In Vivo vs. In Vitro Directed Evolution: A Comprehensive Guide for Biotech and Pharma Research
Directed evolution is a cornerstone of modern protein engineering, but the choice between in vivo and in vitro platforms profoundly impacts the success of R&D projects.
In Vivo vs. In Vitro Directed Evolution: A Comprehensive Guide for Biotech and Pharma Research
Abstract
Directed evolution is a cornerstone of modern protein engineering, but the choice between in vivo and in vitro platforms profoundly impacts the success of R&D projects. This article provides a definitive comparison for researchers and drug development professionals. We explore the foundational principles of both approaches, from the physiological relevance of living systems to the controlled precision of test-tube methods. The review details cutting-edge methodologies, including mutator strains, viral platforms, and DNA shuffling techniques, and offers practical troubleshooting strategies for common challenges like library diversity and host compatibility. By synthesizing validation data and comparative analyses, this guide empowers scientists to select the optimal platform for evolving enzymes, antibodies, and therapeutic proteins, ultimately accelerating the development of novel biologics and biocatalysts.
Core Principles: Defining the Environments of In Vivo and In Vitro Evolution
Directed evolution is a powerful protein engineering methodology that harnesses the principles of natural selection in a controlled laboratory setting to generate biomolecules with novel or enhanced functions. Unlike rational design, which requires extensive prior knowledge of protein structure-function relationships, directed evolution explores vast sequence landscapes through iterative cycles of mutagenesis and screening, often uncovering non-intuitive and highly effective solutions [1]. This approach compresses geological timescales of evolution into weeks or months by intentionally accelerating mutation rates and applying user-defined selection pressures [1]. The profound impact of this technology was recognized with the 2018 Nobel Prize in Chemistry awarded to Frances H. Arnold for establishing directed evolution as a cornerstone of modern biotechnology [1].
Within this field, in vivo directed evolution distinguishes itself by performing the entire evolutionary process within living cellular environments. This stands in contrast to in vitro methods that conduct diversification and screening outside biological systems, or hybrid approaches that combine in vitro mutagenesis with cellular screening [2]. The strategic advantage of in vivo evolution lies in its capacity to leverage the authentic cellular context—including appropriate post-translational modifications, native protein-folding machinery, relevant ionic conditions, and complex protein-interaction networks—all of which are difficult to replicate in artificial systems [2] [3]. This review provides a comprehensive comparison between in vivo and in vitro directed evolution platforms, examining their methodologies, applications, and performance characteristics to inform strategic decision-making in biomedical research and therapeutic development.
Fundamental Principles and Comparative Framework
Core Mechanism of Directed Evolution
The directed evolution workflow functions as a two-part iterative engine that drives a population of protein variants toward a desired functional goal. A typical campaign begins with a parent gene encoding a protein with basal-level activity. This gene undergoes diversification to create a library of variants, which are then subjected to screening or selection to identify individuals with improved performance [1]. The genes from these improved variants are isolated and serve as templates for subsequent rounds of mutagenesis and screening, allowing beneficial mutations to accumulate progressively [1]. The critical distinction from natural evolution is that the selection pressure is decoupled from organismal fitness, with the sole objective being optimization of a specific protein property defined by the experimenter [1].
Key Distinguishing Features of In Vivo Evolution
In vivo directed evolution platforms perform both diversification and selection within living cells, creating a closed system where evolution occurs in a biologically relevant context. These systems can be broadly categorized based on their host organisms:
- Prokaryotic Systems: Primarily utilizing Escherichia coli mutator strains with deficiencies in DNA repair pathways to increase spontaneous mutation frequencies [2].
- Yeast-Based Systems: Leveraging Saccharomyces cerevisiae for its high recombination efficiency, eukaryotic processing capabilities, and suitability for surface display technologies [4].
- Mammalian Systems: Utilizing advanced platforms like PROTEUS (PROTein Evolution Using Selection) that employ chimeric virus-like vesicles to enable extended evolution campaigns in mammalian cells without loss of system integrity [3].
The defining characteristic of in vivo evolution is that the target protein is evolved within the same type of cellular environment where it will ultimately function, ensuring that selected variants are pre-adapted to physiological conditions [2] [3].
Table 1: Core Characteristics of Directed Evolution Platforms
| Feature | In Vivo Evolution | In Vitro Evolution | Hybrid Approaches |
|---|---|---|---|
| Cellular Environment | Full biological context maintained | Artificial conditions | Cellular environment only during screening |
| Post-Translational Modifications | Native processing preserved | Lacks most modifications | Possible if using eukaryotic hosts |
| Diversification Method | Cellular mutagenesis pathways | Error-prone PCR, DNA shuffling | In vitro mutagenesis |
| Library Size Limitations | Transformation efficiency-dependent | Vast libraries possible (~1015) | Transformation efficiency-dependent |
| Throughput | Limited by cellular growth rates | Potentially extremely high | Limited by cellular growth rates |
| Technical Complexity | Variable (prokaryotic to mammalian) | Generally high | Moderate to high |
| Representative Techniques | Mutator strains, PROTEUS, somatic hypermutation | mRNA display, ribosome display, phage display | Phage display, yeast display |
Methodologies and Experimental Protocols
In Vivo Diversification Strategies
In vivo directed evolution employs several sophisticated mechanisms to generate genetic diversity within living cells:
Microbial Mutator Strains: Prokaryotic systems frequently utilize engineered bacterial strains with defective DNA repair machinery to elevate mutation rates. The commercially available XL1-Red E. coli strain, deficient in mutD, mutS, and mutT genes, increases spontaneous mutation frequency to approximately 1 base change per 2,000 nucleotides [2]. This approach was successfully applied to shift the pH optimum of ADH beta-glucuronidase from Lactobacillus gasseri, generating variants with enhanced activity at neutral pH for broader application as a reporter enzyme [2].
Targeted In Vivo Mutagenesis: Recent advances enable more precise targeting of mutagenesis to specific genes of interest. Orthogonal systems utilizing specialized DNA polymerases (e.g., DNA Pol I), pGLK1/2 plasmids, Ty1 retrotransposons, T7RNAP, and CRISPR-based systems restrict mutagenesis to target sequences, minimizing background mutations in the host genome [5]. The EvolvR system, for instance, uses a CRISPR-guided nickase fused to an error-prone polymerase to introduce mutations within a defined window, offering programmable and continuous evolution in living cells [6].
Somatic Hypermutation in Vertebrate Cells: A particularly sophisticated approach harnesses the natural diversification machinery of the vertebrate immune system. Kling-EVOLVE Technology activates activation-induced cytidine deaminase (AID) to induce somatic hypermutation (SHM) in immortalized B cell clones, mimicking the natural process of antibody affinity maturation [7]. This enables directed evolution of therapeutic antibodies ex vivo, allowing researchers to enhance affinity and cross-reactivity against viral escape variants such as SARS-CoV-2 EG.5.1 and JN.1 [7].
Viral Vector-Based Mutagenesis: The PROTEUS platform utilizes chimeric virus-like vesicles (VLVs) based on a modified Semliki Forest Virus replicon [3]. These VLVs carry an error-prone RNA-dependent RNA polymerase that introduces random mutations during replication, with a measured rate of 2.6 mutations per 105 transduced cells [3]. This system enables continuous evolution in mammalian cells while maintaining dependence on host-derived VSVG envelope protein for propagation, creating a tight link between target gene function and viral fitness [3].
Diagram 1: In Vivo Directed Evolution Workflow. The process involves iterative cycles of diversification within living systems followed by selection based on cellular fitness or high-throughput screening.
Selection and Screening Methodologies
Linking genotype to phenotype represents the primary bottleneck in directed evolution, with success dictated by the principle: "you get what you screen for" [1]. In vivo systems employ various selection strategies:
Cellular Fitness Coupling: The most powerful approach directly links desired protein function to host cell survival or growth advantage. In the PROTEUS platform, the target transgene (e.g., tetracycline-controlled transactivator, tTA) is placed in a circuit where its activity drives expression of VSVG envelope protein, which is essential for propagation of the chimeric VLVs [3]. Variants with improved function (e.g., doxycycline resistance) consequently produce more VSVG, granting them a replicative advantage that enables their dominance within the viral population over multiple rounds [3].
Fluorescence-Activated Cell Sorting (FACS): When direct selection is not feasible, FACS provides a high-throughput screening alternative capable of processing >10⁷ variants per day [5]. Cell surface display technologies (yeast, mammalian) present protein variants on the extracellular membrane while retaining the genetic material inside. Labeling with fluorescently tagged ligands enables quantitative assessment of binding affinity, allowing researchers to isolate top-performing clones through sorting [4]. This approach was successfully used to identify peptide mimotopes for FMC63, the scFv domain used in clinical CD19 CAR-T cells, through yeast surface display followed by affinity maturation [4].
