutants [1] | T. aquaticus (Stoffel Fragment) | 2'-Az / 2'-F Mixed Polymers | Not specified in results | High synthesis accuracy | Improved fidelity over previous systems | Synthesis of click-compatible XNAs |
Structural Determinants of XNA Recognition
The following diagram illustrates the structural pathway of nucleotide incorporation by an engineered XNA polymerase, based on crystallographic studies of Kod-RI:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Polymerase-XNA Crystallography Studies
| Reagent / Material | Specifications & Functional Role | Commercial Sources / Production Method |
|---|---|---|
| Engineered Polymerases | B-family polymerases (e.g., from Thermococcus spp.) engineered via directed evolution for XNA recognition. | Custom engineering and heterologous expression in E. coli [52] [53] |
| XNA Nucleoside Triphosphates | Chemically synthesized triphosphates of XNAs (e.g., TNA, pTNA, FANA, 2'-Az). Serve as polymerase substrates. | Custom chemical synthesis [52] [1] [53]; some available from TriLink Biotechnologies, Metkinen [1] |
| Modified Primers/Templates | DNA oligonucleotides with specific modifications (e.g., 5'-Hexynyl, 5'-Cy5). Enable complex formation, phasing, and tracking. | Integrated DNA Technologies (IDT) [52] [1] |
| Crystallization Kits | Sparse-matrix screens for initial crystal condition identification. | Hampton Research, Qiagen [52] |
Application in Synthetic Biology and Therapeutic Development
The structural insights gained from polymerase-XNA complexes are directly enabling next-generation applications in synthetic biology and molecular medicine. Understanding the molecular recognition of XNAs by engineered polymerases is a critical step towards establishing orthogonal genetic systems. These systems, where XNA polymers do not cross-pair with natural DNA or RNA, create a genetic firewall for biocontainment [52]. This is a key safety strategy for creating genetically modified organisms that cannot exchange synthetic genetic information with natural ecosystems.
Furthermore, these structural studies facilitate the in vitro evolution of functional XNA molecules, such as aptamers (target-binding molecules) and catalysts (XNAzymes), for therapeutic and diagnostic applications [2] [15]. XNAs are particularly attractive for these roles due to their high biological stability and nuclease resistance, which can overcome the limitations of natural nucleic acid-based therapeutics [54] [2]. The continuous improvement of XNA polymerases, guided by structural data, is therefore pivotal for advancing the field of synthetic genetics and realizing the full potential of XNA technology in biomedicine.
The directed evolution of xeno-nucleic acid (XNA) polymerases is a cornerstone of synthetic biology, enabling the replication and manipulation of synthetic genetic polymers. A critical step in this process involves the enzymatic synthesis of XNA episomes, where laboratory-evolved polymerases copy DNA information into XNA. Phosphonomethylthreosyl nucleic acid (pTNA), a structurally unique XNA, is of particular interest due to its potential as an orthogonal genetic material for storing synthetic biology information in cells. Its backbone consists of α-l-threofuranosyl sugars connected by 3′→2′ phosphonomethyl linkages, making it structurally distinct from both threose nucleic acid (TNA) and DNA [52] [55]. This application note provides a comparative analysis of the substrate recognition mechanisms of pTNA, TNA, and DNA by engineered polymerases, detailing protocols for evaluating polymerase activity and providing essential tools for researchers in the field.
Structural Comparison of Genetic Polymers
The enzymatic synthesis of XNAs relies on engineered polymerases that can accommodate sugar-modified substrates, often with some sacrifice to protein-folding stability [6]. The distinct chemical structures of the sugar-phosphate backbones in DNA, TNA, and pTNA are the primary determinants of their recognition by these polymerases.