Plate-Based Screening: Traditional but effective, microtiter plate-based assays (typically 96- or 384-well format) allow individual clones to be cultured and assayed for activity using colorimetric or fluorometric substrates read by plate readers [1]. While throughput is limited to 10³-10⁴ variants, these methods provide robust quantitative data on enzyme performance and are particularly useful for validating hits from primary screens [1].
Table 2: Performance Comparison of Directed Evolution Platforms for Specific Applications
| Application | Platform | Key Results | Experimental Data | Reference |
|---|---|---|---|---|
| Antibody Affinity Maturation | In Vivo (B cell SHM) | Enhanced neutralization potency against SARS-CoV-2 variants EG.5.1 and JN.1 | Improved binding affinity and neutralization | [7] |
| Transcription Factor Engineering | PROTEUS (Mammalian) | Evolved tTA with improved doxycycline responsiveness (TetON-4G) | Enhanced sensitivity in gene regulation | [3] |
| Enzyme Thermostability | In Vitro (epPCR) | Significant improvement in subtilisin E thermal tolerance | Retained activity after heat challenge | [5] |
| CAR-T Ligand Discovery | Yeast Surface Display | Identified high-affinity peptide mimotopes for FMC63 scFv | KD measurements via flow cytometry | [4] |
| β-glucosidase Engineering | SEP/DDS (In Vivo) | Simultaneously enhanced activity and organic acid tolerance | 3.5-fold higher tolerance to formic acid | [8] |
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of in vivo directed evolution requires specialized reagents and genetic tools. The following table details key solutions used in the experimental approaches discussed in this review:
Table 3: Essential Research Reagents for In Vivo Directed Evolution
| Reagent/Solution | Function | Example Application |
|---|---|---|
| XL1-Red E. coli | Mutator strain with defective DNA repair pathways | Random mutagenesis of plasmid-borne genes [2] |
| Bcl6/Bcl-xL Retroviral Vector | B cell immortalization through apoptosis inhibition | Creation of stable B cell libraries for antibody discovery [7] |
| pSFV-DE Replicon | Attenuated SFV replicon for viral vector propagation | PROTEUS platform for mammalian directed evolution [3] |
| Error-Prone Pol I | Engineered low-fidelity DNA polymerase I | Targeted mutagenesis of ColE1 plasmid regions in E. coli [2] |
| AID Expression System | Induction of somatic hypermutation in B cells | Ex vivo antibody affinity maturation (Kling-EVOLVE) [7] |
| CRISPR-Directed EvolvR | CRISPR-guided nickase fused to error-prone polymerase | Targeted continuous evolution in living cells [6] |
| Yeast Surface Display Library | Peptide/protein library displayed on yeast surface | Identification of CAR-binding mimotopes [4] |
Comparative Analysis: Strategic Considerations for Platform Selection
Performance Metrics and Limitations
When selecting between in vivo and in vitro directed evolution platforms, researchers must consider several critical performance metrics and inherent limitations:
Library Diversity and Quality: In vitro methods generally provide superior library sizes and diversity. Ribosome and mRNA display systems can theoretically access library sizes of >1015 variants, completely bypassing the transformation efficiency bottleneck that constrains cellular systems to ~108-1011 variants [2]. However, in vivo libraries benefit from biological pre-screening, as proteins that fail to fold properly or are toxic to the host are automatically eliminated, enriching for functional variants [2].
Throughput and Screening Efficiency: In vitro platforms typically offer higher screening throughput, especially when combined with microfluidic droplet sorting or other compartmentalization approaches [5]. However, in vivo selection systems that directly couple desired function to cellular fitness can potentially screen entire libraries in a single step without manual intervention, representing the ultimate throughput when applicable [3].
Biological Relevance: This dimension represents the key advantage of in vivo systems. Mammalian-directed evolution platforms like PROTEUS ensure that evolved proteins are optimized for function within physiologically relevant environments, including appropriate post-translational modifications, native binding partners, and compartmentalization [3]. This is particularly critical for therapeutic proteins like antibodies, where performance in mammalian systems predicts clinical success more accurately than bacterial or yeast expression [7] [3].
Technical Accessibility: Microbial and yeast-based systems generally offer lower technical barriers to implementation, with well-established protocols and reagents. Mammalian systems require more specialized expertise and facilities but provide superior biological relevance for mammalian-targeted applications [2] [3].
Emerging Trends and Future Directions
The field of in vivo directed evolution continues to advance rapidly, with several emerging trends shaping its future trajectory:
Integration with CRISPR Technologies: CRISPR-based systems are revolutionizing in vivo directed evolution by enabling targeted and diversified mutagenesis. Technologies like CasPER (Cas9-mediated Protein Evolution Reaction) and diversifying base editors allow researchers to focus mutations on specific genomic loci while maintaining reading frames, dramatically increasing the efficiency of functional variant generation [6]. These systems are particularly valuable for antibody affinity maturation and membrane protein engineering [6].
Continuous Evolution Platforms: Systems like OrthoRep in yeast and PROTEUS in mammalian cells enable continuous evolution without repeated intervention, allowing for extended evolutionary campaigns that can accumulate complex sets of mutations requiring multiple generations to emerge [3]. These platforms are particularly valuable for tackling challenging engineering problems where improvements require coordinated mutations at distant sites.
Machine Learning Integration: The combination of directed evolution with machine learning creates powerful feedback loops where experimental data trains predictive algorithms that then guide subsequent library design [9]. This approach helps navigate the vast sequence space more efficiently, reducing experimental burden while increasing the probability of discovering high-performing variants [9].
In vivo directed evolution represents a sophisticated methodology for engineering biomolecules within biologically relevant cellular environments. While in vitro platforms maintain advantages in library size and screening throughput, in vivo systems provide the authentic cellular context essential for optimizing complex protein functions, particularly for therapeutic applications. The choice between these platforms ultimately depends on the specific project requirements, with in vivo approaches offering clear advantages for engineering proteins that function within mammalian systems, require specific post-translational modifications, or participate in complex cellular pathways. As technologies like CRISPR-mediated diversification and continuous mammalian evolution platforms mature, in vivo directed evolution is poised to become an increasingly powerful tool for creating next-generation biotherapeutics and engineered enzymes, firmly establishing its role in harnessing living systems for protein optimization.
In Vitro Directed Evolution is a powerful protein engineering method that mimics natural evolution entirely outside of living cells. This approach enables researchers to steer proteins or nucleic acids toward user-defined goals through iterative rounds of mutagenesis, selection, and amplification in a controlled, cell-free environment [10]. By decoupling the evolutionary process from cellular constraints, in vitro methods offer unique advantages in precision, flexibility, and the ability to explore vast sequence landscapes that would be inaccessible or toxic within living organisms [2].
Core Principles and Methodological Framework
The in vitro directed evolution cycle operates as a highly controlled, iterative algorithm for optimizing biomolecules. It compresses evolutionary timescales from millennia to weeks by accelerating mutation rates and applying precise, user-defined selection pressures [1]. This process consists of three fundamental stages, each critical to success.
Diversification begins with creating genetic variation in a parent gene through methods like error-prone PCR (epPCR) or DNA shuffling. epPCR intentionally reduces the fidelity of DNA polymerase through manganese ions and unbalanced nucleotide concentrations to introduce random point mutations [1]. DNA shuffling fragments multiple parent genes and reassembles them through primer-free PCR, creating chimeric genes that recombine beneficial mutations [1]. The generated library of variant genes is then transcribed and translated in vitro using cell-free systems.
The selection phase links each protein variant's function (phenotype) to its genetic code (genotype). mRNA display creates a covalent mRNA-protein linkage via puromycin, allowing isolation of functional proteins through affinity selection [2]. Ribosome display maintains the genotype-phenotype link through non-covalent protein-mRNA-ribosome complexes during in vitro translation [2]. Both methods enable efficient isolation of proteins with desired binding properties.
Amplification completes the cycle, where genetic material from selected variants is recovered and amplified to serve as templates for subsequent evolution rounds. This iterative refinement allows beneficial mutations to accumulate, progressively steering proteins toward enhanced or novel functions [10].
Comparative Analysis: In Vitro vs. In Vivo Platforms
The choice between in vitro and in vivo directed evolution platforms represents a fundamental strategic decision in protein engineering. Each approach offers distinct advantages and suffers from particular limitations, making them suitable for different research objectives and constraints.
Table 1: Platform Comparison Between In Vitro and In Vivo Directed Evolution
| Parameter | In Vitro Directed Evolution | In Vivo Directed Evolution |
|---|---|---|
| Library Size | Extremely large (up to 1015 variants) [10] | Limited by transformation efficiency (typically 106-109 variants) [2] |
| Selection Environment | Controlled, customizable conditions (solvents, temperature, pH) [2] | Cellular environment with inherent constraints [2] |
| Toxic Proteins | Compatible [2] | Problematic [2] |
| Throughput | Very high for binding/affinity selection [2] | Lower throughput for screening [5] |
| Genotype-Phenotype Linkage | Covalent (mRNA display) or complex-based (ribosome display) [2] | Cellular compartmentalization [10] |
| Functional Complexity | Limited to single molecules or simple interactions [2] | Suitable for complex pathways and cellular functions [2] |
| Post-translational Modifications | Lacks native cellular modification machinery | Supports native folding and modifications [2] |
The critical distinction lies in their operational environments. In vitro evolution occurs in cell-free systems, offering control over selection conditions and access to enormous library diversity. This comes at the cost of biological relevance, particularly for proteins requiring specific cellular environments for proper function [2]. In vivo evolution occurs within living cells, preserving native contexts but limiting library size and environmental control [2] [10].
Experimental Protocols and Case Studies
mRNA and Ribosome Display Methodologies
mRNA Display Protocol begins with in vitro transcription of a diversified DNA library to create mRNA molecules. These are then ligated to puromycin, a molecule that mimics aminoacyl-tRNA and can enter the ribosome's A-site. During in vitro translation, when the ribosome reaches the mRNA-puromycin junction, puromycin covalently attaches to the nascent polypeptide chain, creating a stable mRNA-protein fusion. This covalent linkage enables stringent affinity selection using immobilized targets, including denaturing conditions. After selection, bound complexes are dissociated, mRNA is reverse transcribed, and the resulting cDNA is amplified for subsequent rounds or analysis [2].
Ribosome Display Protocol utilizes the stability of ribosomal complexes during in vitro translation. The DNA library must lack a stop codon, preventing ribosomal dissociation after protein synthesis. This results in stable ternary complexes of mRNA, ribosome, and synthesized protein. These complexes can be directly used for selection against immobilized targets. The mRNA from selected complexes is then isolated, reverse transcribed to cDNA, and amplified. Ribosome display typically uses longer mRNA constructs with stem-loop structures to protect against degradation, and selections are performed under conditions that stabilize the ribosomal complexes [2].
Quantitative Performance Metrics
Table 2: Experimental Data from Directed Evolution Applications
| Evolved Protein | Evolution Platform | Key Improvement | Fold Improvement | Selection Method |
|---|---|---|---|---|
| GFP from Aequorea victoria | Machine learning-guided in vitro evolution | Fluorescence activity at 488 nm | 74.3-fold [11] | FACS-based screening |
| TEM-1 β-lactamase | In vivo mutator strain (error-prone Pol I) | Resistance to antibiotic aztreonam | 150-fold [2] | Bacterial survival selection |
| Esterase from Pseudomonas fluorescens | In vivo (XL1-Red mutator strain) | Hydrolysis of sterically hindered 3-hydroxy ester | Functional shift [2] | Colorimetric colony screening |
| Virus-like particles (eVLPs) | In vivo barcoded evolution | Delivery potency in mammalian cells | 2-4 fold [12] | Barcode sequencing selection |
Recent advances demonstrate how in vitro evolution is being enhanced with computational approaches. The DeepDE algorithm exemplifies this trend, using supervised learning on approximately 1,000 mutants to guide GFP evolution, achieving a remarkable 74.3-fold activity increase in just four rounds [11]. This highlights how machine learning can dramatically accelerate the in vitro evolution process by intelligently navigating sequence space.
Essential Research Reagent Solutions
Successful in vitro directed evolution requires specialized reagents and systems to execute the key process steps outside of cellular environments.
Table 3: Key Research Reagents for In Vitro Directed Evolution
| Reagent/Solution | Function | Application Examples |
|---|---|---|
| Error-Prone PCR Kits | Introduces random mutations during gene amplification | Commercial systems with optimized manganese concentrations and nucleotide biases [1] |
| Cell-Free Translation Systems | Protein synthesis without cellular constraints | Wheat germ, rabbit reticulocyte, or E. coli extracts for in vitro transcription/translation [2] |
| Puromycin Linkers | Creates covalent mRNA-protein fusions | Critical for mRNA display platforms [2] |
| Immobilized Ligands | Selection matrix for affinity-based isolation | Streptavidin beads for biotinylated targets, nickel-NTA for His-tagged proteins [2] |
| Barcoded sgRNA Libraries | Encodes variant identity in complex evolution schemes | Enables tracking of eVLP variants in sophisticated in vivo/in vitro hybrid systems [12] |
Emerging Frontiers and Integrative Approaches
The field of in vitro directed evolution is rapidly advancing through integration with cutting-edge technologies. Machine learning platforms are now being coupled with automated laboratory systems to create closed-loop evolution environments that continuously propose, synthesize, and test protein variants [13]. These systems significantly reduce experimental bottlenecks and enable more efficient exploration of sequence-function relationships.
CRISPR-based tools have also revolutionized diversification strategies, with systems like MutaT7 and EvolvR enabling targeted mutagenesis of specific genomic regions [6]. When combined with in vitro selection methods, these precise diversification tools create powerful hybrid platforms that leverage the benefits of both targeted and random mutagenesis approaches.
Additionally, novel compartmentalization strategies using water-in-oil emulsions allow ultra-high-throughput screening by creating artificial cellular environments that maintain genotype-phenotype linkages while enabling in vitro conditions [10]. These advancements collectively expand the scope and efficiency of in vitro directed evolution, opening new possibilities for engineering complex protein functions.
In vitro directed evolution provides an unparalleled platform for protein engineering in precisely controlled environments, free from cellular constraints. Its capacity to generate extraordinary library diversity and withstand stringent selection conditions makes it indispensable for optimizing molecular binding, stability, and activity. While the choice between in vitro and in vivo platforms remains context-dependent, ongoing integrations with machine learning, automation, and CRISPR technologies continue to expand the capabilities and applications of in vitro methodologies. As these tools mature, they promise to accelerate the discovery of novel biocatalysts, therapeutic proteins, and functional biomaterials for diverse biotechnology applications.
Directed evolution stands as a cornerstone technique in modern protein engineering, mimicking the principles of natural selection to develop biomolecules with enhanced or novel functions. The methodology primarily branches into two distinct platforms: in vivo (within living cells) and in vitro (in a cell-free environment). The choice between these platforms often centers on a fundamental trade-off: the physiological complexity inherent to living systems versus the precise experimental control afforded by test-tube reactions. This guide provides an objective comparison of these platforms, detailing their performance, supported by experimental data and methodologies, to inform decision-making for researchers in scientific and drug development fields.
Core Principles and Comparative Workflows
At its core, directed evolution involves iterative cycles of diversification (creating genetic variants), selection (isolating variants with desired traits), and amplification (producing templates for the next cycle) [10]. The environment in which this cycle is executed defines the platform's characteristics.
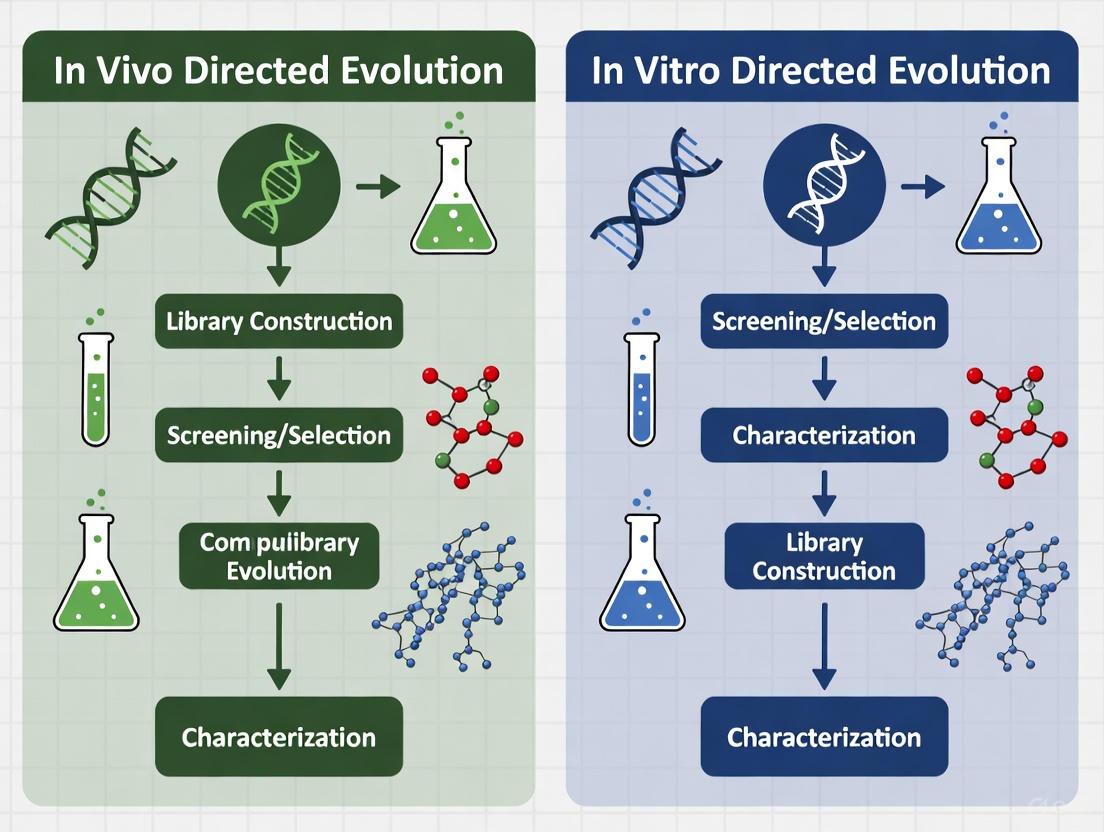
The workflows for in vivo and in vitro directed evolution differ significantly in their execution and compartmentalization, as illustrated below.