Table 1: Structural Characteristics of DNA, TNA, and pTNA
| Feature | DNA | TNA | pTNA |
|---|---|---|---|
| Sugar Moisty | Deoxyribose (5-carbon) | Threose (4-carbon) | α-l-threofuranosyl (4-carbon) |
| Backbone Linkage | 3′→5′ phosphodiester | 3′→2′ phosphodiester | 3′→2′ phosphonomethyl |
| Helical Sense | Right-handed | Not fully characterized | Not fully characterized |
| Cross-pairing with DNA/RNA | Yes | Yes | Orthogonal (No) [52] |
| Nuclease Resistance | Low | High | High [55] |
The following diagram illustrates the structural relationships and polymerase recognition pathways for these three genetic polymers:
Polymerase Recognition Pathways. The diagram illustrates the relationship between natural DNA and the synthetic genetic polymers TNA and pTNA, and their recognition pathways by natural and engineered polymerases.
Key Structural Insights from Crystallography
High-resolution crystal structures of post-catalytic complexes reveal that engineered polymerases mediate Watson-Crick base pairing between extended pTNA adducts and a DNA template, despite pTNA's backbone being structurally more similar to TNA than to DNA [52]. This is a key finding, as it demonstrates that stable Watson-Crick pairing between the primer and template is not an absolute requirement for the enzymatic synthesis of orthogonal XNAs. Comparative analysis shows that the sugar conformation and backbone position of pTNA are more similar to TNA than to DNA, even though pTNA and DNA share the same six-atom backbone repeat length [52].
Quantitative Analysis of Polymerase Performance
Evaluating the efficiency and fidelity of engineered polymerases is crucial for their application in synthetic biology. The following protocol and data analysis framework allow for a direct comparison of polymerase activity across different substrates.
Protocol 1: Primer Extension Assay for Polymerase Activity
Purpose: To assess the ability of an engineered polymerase to synthesize a strand of pTNA, TNA, or DNA from a DNA template.
Reagents:
- Engineered Polymerase (e.g., TgoT-EPFLH for pTNA, Kod-RI or Kod-RSGA for TNA) [52] [55]
- DNA Template (e.g., 5′-Cy5-labeled for detection)
- IR-labeled DNA Primer
- Nucleotides (dNTPs, tNTPs, or ptNTPs, depending on the assay)
- 10x ThermoPol Reaction Buffer (e.g., from New England Biolabs)
- MnCl₂ (often required for XNA synthesis)
- Stop Buffer (e.g., containing formamide and EDTA)
Procedure:
- Annealing: Combine 0.5 µM of the IR-labeled primer with 0.5 µM of the DNA template in 1x ThermoPol reaction buffer. Heat the mixture to 95°C for 5 minutes and then cool on ice for 5 minutes.
- Reaction Setup: To the annealed duplex, add 100 µM of the appropriate nucleotides (dATP, ptADP, etc.).
- Initiation: Add 0.5 µM of the engineered polymerase that has been pre-incubated with 1 mM MnCl₂.
- Incubation: Incubate the reaction at 55°C for a specified time (e.g., 30-60 minutes).
- Termination: Quench the reaction by adding an equal volume of stop buffer and denature at 95°C for 10 minutes.
- Analysis: Resolve the products by 15% denaturing polyacrylamide gel electrophoresis (PAGE). Image the gel using a fluorescence scanner (e.g., LI-COR Odyssey CLx). [52]
The data obtained from primer extension assays can be quantified to compare key performance metrics across different genetic polymers and polymerase variants.
Table 2: Comparative Polymerase Performance Metrics
| Polymerase | Substrate | Maximum Synthesis Length (nt) | Relative Processivity | Cofactor Requirement |
|---|---|---|---|---|
| TgoT-EPFLH | pTNA | ~57 [52] | Low | Mn²⁺ [52] |
| Kod-RI | TNA | Variable | Medium | Mn²⁺ |
| Kod-RSGA | TNA | Variable | High | Mn²⁺ |
| Bst DNA Pol | DNA | >1000 | High | Mg²⁺ |
Structural Analysis of Polymerase-Substrate Complexes
Understanding the molecular details of how engineered polymerases recognize non-canonical substrates is fundamental to directing their further evolution. X-ray crystallography is the primary method for obtaining this high-resolution structural information.