Diagram 1: Comparative workflows of in vivo and in vitro directed evolution.
Performance Comparison: Quantitative Data
The following tables summarize key performance metrics and application profiles for the two platforms, based on current literature and experimental data.
Table 1: Performance and Operational Metrics Comparison
| Parameter | In Vivo Directed Evolution | In Vitro Directed Evolution |
|---|---|---|
| Typical Library Size | Limited by transformation efficiency (often 10^6 - 10^9 variants) [2] [5] | Very large, up to 10^15 variants possible [10] [2] |
| Mutagenesis Rate | Can be tightly controlled; e.g., ~600-fold increase over background with engineered systems [14] | Fully user-defined and controllable |
| Throughput | High, especially when coupled with FACS or biosensors [14] | Ultra-high-throughput, compatible with microfluidic droplet screening [14] |
| Experimental Duration | Can be longer due to cell growth and transformation steps | Often faster, bypassing cell culture and transformation [14] |
| Representative Mutation Rate | ITMU system: 1.18 × 10^5-fold increase over host genome [15] | N/A (fully user-defined) |
Table 2: Application Scope and Functional Characteristics
| Characteristic | In Vivo Directed Evolution | In Vitro Directed Evolution |
|---|---|---|
| Physiological Relevance | High (native folding, PTMs, cellular environment) [2] | Low (lacks complex cellular milieu) |
| Experimental Control | Lower (constrained by cellular metabolism and homeostasis) | High (full control over reaction conditions) [10] [2] |
| Toxic Product/Protein Tolerance | Low [2] | High [2] |
| Ideal For | Engineering metabolic pathways, complex multi-protein interactions, proteins requiring PTMs [2] [14] | Engineering isolated enzymes, toxic proteins, and under non-physiological conditions (harsh solvents, extreme pH) [10] [2] |
| Key Limitation | Difficulty in coupling desired activity directly to cell survival (non-selectable traits) [14] | Difficult to reproduce complex cellular interactions or select for activities that require a cellular context [2] |
Experimental Protocols in Practice
In Vivo Protocol: Temperature-Controlled Continuous Evolution inE. coli
This protocol leverages a thermal-responsive repressor for tunable mutagenesis [14].
- System Construction: A two-plasmid system is used.
- Mutator Plasmid (pSC101): A low-copy plasmid carrying a gene for an error-prone DNA polymerase I (Pol I) under the control of a λPR promoter, which is regulated by an engineered thermal-sensitive repressor, cI857.
- Target Plasmid (pET28a): A high-copy ColE1-based plasmid containing the gene of interest (GOI). The replication of this plasmid specifically depends on Pol I* activity.
- Mutagenesis Cycle:
- Repression Phase: Cultures are grown at 30°C, where cI857* represses Pol I* expression, minimizing background mutation and allowing library expansion.
- Mutagenesis Phase: Temperature is shifted to 37°C, inactivating the repressor and inducing Pol I* expression. This polymerase erroneously replicates the target plasmid, introducing mutations primarily in the GOI.
- Mutation Fixation: The system can be combined with a genomic MutS mutation (defective in DNA mismatch repair) to enhance the fixation of generated mutations [14].
- Screening/Selection: The mutated library is screened using Fluorescence-Activated Cell Sorting (FACS) if a biosensor is available, or via microfluidic droplet assays for secreted enzymes. For selectable traits (e.g., antibiotic resistance), cells are plated on selective media.
In Vitro Protocol: Emulsion-Based Compartmentalization and Screening
This protocol establishes a strong genotype-phenotype link without cells [5] [16].
- Library Generation: The gene library is created in vitro using methods like error-prone PCR or DNA shuffling [10].
- In Vitro Transcription-Translation (IVTT): The DNA library is expressed using a cell-free protein synthesis system.
- Compartmentalization: The IVTT reaction mixture, along with substrates and reagents, is emulsified in water-in-oil droplets. This creates billions of picoliter-scale compartments, each ideally containing a single gene variant and the proteins it encodes.
- Incubation & Selection: The emulsion is incubated to allow enzymatic activity. The product of the reaction is coupled to a selectable signal, such as the formation of a fluorescent product or the capture of the gene itself.
- For example, in "CSR" (Compartmentalized Self-Replication), active DNA polymerase variants replicate their own genes within the droplet [16].
- Recovery and Amplification: Droplets containing the desired activity (e.g., fluorescence) are sorted using flow cytometry, or the entire emulsion is broken and the enriched genes from active variants are recovered and amplified by PCR for the next round of evolution.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Their Functions in Directed Evolution
| Reagent / Tool | Primary Function | Platform |
|---|---|---|
| Error-prone Pol I (e.g., Pol I*) | Engineered DNA polymerase for targeted, continuous mutagenesis of plasmids in host cells [14]. | In Vivo |
| Mutator Strains (e.g., XL1-Red) | E. coli strains deficient in DNA repair pathways to increase global mutation rates [2]. | In Vivo |
| Orthogonal DNA Replication System (OrthoRep) | A system in yeast that replicates a target plasmid with a high error rate, keeping mutagenesis separate from the genome [17]. | In Vivo |
| Phage-Assisted Continuous Evolution (PACE) | Links protein function to viral propagation, enabling continuous evolution in a chemostat with minimal intervention [17]. | In Vivo |
| Transcription Factor-based Biosensors | Converts the concentration of a target metabolite into a fluorescent signal, enabling high-throughput screening via FACS [14]. | In Vivo |
| Error-prone PCR | A standard method to introduce random point mutations across a gene during amplification [10] [5]. | In Vitro |
| DNA Shuffling | Fragments and reassembles homologous genes to create chimeric libraries, mimicking recombination [10] [5]. | In Vitro |
| mRNA/Ribosome Display | Links a protein to its mRNA (genotype-phenotype link) for affinity-based selection without cells [2] [5]. | In Vitro |
| In Vitro Transcription-Translation (IVTT) | Cell-free system for protein synthesis from DNA templates [10]. | In Vitro |
| Microfluidic Droplet Generators | Encapsulates single genes/cells into droplets for ultra-high-throughput screening [14]. | Both |
The distinction between in vivo and in vitro directed evolution platforms is not a matter of superiority, but of strategic alignment with research goals. In vivo platforms offer the critical advantage of a biologically complex environment, making them indispensable for engineering proteins whose function is inextricably linked to cellular context, such as metabolic pathway enzymes or proteins requiring specific post-translational modifications. Conversely, in vitro platforms provide unparalleled experimental control and the ability to generate and screen vast molecular diversity, ideal for optimizing isolated enzyme properties or evolving proteins toxic to cells. The ongoing development of advanced tools, such as orthogonal replication systems and sophisticated biosensors, continues to push the boundaries of both platforms. The most effective approach often lies in a complementary strategy, leveraging the unique strengths of each system to navigate the complex fitness landscape of protein engineering.
Directed evolution (DE) stands as a cornerstone of modern protein engineering, harnessing the principles of natural selection—variation, selection, and heredity—to optimize enzymes and proteins for human-defined applications in therapeutics, industrial biocatalysis, and basic research [1] [10]. The core process is an iterative cycle of creating genetic diversity in a gene of interest and identifying improved variants [10]. This fundamental algorithm, or "Evolutionary Cycle," provides a universal framework for comparing the two primary experimental platforms for DE: in vivo (within living cells) and in vitro (in a cell-free system) [2] [10]. The choice between these platforms represents a critical strategic decision, as each offers distinct advantages and imposes specific constraints on the evolutionary experiment [2]. This guide provides an objective comparison of these platforms, focusing on their performance, supported by experimental data and detailed methodologies.
Deconstructing the Universal Evolutionary Cycle
The universal evolutionary cycle in directed evolution consists of three fundamental, iterative steps. The workflow below illustrates how this core process is implemented across different platforms.
Core Step 1: Generating Genetic Diversity
The first step involves creating a vast library of genetic variants from a parent gene [10]. The methods for achieving this can be grouped into several categories, as shown in the table below.
Table 1: Common Methods for Genetic Diversification in Directed Evolution
| Method | Principle | Key Advantage | Key Limitation |
|---|---|---|---|
| Error-Prone PCR (epPCR) [1] | Reduces DNA polymerase fidelity during gene amplification. | Simple; does not require prior structural knowledge. | Biased towards transition mutations; limited amino acid sampling. |
| DNA Shuffling [1] | Fragments homologous genes and reassembles them. | Recombines beneficial mutations from multiple parents. | Requires high sequence homology (>70-75%) between parents. |
| Site-Saturation Mutagenesis [1] | Targets specific codons to encode all 20 amino acids. | Enables deep exploration of key "hotspot" residues. | Practical for only a small number of positions at a time. |
| Mutator Strains [2] | Uses engineered cells with defective DNA repair. | Simple in vivo system; continuous mutagenesis. | Mutagenesis is genome-wide and not restricted to the target gene. |
| Orthogonal Systems (e.g., MutaT7) [18] | Uses targeted in vivo mutagenesis systems. | Restricts mutations to the plasmid-borne gene of interest. | Can be limited by mutation spectrum and target size. |
Core Step 2 & 3: Selection, Screening, and Amplification
After a library is created, the functional variants must be identified (Selection/Screening) and their genes harvested (Amplification).
- Selection involves coupling the desired protein function directly to host cell survival or replication, automatically enriching for improved variants [18] [10]. This method is extremely high-throughput, limited only by the number of cells that can be cultivated, but can be difficult to design and may not provide quantitative data on individual variants [1].
- Screening involves assaying each variant individually (e.g., using colorimetric or fluorescent assays) and selecting the best performers based on a quantitative threshold [10]. While lower in throughput than selection, screening provides rich data on the performance of each variant [1].
Finally, the genes encoding the top-performing variants are amplified via PCR or host cell cultivation to serve as the template for the next round of evolution [10].
Platform Comparison: In Vivo vs. In Vitro Directed Evolution
The universal cycle is implemented differently depending on whether the experiment is conducted inside living cells (in vivo) or in a test tube (in vitro). The table below summarizes the core differentiators.
Table 2: Objective Comparison of In Vivo and In Vitro Directed Evolution Platforms
| Parameter | In Vivo Platform | In Vitro Platform |
|---|---|---|
| Experimental Environment | Living cells (e.g., E. coli, yeast) [2]. | Cell-free systems (e.g., emulsion droplets, ribosome display) [2] [10]. |
| Throughput (Library Size) | Limited by transformation efficiency, typically 10^8 - 10^11 variants [2]. | Not limited by transformation; can reach 10^13 - 10^15 variants [10]. |
| Functional Context | Cellular environment; tests protein folding, solubility, and function under physiological conditions [2]. | Flexible conditions; can use harsh solvents or extreme temperatures [2]. |
| Toxic Proteins | Difficult to express without harming the host [2]. | Amenable, as there is no host to kill [2]. |
| Genotype-Phenotype Linkage | Automatic via cellular compartmentalization [10]. | Requires engineering (e.g., mRNA display, emulsion compartments) [10]. |
| Typical Selection Pressure | Growth-coupled selection [18]. | Affinity binding (e.g., phage display) [10] or in vitro compartmentalization [10]. |
Key Performance Differentiators and Experimental Data
- Library Size and Diversity: The in vitro platform holds a decisive advantage in raw library size, bypassing the bottleneck of cellular transformation [2]. This allows for a more exhaustive exploration of sequence space.
- Functional Context and Folding: The in vivo platform provides a significant advantage by ensuring that evolved proteins are functional in a natural cellular environment, complete with native folding chaperones and post-translational modifications [2]. This is critical for engineering therapeutic proteins like antibodies.
- Applicability to Complex Traits (Growth-Coupling): In vivo platforms uniquely enable growth-coupled selection, where improved enzyme activity directly enhances host cell fitness. A recent study demonstrating Growth-coupled Continuous Directed Evolution (GCCDE) achieved automated evolution of over 10^9 variants per culture. In this system, the activity of a target enzyme (CelB) was linked to the ability of E. coli to utilize lactose as a sole carbon source. Variants with higher activity promoted faster growth and automatically outcompeted others, requiring no manual intervention for screening [18].
Experimental Deep Dive: A Growth-Coupled In Vivo Evolution Protocol
The GCCDE study [18] serves as an exemplary case for a high-performance in vivo evolution protocol.
Detailed Experimental Methodology
Step 1: System Construction
- Host Strain: An E. coli Dual7 strain, derived from DH10B, with mutations rendering its native β-galactosidase activity negligible and chromosomally integrating the MutaT7 mutagenesis system [18].
- Target Gene & Plasmid: The celB gene from Pyrococcus furiosus was cloned into a low-copy-number plasmid under a hybrid promoter (P_tetO) for regulated expression [18].
- Diversification: An initial library was created using error-prone PCR on the celB plasmid. This library was then transformed into the Dual7 strain, where continuous in vivo mutagenesis was driven by the MutaT7 system (induced by lactose) [18].
Step 2: Continuous Evolution and Selection
- Culture System: The transformed library was cultivated in a continuous culture system (chemostat) with a minimal medium where lactose was the sole carbon source [18].
- Growth Coupling: Only cells expressing CelB variants with sufficient β-galactosidase activity could hydrolyze lactose into glucose and galactose for energy, thereby growing faster [18].
- Selective Pressure: The temperature was gradually lowered from 37°C to 27°C to selectively favor CelB variants with improved activity at lower temperatures [18]. Over approximately 200 hours of continuous evolution, faster-growing cells outcompeted and replaced slower-growing ones.
Step 3: Analysis and Validation
- Screening: Post-evolution, cultures were plated and subjected to blue-white screening using X-gal [18].
- Characterization: Dark-blue colonies were picked, and their CelB activity was quantitatively measured using a chlorophenol red-β-D-galactopyranoside (CPRG) assay in 96-well plates [18].
- Result: The top variants (e.g., AA10, T1, W10) showed a ~70% increase in enzymatic activity compared to the wild-type enzyme while retaining thermostability [18].
The logical flow of the GCCDE experiment is summarized below.
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and their functions in a typical directed evolution campaign, particularly for in vivo growth-coupled experiments.
Table 3: Essential Research Reagents for Directed Evolution
| Reagent / Solution | Function / Application | Example from GCCDE Study [18] |
|---|---|---|
| Mutator Strains / Systems | Provides continuous in vivo mutagenesis of the target gene. | E. coli Dual7 strain with MutaT7 system. |
| Specialized Host Strains | Provides a genetic background devoid of the target enzyme's native activity. | DH10B-derived strain with lacZ mutation. |
| Selective Growth Media | Creates a direct link between enzyme activity and survival/growth. | Lactose minimal medium as the sole carbon source. |
| Reporters for Screening | Enables visual or quantitative identification of improved variants. | X-gal for blue-white screening on plates. |
| Assay Substrates | Allows quantitative measurement of enzymatic activity. | CPRG (Chlorophenol red-β-D-galactopyranoside) for 96-well plate assays. |
| Expression Vectors | Carries the gene of interest and allows regulated expression. | Low-copy-number plasmid with P_tetO promoter induced by aTc. |
The universal evolutionary cycle provides a robust framework for comparing directed evolution platforms. The in vitro platform is unparalleled in its ability to screen vast libraries and evolve proteins under non-physiological conditions or those that are toxic to cells. In contrast, the in vivo platform excels in its ability to select for function in a cellular environment, particularly through automated, growth-coupled systems like GCCDE, which dramatically reduce manual labor and enable real-time selection.
The choice between platforms is not mutually exclusive. A powerful emerging strategy is to use in vitro methods for initial deep diversification, followed by in vivo platforms for functional screening and final optimization in a relevant biological context [18]. Furthermore, the integration of AI-informed protein design [19] with high-throughput experimental validation promises to create hybrid "semi-rational" approaches that are faster and more efficient than either method alone. For the drug development professional, this evolving toolkit offers increasingly precise and powerful means to engineer next-generation biologics and biocatalysts.
Historical Context and the Rise of Modern Directed Evolution
Directed evolution (DE) is a cornerstone technique in protein engineering that mimics natural selection to steer biomolecules toward user-defined goals. [10] The field has matured from early experiments in the 1960s, such as Spiegelman's work on evolving RNA molecules, into a sophisticated discipline integral to industrial and medical innovation. [10] A pivotal moment in its recognition was the awarding of the 2018 Nobel Prize in Chemistry for the directed evolution of enzymes and the phage display of peptides and antibodies. [10] This review will objectively compare the performance of modern in vivo (within living cells) and in vitro (in an artificial cell-free environment) directed evolution platforms, framing the analysis within a broader thesis on their respective applications in contemporary research.