Protocol 2: Crystallizing Post-Catalytic Polymerase Complexes
Purpose: To obtain high-resolution crystal structures of engineered polymerases bound to DNA primer-template duplexes extended with pTNA or DNA nucleotides.
Reagents:
- Purified Engineered Polymerase (e.g., Kod-RI, Kod-RSGA) at ≥10 mg/mL
- DNA Primer-Template Duplex (e.g., Primer Pc and Cy5-labeled Template Tc)
- Nucleotides (dATP or ptADP)
- Crystallization Buffers (e.g., from Hampton Research or Qiagen screens)
- Kod Buffer (e.g., 20 mM Tris-HCl pH 7.5, 50 mM NaCl, 0.1% Triton X-100, 1 mM DTT) supplemented with 20 mM MgCl₂
Procedure:
- Duplex Annealing: Anneal equal amounts of the DNA primer and the Cy5-labeled template in Kod buffer with 20 mM MgCl₂ by heating to 95°C for 5 minutes and then slowly cooling to 10°C.
- Binary Complex Formation: Incubate 1.5 molar equivalents of the annealed duplex with 5 mg/mL of the polymerase at 37°C for 30 minutes.
- Post-Catalytic Complex Formation: Add 5 molar equivalents of the desired nucleotide (dATP or ptADP) to the binary complex and incubate at 37°C for another 30 minutes.
- Crystallization Setup: Screen for crystallization conditions using a pipetting robot to set up 0.5 µL hanging drops with equal volumes of the protein complex and crystallization solution.
- Optimization: Optimize initial crystal hits over pH and precipitant concentration ranges in 24-well trays.
- Data Collection and Analysis: Flash-freeze crystals in liquid nitrogen and collect X-ray diffraction data at a synchrotron facility. Solve the structure by molecular replacement. [52]
The workflow for preparing and analyzing these complexes is methodically outlined below:
Structural Analysis Workflow. The diagram shows the key steps involved in preparing and solving the crystal structure of a post-catalytic polymerase complex bound to a synthetic genetic polymer.
The Scientist's Toolkit: Research Reagent Solutions
Successful experimentation in XNA polymerase engineering requires a specific set of reagents and tools. The following table catalogues essential materials for research in this field.
Table 3: Essential Research Reagents for XNA Polymerase Studies
| Reagent / Material | Function / Application | Examples / Specifications |
|---|---|---|
| Engineered XNA Polymerases | Enzymatic synthesis and reverse transcription of XNAs. | TgoT-EPFLH (pTNA synthesis), Kod-RSGA (TNA synthesis) [52] |
| Chemically Synthesized XNAs | Substrates for polymerase assays; templates for reverse transcription. | pTNA nucleotides (ptNTPs), TNA nucleotides (tNTPs) [52] |
| Fluorescently-Labeled Oligos | For detection and quantification in primer extension assays. | IRDye-labeled primers, 5'-Cy5-labeled templates [52] |
| Divalent Metal Cofactors | Essential cofactors for polymerase activity; Mn²⁺ often crucial for XNA synthesis. | MnCl₂, MgCl₂ [52] |
| Expression Plasmid System | Production of engineered polymerases in a host system. | pET21a vector in E. coli expression systems [52] |
This application note provides a detailed comparative framework for analyzing the substrate recognition of pTNA, TNA, and DNA by engineered polymerases. The structural and quantitative data presented highlight the unique challenge of synthesizing orthogonal genetic polymers like pTNA, which does not cross-pair with natural nucleic acids but can still be synthesized by evolved enzymes utilizing Watson-Crick base pairing [52]. The provided protocols for primer extension assays and protein crystallography, alongside the essential reagent toolkit, offer researchers a solid foundation for driving the next phase of polymerase engineering. Future directions will focus on evolving polymerases with enhanced fidelity and processivity for pTNA synthesis, ultimately advancing the use of XNAs as robust informational and functional biopolymers in synthetic biology.
In the expanding field of synthetic biology, the directed evolution of xenobiotic nucleic acid (XNA) polymerases represents a frontier for creating novel biocatalysts that manipulate genetic information beyond the constraints of natural DNA [56]. The performance of these enzymes is primarily benchmarked against two critical parameters: fidelity, which refers to the accuracy of nucleotide incorporation, and activity, which encompasses catalytic efficiency, processivity, and robustness under various conditions [57] [58]. Understanding the balance between these parameters is essential for applications ranging from precise diagnostic assays to the synthesis of artificial genetic systems [56]. This application note provides a structured framework for benchmarking both evolved and natural polymerases, featuring standardized protocols and quantitative comparisons to guide researchers in selecting and engineering enzymes for advanced synthetic biology applications.
Key Performance Metrics: Fidelity and Activity
DNA Polymerase Fidelity
DNA polymerase fidelity is a measure of the enzyme's accuracy during DNA synthesis. It is maintained by two primary catalytic activities: a DNA-dependent DNA polymerase activity for correct nucleotide incorporation and a proofreading 3'→5' exonuclease activity that removes misincorporated nucleotides [57]. Fidelity is quantified as an error rate, representing the number of errors made per base synthesized. Natural high-fidelity replicative DNA polymerases achieve remarkably low error rates, often in the range of 10⁻⁶ to 10⁻⁸ errors per base pair, which is crucial for maintaining genome stability [57].
Modern methods for assessing fidelity have moved beyond low-throughput techniques like the plasmid-based LacZα assay. The development of high-throughput sequencing workflows, particularly those utilizing Pacific Biosciences' single-molecule real-time (SMRT) sequencing, allows for the direct measurement of both error rates and error profiles (the specific types of mutations a polymerase tends to make) without the need for PCR amplification, which can itself introduce artifacts [57].
DNA Polymerase Activity
Activity encompasses several functional attributes of a DNA polymerase:
- Catalytic Efficiency: The rate of nucleotide incorporation and the ability to synthesize full-length products [58].
- Processivity: The average number of nucleotides incorporated per enzyme-binding event. High processivity is vital for amplifying long and complex DNA templates [58].
- Thermostability: The ability to retain structure and function at high temperatures (e.g., 95°C), which is a prerequisite for PCR [59].
- Substrate Tolerance: The enzyme's ability to utilize modified nucleotides, such as dUTP, or to synthesize XNA [56] [58]. A key engineering goal is to create polymerases that can efficiently use XNA substrates while maintaining high fidelity and processivity [56].
The Fidelity-Activity Trade-off
Protein engineering often involves balancing fidelity with other desirable traits. For instance, the fusion of a non-specific DNA binding domain (e.g., Sso7d) to a polymerase can significantly enhance its processivity and efficiency. However, this modification can sometimes come at the cost of reduced fidelity. The engineered Neq2X7 polymerase, a fusion of a Nanoarchaeum equitans polymerase with an Sso7d domain, demonstrates this trade-off; while it exhibits high activity and can amplify long, GC-rich targets with short extension times, its error rate was measured to be approximately 100-fold higher than its parental variant [58].
Experimental Protocols for Benchmarking
Protocol 1: Measuring Fidelity via High-Throughput Sequencing
This protocol uses PacBio Circular Consensus Sequencing (CCS) to achieve high-accuracy fidelity measurements without amplification bias [57].
- Principle: A primer extension assay is performed by the polymerase of interest on a defined template. The synthesized products are then sequenced using long-read, PCR-free SMRT sequencing. Repeated sequencing of the same molecule (CCS) yields highly accurate consensus sequences, allowing for the identification of synthesis errors made by the polymerase versus sequencing errors [57].
- Workflow:
- Primer Extension: Incubate the target polymerase with a single-stranded DNA template and dNTPs under desired buffer conditions.
- Library Preparation: Prepare the extended dsDNA product for PacBio sequencing according to the manufacturer's instructions.