Principles and Methodologies of Directed Evolution
The fundamental cycle of directed evolution consists of three iterative steps: diversification (creating a library of gene variants), selection or screening (isolating variants with the desired function), and amplification (generating a template for the next round). [10] The success of any DE experiment is directly tied to the total library size, as screening more mutants increases the odds of finding one with enhanced properties. [10]
A key distinction between platforms lies in how the "fitness" of a variant is measured. Selection directly couples protein function to the survival of its gene, forcing the host organism to rely on the protein's activity to live or die. [10] Screening, conversely, involves individually assaying each variant (e.g., via a colorimetric or fluorescent signal) and ranking their performance. While screening provides rich, quantitative data on each variant, selection systems are typically higher in throughput, limited only by the transformation efficiency of the host cells. [10]
In Vivo vs. In Vitro Directed Evolution: A Platform Comparison
The choice between conducting DE in living cells or in a test tube has profound implications on the experimental workflow, library diversity, and types of proteins that can be evolved. The table below summarizes the core distinctions.
| Feature | In Vivo Directed Evolution | In Vitro Directed Evolution |
|---|---|---|
| Cellular Environment | Uses living organisms (e.g., bacteria, yeast, mammalian cells). [10] | Performed in cell-free systems (e.g., free solution, artificial microdroplets). [10] |
| Library Size | Limited by host transformation efficiency. [10] | Can generate vastly larger libraries (up to (10^{15}) variants). [10] |
| Selection Conditions | Constrained by cellular viability; reflects a natural cellular environment. [10] | Highly versatile; allows for extreme conditions (e.g., high temperature, organic solvents). [10] |
| Protein Expression | Can express toxic proteins, but this may impact host health. | Can readily express proteins that would be toxic to living cells. [10] |
| Key Advantage | Ideal for evolving proteins that function in a complex biological context with native post-translational modifications. [20] | Superior for exploring a wider sequence space and evolving proteins for non-biological applications. [10] |
| Key Limitation | Low throughput can be a bottleneck for library size. [10] | Lacks the complex cellular machinery and environment of a living cell. [10] |
Modern In Vivo Platforms: Experimental Protocols and Data
Recent advances have led to specialized in vivo platforms that address the unique challenges of evolving proteins within mammalian and plant cells.
PROTEUS: A Mammalian Cell Evolution Platform
The PROTein Evolution Using Selection (PROTEUS) system uses chimeric virus-like vesicles (VLVs) to enable extended directed evolution campaigns in mammalian cells. [20]
- Core Methodology: The gene of interest is cloned into a modified Semliki Forest Virus (SFV) replicon. The host cell's production of the vesiculovirus G (VSVG) envelope protein is made dependent on the activity of the evolved protein, creating a direct link between function and viral propagation. [20]
- Experimental Workflow:
- Diversification: The target gene is mutagenized.
- Packaging: The variant library is packaged into VLVs in specialized producer cells.
- Selection: Naive host cells, engineered to express VSVG only if the protein variant performs the desired function, are transduced with the VLV library. Only functional variants produce new VLVs.
- Amplification: The supernatant containing enriched VLVs is used to infect new host cells, repeating the cycle.
- Performance Data: In a model selection, PROTEUS was able to enrich for VLVs carrying a circuit-activating transgene even when they were outnumbered 1000:1 by neutral VLVs, with complete takeover of the population within three rounds. [20] The system's error-prone RNA polymerase provides an inherent mutation rate, measured at approximately 2.6 mutations per 100,000 transduced cells, enabling continuous evolution. [20]
GRAPE: A Plant Cell Evolution Platform
The Geminivirus Replicon-Assisted in Planta Directed Evolution (GRAPE) platform enables rapid evolution directly in plant cells. [21]
- Core Methodology: Gene variants are inserted into an artificial geminivirus replicon. The desired gene activity is linked to the virus's rolling circle replication (RCR), leading to the selective amplification of the best-performing variants. [21]
- Experimental Workflow:
- A library of mutagenized genes of interest (GOIs) is cloned into geminivirus replicons.
- The replicon library is delivered into plant leaves (e.g., Nicotiana benthamiana).
- Inside plant cells, variants that enhance or create a desired activity (e.g., disease resistance) stimulate viral replication.
- Beneficial variants are massively enriched within the viral pool, while non-functional ones are diluted out.
- Performance Data: A full cycle of selection in the GRAPE platform can be completed on a single leaf in just four days. [21] This system has successfully evolved immune receptors in rice to recognize a broader range of pathogen effectors, demonstrating its practical application in crop engineering. [21]
Advanced In Vitro and Hybrid Platforms
For applications requiring massive library sizes or delivery of gene-editing machinery, in vitro and hybrid approaches are paramount.
Directed Evolution of Engineered Virus-like Particles (eVLPs)
eVLPs are engineered to deliver proteins and RNAs transiently, offering a promising modality for gene therapy. A recent breakthrough involved a directed evolution system for eVLP capsids to improve production and delivery efficiency. [12] [22]
- Core Methodology: A key innovation was solving the genotype-phenotype link in DNA-free eVLPs. Each eVLP variant packages ribonucleoproteins (RNPs) loaded with a barcoded sgRNA unique to that variant's genetic code. [12] [22]
- Experimental Workflow:
- A library of eVLP capsid mutants is created.
- Each mutant is produced in a cell along with its unique barcoded sgRNA, which gets packaged into the eVLP.
- The eVLP library is subjected to a selection pressure (e.g., transduction into human cells).
- eVLPs that successfully transduce target cells are lysed, and their barcoded sgRNAs are sequenced to identify which capsid variants are enriched.
- Performance Data: Through this method, fifth-generation (v5) eVLPs were developed. They demonstrated a 2-to-4-fold increase in delivery potency in cultured mammalian cells compared to the previous-best (v4) eVLPs. [12] [22]
Diagram of the barcoded eVLP directed evolution workflow.
The Scientist's Toolkit: Key Research Reagents
Successful directed evolution campaigns rely on a suite of specialized reagents and tools. The following table details essential components for establishing these platforms.
| Reagent / Tool | Function in Directed Evolution |
|---|---|
| Error-Prone PCR | A common method for introducing random point mutations across the gene of interest to create initial library diversity. [10] |
| Yeast Surface Display | A platform for displaying protein variants on the yeast cell surface, enabling screening for binding interactions using flow cytometry. [4] |
| Barcoded sgRNA | Serves as a heritable, sequenceable tag that links a variant's identity to its function in systems that lack packaged DNA (e.g., eVLPs). [12] [22] |
| Viral Replicons (SFV, Geminivirus) | Engineered viral genomes that lack key structural genes. They serve as vectors for gene expression and replication within host cells, and their propagation can be tied to a protein's function. [20] [21] |
| CRISPR-Cas Systems | Enables precise and efficient gene targeting for creating focused mutant libraries. RNA-guided nucleases like Cas9 can be used to introduce targeted double-strand breaks, which are repaired with introduced mutations. [6] |
Core cycle of a directed evolution experiment.
The dichotomy between in vivo and in vitro directed evolution is not a matter of one platform being superior to the other. Instead, the choice is dictated by the biological question and the desired application. In vivo platforms like PROTEUS and GRAPE are indispensable for evolving proteins that must function within the complex, native context of a mammalian or plant cell, complete with their unique signaling networks and post-translational modifications. [20] [21] Conversely, in vitro and hybrid platforms, exemplified by the evolved eVLPs, provide unmatched library diversity and control over selection conditions, making them powerful for optimizing molecular delivery vehicles and enzymes for industrial processes. [12] [10] [22] The ongoing integration of these methods with cutting-edge tools like CRISPR base editors [6] and computational design ensures that directed evolution will continue to be a foundational technology for engineering biology.
Platforms in Practice: Techniques and Real-World Applications
Directed evolution has long served as a powerful methodology for engineering biomolecules with novel functions, traditionally relying on in vitro systems or microbial hosts [23] [24]. However, when the goal is to develop tools for mammalian biology or therapeutics, a significant compatibility gap often emerges. Proteins evolved in bacteria or yeast may misfold, lack proper post-translational modifications, or fail to integrate with unique mammalian signaling pathways when transferred into mammalian cells [24]. This fundamental limitation has driven the development of sophisticated in vivo directed evolution platforms that perform the entire evolutionary cycle—diversification, selection, and amplification—within the complex cellular environment where the biomolecule must ultimately function [23] [24] [3].