- SMRT Sequencing: Sequence the library on a PacBio platform to generate long CCS reads.
- Bioinformatic Analysis: Align the consensus reads to the reference template sequence. Identify and categorize mismatches, insertions, and deletions to calculate the overall error rate and specific error profile.
- Applications: Ideal for comparing error rates and spectra across different polymerase families (A, B, C, D) or for characterizing the effect of active-site mutations on fidelity [57].
Protocol 2: Assessing Activity and Processivity
This protocol benchmarks polymerase performance through quantitative PCR and long-range amplification assays.
- Principle: The polymerase's activity is measured by its efficiency in amplifying targets of varying lengths and complexity under different reaction conditions.
- Workflow:
- Standard Activity Assay: Use a fluorescence-based DNA polymerase assay that measures the incorporation of fluorescently labeled nucleotides into a primed single-stranded DNA template (e.g., M13mp18). Activity is calculated in units/mg of enzyme [58].
- Long-Range and GC-Rich PCR: Perform PCR amplification on a series of DNA templates (e.g., 3 kb, 6 kb, 12 kb). Test the polymerases with standard (1 min/kb) and shortened (15 s/kb) extension times to assess processivity [58].
- Analysis: Analyze PCR products by agarose gel electrophoresis. A polymerase with high processivity and activity will produce strong, specific bands even for long amplicons and with short extension times.
- Applications: Directly compare the performance of engineered and natural polymerases; identify variants suitable for long-range PCR or rapid amplification.
Protocol 3: Evaluating Reverse Transcriptase Activity in Engineered DNA Polymerases
This protocol tests the ability of engineered thermostable DNA polymerases to perform coupled reverse transcription-PCR (RT-PCR), a key application for diagnostic enzymes [59] [60].
- Principle: Engineered Taq polymerase variants (e.g., RevTaq, OmniTaq2) have been developed to possess inherent reverse transcriptase activity, enabling single-enzyme RT-PCR.
- Workflow:
- Reaction Setup: Prepare a single-tube reaction containing the engineered DNA polymerase, target RNA (e.g., SARS-CoV-2 RNA or endogenous mRNA), dNTPs, and gene-specific primers.
- RT-PCR Amplification: Run a thermocycling program that combines a cDNA synthesis step (e.g., 45–60°C for 10–30 min) with standard PCR cycling.
- Detection: Use end-point detection (gel electrophoresis) or real-time detection (SYBR Green or TaqMan probes) to quantify the output.
- Comparison: Benchmark the engineered polymerase against a conventional two-enzyme system (e.g., M-MLV reverse transcriptase mixed with Taq polymerase).
- Applications: Validate the performance of novel single-enzyme RT-PCR systems for molecular diagnostics and multiplex RNA detection [59] [60].
The following diagram illustrates the logical relationship and workflow for the key benchmarking protocols.