This guide compares the established workhorses of in vivo evolution, such as bacterial mutator strains, with groundbreaking mammalian platforms like PROTEUS [3], VEGAS [25] [26], and OrthoRep [23]. We objectively evaluate their performance based on experimental data, detailing their operational principles to provide a clear resource for researchers selecting a platform for specific projects.
Platform Comparison at a Glance
The table below summarizes the core characteristics and performance metrics of major in vivo directed evolution systems.
Table 1: Comparison of Key In Vivo Directed Evolution Platforms
| Platform | Host Organism | Mutation Mechanism | Typical Mutation Rate | Unit of Selection | Key Advantages |
|---|---|---|---|---|---|
| Bacterial Mutator Strains [2] | E. coli | Defective DNA repair (e.g., XL1-Red) or error-prone DNA Pol I [2] | ~1 in 2,000 bases (XL1-Red) [2] | Cell | Simple setup, cost-effective |
| OrthoRep [23] | Yeast | Orthogonal error-prone DNA polymerase replicating a linear plasmid [23] | Not Specified | Cell | Durable, genome not mutated [23] |
| MutaT7 [23] | E. coli, Yeast, Mammals | T7 RNAP-fused deaminase causing transcription-coupled mutagenesis [23] | Not Specified | Cell | Easy implementation, broad host range [23] |
| EvolvR [23] | E. coli, Yeast, Mammals | Nickase Cas9 fused to error-prone DNA polymerase [23] | Not Specified | Cell | Programmable targeting via gRNA [23] |
| VEGAS [25] [26] | Mammalian Cells | Error-prone replication of Sindbis virus RNA genome [26] | >10(^{-3}) per base per round [26] | Virus | One-day cycles, complex signaling outputs [25] |
| PROTEUS [3] | Mammalian Cells | Error-prone replication of engineered Semliki Forest Virus (SFV) replicon [3] | 2.6 mutations per 10^5 transduced cells [3] | Virus (VLV) | Stable, low cheater particle formation [3] |
Table 2: Documented Experimental Outcomes from Platform Applications
| Platform | Evolved Target | Selection Pressure | Outcome | Timeline |
|---|---|---|---|---|
| Bacterial Mutator Strains [2] | TEM-1 β-lactamase | Aztreonam resistance | 150-fold increase in resistance [2] | Not Specified |
| OrthoRep [23] | Drug-activatable dihydrofolate reductase (DHFR) | Growth in media with drug | Not Specified | Not Specified |
| VEGAS [26] | GPCRs (e.g., ADORA2B), Nanobodies | Transcriptional activation of a reporter gene | New signaling functions, allosteric nanobodies [26] | < 1 week [26] |
| PROTEUS [3] | Tetracycline-controlled transactivator (tTA) | Doxycycline resistance | tTA-4G variant with altered doxycycline responsiveness [3] | Not Specified |
Platform Methodologies and Experimental Protocols
Bacterial Mutator Strains
Principles and Workflow: These systems use engineered E. coli strains with defective DNA repair pathways (e.g., lacking mutS, mutD, and mutT functions) or expressing error-prone DNA polymerases. This leads to genome-wide mutagenesis as the culture grows, eliminating the need for external library generation [2]. The gene of interest (GOI) is typically hosted on a plasmid. Cells carrying beneficial mutations in the GOI are selected based on a growth advantage, such as antibiotic resistance or survival on minimal media [2].
Detailed Protocol:
- Clone GOI: Insert the gene of interest into an appropriate plasmid vector.
- Transform Mutator Strain: Introduce the plasmid into a mutator strain like E. coli XL1-Red [2].
- Propagate for Mutagenesis: Grow the transformed culture for multiple generations (e.g., 24-48 hours) to allow for the accumulation of random mutations throughout the genome and plasmid.
- Apply Selection: Plate the culture on solid media or grow in liquid media containing the selective agent (e.g., an antibiotic).
- Screen and Isolate: Pick surviving colonies and screen for the desired, improved protein function.
- Iterate: Use the plasmid from improved variants to retransform fresh mutator cells and repeat steps 3-5 for further optimization.
Mammalian Viral Platforms: PROTEUS and VEGAS
Principles and Workflow: These systems decouple the unit of evolution (the virus) from the unit of production (the host cell). The GOI is placed within the genome of an engineered RNA virus. The virus's natural error-prone replication provides diversification. A key feature is that viral propagation is made dependent on the GOI's function through a synthetic circuit, creating a direct link between function and fitness [3] [26].
Detailed Protocol for PROTEUS [3]:
- Circuit Design: Engineer a genetic circuit where the expression of a viral envelope protein (e.g., VSVG) is controlled by the activity of the GOI.
- Clone into Replicon: Insert the GOI into the pSFV-DE replicon vector, an engineered Semliki Forest Virus genome.
- Initial Packaging: Co-transfect packaging cells (BHK-21) with the replicon vector and a helper plasmid constitutively expressing VSVG to produce the initial pool of Virus-Like Vesicles (VLVs).
- Evolution Cycles (Repeated Rounds):
- Transduction: Infect fresh, naive BHK-21 cells that have been transfected with the VSVG-expressing plasmid. Only VLVs carrying a functional GOI will activate the circuit and produce new VSVG, enabling the production of new VLVs.
- Harvest: Collect supernatant containing the evolved VLV progeny after ~24-48 hours.
- Diversification: The error-prone viral RNA polymerase introduces mutations during each replication cycle.
- Isolation and Validation: After multiple rounds, harvest the evolved VLV genomes, clone the GOI into a standard expression vector, and characterize the function of the evolved variants.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these platforms requires specific genetic tools and reagents.
Table 3: Key Reagents for In Vivo Directed Evolution Platforms
| Platform | Essential Reagents | Function |
|---|---|---|
| Bacterial Mutator Strains [2] | E. coli XL1-Red strain | Engineered mutator strain with defective DNA repair for random mutagenesis. |
| Plasmid with target gene | Vector that harbors the gene of interest for mutagenesis and selection. | |
| Selective media (e.g., antibiotics) | Applies pressure to enrich for cells with improved GOI function. | |
| OrthoRep [23] | Engineered yeast strain | Host organism containing the orthogonal DNAP and linear plasmid. |
| Orthogonal DNA Polymerase (DNAP) | Error-prone polymerase that specifically replicates the linear plasmid. | |
| Linear plasmid (p1) | Special plasmid encoding the GOI, replicated exclusively by the orthogonal DNAP. | |
| MutaT7 [23] | T7 RNA Polymerase-deaminase fusion | Enzyme that targets mutagenesis to genes under a T7 promoter. |
| Plasmid with T7 promoter-GOI | Vector where the GOI is placed downstream of a T7 promoter for targeted hypermutation. | |
| EvolvR [23] | nCas9-Error-prone DNAP fusion | Enzyme complex that introduces localized mutations at a gRNA-specified site. |
| Guide RNA (gRNA) | RNA molecule that directs the EvolvR complex to a specific DNA locus. | |
| PROTEUS [3] | pSFV-DE replicon vector | Engineered SFV genome backbone for hosting the GOI and viral replication. |
| pCMV_VSVG plasmid | Plasmid for expressing the VSVG envelope protein, making viral propagation host-dependent. | |
| BHK-21 cells | Mammalian cell line used for packaging and propagating the chimeric VLVs. | |
| VEGAS [26] | pTSin plasmid | Sindbis virus-based vector for encoding the GOI. |
| pCMV-SSG plasmid | Plasmid expressing the Sindbis structural proteins for virus packaging. | |
| HEK293T cells | Mammalian cell line commonly used for Sindbis virus production and evolution. |
The data from these platforms reveal a clear trade-off between simplicity and environmental relevance. Bacterial mutator strains offer a straightforward, low-cost entry into in vivo evolution and are highly effective for optimizing proteins that function well in prokaryotes [2]. However, mammalian viral platforms like PROTEUS and VEGAS, while more complex to establish, provide a decisive advantage for targets that require an authentic mammalian cellular environment. They directly select for functions within complex signaling networks and can evolve sophisticated phenotypes, such as allosteric control and specific pathway activation, on timescales of a week or less [3] [26].
The choice of system should be guided by the biological question. For enzyme evolution where prokaryotic expression is sufficient, bacterial systems remain a powerful tool. For evolving therapeutic proteins, signaling receptors (like GPCRs), or intracellular biosensors intended for human cell application, mammalian platforms are increasingly the superior option. They minimize the "translation gap" that occurs when moving molecules from microbial systems to mammalian settings, thereby accelerating the development of more effective research tools and therapeutics [24] [3].
Directed evolution stands as a cornerstone of modern protein engineering, enabling researchers to mimic natural selection in laboratory settings to develop biomolecules with enhanced or novel functions. The in vitro toolkit for generating genetic diversity is foundational to this process, offering precise control over mutagenesis conditions and library construction. Among the most established and powerful techniques are error-prone PCR (epPCR), DNA shuffling, and display technologies. These methods have consistently proven their value for evolving proteins, enzymes, and other biomolecules, independent of cellular transformation efficiency. This guide provides a detailed, objective comparison of these core in vitro technologies, framing them within the broader context of directed evolution platform selection for research and therapeutic development.