Quantitative Data Comparison
Table 1: Comparison of Natural and Evolved DNA Polymerase Performance Characteristics
| Polymerase | Type / Origin | Key Features / Mutations | Reported Error Rate (bp⁻¹) | Key Activity & Processivity Findings |
|---|---|---|---|---|
| Replicative DNAPs (A,B,C,D Families) [57] | Natural | Polymerase + Proofreading (DnaQ-like or PDE) exonuclease activity. | ~10⁻⁶ – 10⁻⁸ (estimated) | Diverse, family-specific error profiles. Fidelity is influenced by dNTP ratios and replisome components. |
| Neq2X [58] | Natural (Archaeal) N. equitans with two fidelity mutations (A523R/N540R). | High natural processivity, lacks uracil-stalling domain. | ~2×10⁻⁶ (comparable to Pfu) | Baseline for comparison. Lower processivity than fused variant. |
| Neq2X7 [58] | Evolved (Engineered Fusion) | Neq2X with C-terminal Sso7d DNA-binding domain. | < 2×10⁻⁵ (≈100x lower fidelity than Neq2X) | 8x higher activity than Neq2X. Amplifies 12 kb fragment with 15 s/kb extension; works with dUTP. |
| RevTaq [59] [60] | Evolved (Engineered Taq) | Four mutations: L459M, S515R, I638F, M747K. | Not explicitly quantified | Enables single-enzyme RT-PCR. Compatible with real-time and probe-based detection. |
| OmniTaq2 [59] | Evolved (Engineered Taq) | Single mutation: D732N. Confers strand displacement and RT activity. | Not explicitly quantified | Enables single-enzyme RT-PCR. Strand displacement aids in resolving RNA secondary structure. |
Table 2: Performance of Engineered Polymerases in Specific Functional Assays
| Functional Assay | Polymerase | Performance Outcome | Experimental Details |
|---|---|---|---|
| Long-Range PCR (12 kb) [58] | Neq2X7 | Success with 15 s/kb extension | Demonstrated high processivity and catalytic speed. |
| Neq2X, PfuX7 | Failure with 15 s/kb extension | Required standard 1 min/kb extension time. | |
| dUTP-tolerance in PCR [58] | Neq2X7 | Success for long amplicons with dUTP | Essential for USER cloning and contamination-free diagnostics. |
| PfuX7 | Limited success | Requires engineered uracil-binding pocket mutation. | |
| Single-Enzyme RT-PCR [59] [60] | RevTaq, OmniTaq2, ReverHotTaq | Success in SARS-CoV-2 detection | Effective in endpoint and real-time RT-PCR; simplified workflow vs. two-enzyme systems. |
| Conventional M-MLV/Taq Mix | Success (benchmark) | Standard method, requires two enzymes and potential buffer optimization. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for Polymerase Benchmarking Experiments
| Reagent / Kit | Function / Description | Example Use in Protocols |
|---|---|---|
| PacBio SMRT Sequencing | Long-read, PCR-free sequencing platform for high-accuracy fidelity analysis. | Protocol 1: Determining polymerase error rates and error profiles [57]. |
| Single-Enzyme RT-PCR Polymerases (e.g., RevTaq, OmniTaq2) | Engineered thermostable DNA polymerases with reverse transcriptase activity. | Protocol 3: Enabling one-tube, one-enzyme reverse transcription and PCR amplification [59] [60]. |
| Fluorescence-Based Polymerase Assay Kit | Measures real-time nucleotide incorporation using a fluorescent DNA intercalating dye. | Protocol 2: Quantifying polymerase catalytic activity in units/mg [58]. |
| UNG (Uracil-N-glycosylase) | Enzyme that degrades uracil-containing DNA to prevent PCR carryover contamination. | Used with dUTP-tolerant polymerases (e.g., Neq2X7) for contamination control in diagnostics [58]. |
| GC-Rich & Long-Range DNA Templates | Challenging templates to assess polymerase processivity and robustness. | Protocol 2: Benchmarking polymerase performance under demanding conditions [58]. |
The systematic benchmarking of activity and fidelity is a cornerstone in the directed evolution of DNA polymerases for synthetic biology. The data clearly demonstrate that while protein engineering can successfully confer new activities—such as reverse transcriptase function or enhanced processivity—these gains can involve trade-offs, most notably a potential reduction in fidelity.
The future of polymerase engineering lies in developing sophisticated high-throughput screening methods that can simultaneously select for high fidelity, desired activity (e.g., XNA synthesis), and robustness. The integration of advanced sequencing techniques [57] and structure-guided design [56] will be crucial. As the field progresses, the ability to rationally design or evolve "multi-purpose" enzymes that do not sacrifice accuracy for function will be paramount for realizing the full potential of synthetic biology, from the creation of semi-synthetic organisms to the development of ultrasensitive molecular diagnostics.
Conclusion
Directed evolution has proven to be a powerful strategy for engineering DNA polymerases that can work with XNA substrates, pushing the boundaries of synthetic biology. The methodologies outlined enable the creation of polymerases that efficiently synthesize, reverse transcribe, and amplify diverse XNAs, including complex mixed polymers with click-chemistry handles like 2′-azide. The rigorous optimization of selection conditions and validation through structural and sequencing analysis are paramount for generating high-fidelity, functional enzymes. Looking forward, these engineered XNA polymerases are poised to revolutionize biomedical research by enabling the development of nuclease-resistant therapeutic aptamers, novel catalytic XNAzymes, and orthogonal genetic systems for secure cellular information storage, thereby creating a robust genetic firewall for next-generation biotechnologies.
References
- 1. Enzymatic Synthesis of Mixed XNA Polymers Containing 2 [https://pmc.ncbi.nlm.nih.gov/articles/PMC12593958/]
- 2. Modified nucleic acids: replication, evolution, and next ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-020-00803-6]
- 3. Directed evolution of artificial enzymes (XNAzymes) from ... [https://pubmed.ncbi.nlm.nih.gov/26401917/]
- 4. Engineering and enhancement of Bst DNA polymerase ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813025092487]
- 5. Polyamines promote xenobiotic nucleic acid synthesis by ... [https://www.sciencedirect.com/org/science/article/pii/S263306792400036X]
- 6. Functional Comparison of Laboratory-Evolved XNA ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/34029459/]
- 7. Xenobiotic Nucleic Acid (XNA) Synthesis by Phi29 DNA ... [https://pubmed.ncbi.nlm.nih.gov/29927114/]
- 8. A microbiological system for screening the interference of XNA ... [https://pubs.rsc.org/en/content/articlehtml/2023/ra/d3ra06172h]
- 9. Measuring XNA polymerase fidelity in a hydrogel particle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11775589/]
- 10. Oligonucleotides and Nucleic... | Thermo Fisher Scientific - US Labeling [https://www.thermofisher.com/us/en/home/references/molecular-probes-the-handbook/nucleic-acid-detection-and-genomics-technology/labeling-oligonucleotides-and-nucleic-acids.html]
- 11. Natural, modified and conjugated carbohydrates in nucleic ... [https://pubs.rsc.org/en/content/articlelanding/2025/cs/d4cs00799a]
- 12. Xeno nucleic acid - Wikipedia [https://en.wikipedia.org/wiki/Xeno_nucleic_acid]
- 13. Directed evolution of a highly efficient TNA polymerase achieved by... [https://www.nature.com/articles/s41929-024-01233-1?error=cookies_not_supported]
- 14. Evaluating the Rate and Substrate of Laboratory Evolved... Specificity [https://pubmed.ncbi.nlm.nih.gov/29148714/]
- 15. XNA enzymes by evolution and design [https://www.sciencedirect.com/science/article/pii/S2666246921000124]
- 16. Engineering polymerases for applications in synthetic biology [https://pubmed.ncbi.nlm.nih.gov/32715992/]
- 17. new materials from synthetic genetic polymers - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4039137/]
- 18. Structural insights into a DNA polymerase reading the xeno ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11724289/]
- 19. Introduction to the themed collection on XNA xeno-nucleic acids [https://pubs.rsc.org/en/content/articlehtml/2022/cb/d2cb90036j]
- 20. Expanding the Horizon of the Xeno Nucleic Acid Space [https://astrobiology.com/2024/12/expanding-the-horizon-of-the-xeno-nucleic-acid-space.html]
- 21. Enzymes as green and sustainable tools for DNA data storage [https://pubs.rsc.org/fi/content/articlehtml/2025/cc/d4cc06351a]
- 22. Compartmentalized partnered replication for the directed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6053311/]
- 23. Engineering of biomolecules by bacteriophage directed ... [https://www.sciencedirect.com/science/article/pii/S0958166917301350]
- 24. Microfluidic Compartmentalized Directed Evolution - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2912841/]
- 25. sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology... [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/phage-display]
- 26. mRNA Display Selections in Protein–Protein Interactions - Page 3 [https://www.medscape.com/viewarticle/745131_3]
- 27. ESCape: an Phage - emulsion approach for the selection of... based [https://pubmed.ncbi.nlm.nih.gov/20932970/]
- 28. Discovery and evolution of RNA and XNA reverse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7116287/]
- 29. Mutant polymerases capable of 2′ fluoro-modified nucleic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9347352/]
- 30. Navigating directed evolution efficiently: optimizing ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2024.1439259/full]
- 31. Beneath the XNA world: Tools and targets to build novel ... [https://www.sciencedirect.com/science/article/abs/pii/S245231002030055X]
- 32. Nucleic acid joining enzymes: biological functions and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12133292/]
- 33. Navigating directed efficiently: optimizing selection... evolution [https://pmc.ncbi.nlm.nih.gov/articles/PMC11493728/]
- 34. GitHub - versioned-science/DNA_ polymerase _ directed _ evolution ... [https://github.com/versioned-science/DNA_polymerase_directed_evolution]
- 35. The effect of different divalent cations on the kinetics and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5992921/]
- 36. of a highly efficient TNA Directed ... | CoLab evolution polymerase [https://colab.ws/articles/10.1038%2Fs41929-024-01233-1]
- 37. Digital PCR: from early developments to its future application ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00055f]
- 38. Rapid and reliable species-level identification from clinical ... [https://www.nature.com/articles/s41598-025-14071-3]
- 39. Droplet diameter, energy efficiency and capacity [https://www.sciencedirect.com/science/article/pii/S0255270124002198]
- 40. 5′ exonuclease activity to the high fidelity of nucleotide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4267634/]
- 41. Efficiency of Correct Nucleotide Insertion Governs DNA ... [https://www.sciencedirect.com/science/article/pii/S0021925819714719]
- 42. Polymerization and editing modes of a high-fidelity DNA ... [https://www.nature.com/articles/s41467-020-19165-2]
- 43. Synthetic genetic polymers capable of heredity and evolution [https://pmc.ncbi.nlm.nih.gov/articles/PMC3362463/]
- 44. Quantitative methods to study helicase, DNA polymerase, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9933136/]
- 45. Overview of High Throughput Sequencing Technologies to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3831009/]
- 46. Empirical assessment of sequencing errors for high throughput pyrosequencing data - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3852801/]
- 47. Error correction enables use of Oxford Nanopore technology for reference-free transcriptome analysis | Nature Communications [https://www.nature.com/articles/s41467-020-20340-8]
- 48. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing - PubMed [https://pubmed.ncbi.nlm.nih.gov/24243955/]
- 49. Calculating the quality of public high-throughput ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5459929/]
- 50. Chapter 3 High throughput sequencing | Omics Data Analysis [https://uclouvain-cbio.github.io/WSBIM2122/sec-hts.html]
- 51. Productive visualization of high-throughput sequencing ... [https://www.nature.com/articles/s41598-021-98889-7]
- 52. Crystallographic analysis of engineered polymerases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9508818/]
- 53. Structural basis for TNA synthesis by an engineered ... [https://www.nature.com/articles/s41467-017-02014-0]
- 54. The XNA alphabet - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12255307/]
- 55. Threose nucleic acid as a primitive genetic polymer and ... [https://www.sciencedirect.com/science/article/abs/pii/S0045206823007101]
- 56. Nucleic acid joining enzymes: biological functions and ... [https://pubmed.ncbi.nlm.nih.gov/39840831/]
- 57. The A, B, C, D's of replicative DNA polymerase fidelity [https://pmc.ncbi.nlm.nih.gov/articles/PMC12634111/]
- 58. Neq2X7: a multi-purpose and open-source fusion DNA ... [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/s12896-024-00844-7]
- 59. Comparison of Commercially Available Thermostable DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11858481/]
- 60. Engineering of novel DNA polymerase variants for single ... [https://www.nature.com/articles/s41598-025-10211-x]