Core Technology Comparison
The following table summarizes the key operational parameters, outputs, and applications of the three primary in vitro directed evolution technologies.
| Technology | Key Mechanism | Typical Mutation Rate/Frequency | Primary Mutation Types | Key Advantages | Common Applications |
|---|---|---|---|---|---|
| Error-Prone PCR (epPCR) | Low-fidelity PCR using mutagenic conditions (e.g., error-prone polymerases, biased dNTP pools, manganese ions) [27] [28]. | Varies by protocol; ~0.05%–0.17% total mutation frequency reported for epADS, a related synthesis method [27]. | Primarily base substitutions; potential for indels [27]. | Technically simple; rapid library generation; no requirement for structural knowledge [2] [8]. | Optimizing enzyme activity, stability, and stereoselectivity; creating starting libraries for aptamer development (e.g., whole-cell SELEX) [28] [8]. |
| DNA Shuffling | In vitro homologous recombination of DNA fragments from related parent sequences [27] [6]. | Dependent on parental diversity and recombination efficiency. | Combines point mutations from parents; can introduce crossovers and new combinations. | Recombines beneficial mutations from multiple parents; explores a larger sequence space than point mutagenesis alone [8]. | Rapid evolution of proteins & enzymes; metabolic pathway engineering; family shuffling to evolve protein families [27]. |
| Display Technologies | In vitro physical coupling of genotype (DNA/RNA) to phenotype (protein/peptide) [2]. | Governed by the input library (often created by epPCR or DNA shuffling). | Governed by the input library. | Extremely high library diversity (up to 1016 variants) [28]; direct selection based on binding affinity. | Isolating high-affinity binding peptides (phage display), antibodies (ribosome display), or aptamers (mRNA display) [2]. |
Comparative Experimental Data and Performance
To objectively compare performance, the following table consolidates quantitative data and observed outcomes from documented applications of these technologies.
| Technology | Documented Experimental Outcomes | Required Screening Throughput | Technical & Resource Considerations |
|---|---|---|---|
| Error-Prone PCR (epPCR) | - Diversification: Achieved 200–4000-fold diversification in fluorescent protein strength via epADS [27].- Bias: Traditional epPCR shows biased mutations (e.g., transitions over transversions); combining polymerases (Taq + Mutazyme II) reduces bias [29].- Specific Application: Inosine-epPCR successfully created functional starting libraries for 10 parallel whole-cell SELEX campaigns [28]. | Lower throughput sufficient for enzyme activity screens; higher throughput needed for binding affinity. | Low cost and technically simple. High-fidelity polymerases unsuitable; requires optimization of mutagenesis rate [28] [29]. |
| DNA Shuffling | - Directed DNA Shuffling (DDS): Co-evolved β-glucosidase for both enhanced activity and organic acid tolerance, minimizing negative/reverse mutations [8].- Segmental epPCR (SEP): Effectively mutates large genes by dividing them into smaller, more manageable fragments [8]. | Medium to High, depending on the complexity of the pathway or protein being evolved. | Moderate complexity. Can be laborious; risk of reverse mutations in traditional protocols; DDS/SEP addresses some limitations [8]. |
| Display Technologies | - Library Size: Can routinely generate libraries with diversities of >1013 unique members, far exceeding transformation limits [2].- Affinity Maturation: Capable of selecting binders with picomolar to nanomolar affinities, rivaling antibodies [28]. | Extremely High (library size >1013). | High complexity and specialized expertise required. A pure in vitro system; no host cell transformation needed [2]. |
Essential Methodologies: Key Experimental Protocols
Error-Prone PCR (epPCR) using Inosine
This protocol, revisited for aptamer development, uses deoxyinosine triphosphate (dITP) to introduce targeted mutations and increase GC content [28].
- Step 1: Initial Mutagenic PCR
- Template: A single-stranded DNA aptamer or gene of interest.
- Reaction Setup: Standard PCR mixture incorporating dITP. Inosine acts as a universal base during amplification.
- Cycling Conditions: Standard PCR cycles suitable for the primers and template.
- Step 2: High-Fidelity Amplification
- Template: The product from Step 1.
- Polymerase: Use a high-fidelity polymerase to amplify the mutated sequences. During this step, inosine is preferentially read as guanine or cytosine, increasing GC content and introducing focused mutations.
- Step 3: Library Preparation
- The final PCR product is converted to single-stranded DNA for use in downstream selection processes like SELEX [28].
Segmental Error-Prone PCR (SEP) and Directed DNA Shuffling (DDS)
This combined approach is designed for the directed evolution of large genes, such as the gene for Penicillium oxalicum 16 β-glucosidase (16BGL) [8].
- Step 1: SEP – Fragmenting and Mutagenizing
- The target gene is divided into several smaller, overlapping segments.
- Each segment is independently mutagenized using standard epPCR protocols.
- Step 2: DDS – Reassembling Mutations
- Mutated fragments from improved variants ("positive variants") identified in initial screening are selectively amplified.
- These positive fragments are mixed with other unmutated or differently mutated fragments.
- The mixture is introduced into Saccharomyces cerevisiae, which uses its high innate homologous recombination efficiency to assemble the fragments into full-length, chimeric genes containing cumulative beneficial mutations [8].
Visualizing the In Vitro Directed Evolution Workflow
The following diagram illustrates the general workflow and logical relationship between the core in vitro technologies discussed, highlighting their role in a typical directed evolution campaign.
The Scientist's Toolkit: Key Research Reagents & Solutions
A successful directed evolution campaign relies on a suite of specialized reagents and tools. The following table details essential components for implementing the featured in vitro technologies.
| Reagent/Solution | Critical Function | Example Applications & Notes |
|---|---|---|
| Low-Fidelity DNA Polymerases | Catalyzes DNA amplification while introducing random base substitutions. | Taq polymerase: Naturally lower fidelity; often used with Mn2+ to increase error rate [29]. Mutazyme II: An engineered polymerase with a mutational spectrum complementary to Taq, used to reduce bias [29]. |
| Mutagenic Nucleotide Mixes | Unbalanced dNTP ratios or inclusion of nucleotide analogs to promote misincorporation. | dITP (Deoxyinosine triphosphate): A nucleotide analog that base-pairs non-specifically, used in inosine-epPCR to create diversity [28]. |
| Homologous Recombination Host | Assembles overlapping DNA fragments into full-length genes in vivo. | Saccharomyces cerevisiae: A preferred host due to its highly efficient homologous recombination system, used in DNA shuffling and SEP/DDS protocols [8]. |
| Selection Matrix | The solid phase or tag to which a target ligand is immobilized for panning display libraries. | Streptavidin-coated beads: Commonly used if the target is biotinylated. Immobilized protein/peptide: Used for selecting binders against specific antigens or receptors. |
Error-prone PCR, DNA shuffling, and display technologies form a powerful, complementary toolkit for in vitro directed evolution. epPCR remains the go-to for simplicity and rapid library generation, while DNA shuffling excels at recombining beneficial mutations. Display technologies offer unparalleled library diversity and are unmatched for affinity-based selection. The choice of technique is not mutually exclusive; they are often used in an iterative fashion or even combined, as with SEP and DDS. When selecting a platform, researchers must weigh factors such as the starting genetic diversity, desired mutation types, available screening capacity, and project resources. These in vitro "workhorses" provide a robust and controlled environment for exploring sequence-function relationships, continuing to be indispensable for advancing protein engineering, therapeutic development, and fundamental biological research.
The field of genetic engineering is rapidly evolving beyond basic CRISPR-Cas9 systems toward sophisticated hybrid platforms that integrate multiple technological advancements. These emerging systems represent a significant paradigm shift in how researchers approach genetic screening, therapeutic development, and functional genomics. Advanced CRISPR platforms now combine the precision of gene editing with the scalability of high-throughput screening, enabling unprecedented investigation of complex biological systems [30]. The evolution from single-gene editing to multiplexed genome engineering has been particularly transformative, allowing simultaneous manipulation of multiple genetic targets within the same system [31].
The development of these sophisticated tools is largely driven by the limitations of conventional CRISPR systems when applied to complex biological contexts. Traditional in vitro models often fail to recapitulate the intricate cellular environments found in living organisms, creating a critical need for platforms that can maintain high-resolution genetic screening capabilities in physiologically relevant settings [30]. The emergence of ITMU (In Vivo Tracking of Multiplexed Understanding) platforms represents the cutting edge of this evolution, combining computational frameworks with experimental innovations to overcome previous constraints in genetic research.
Comparative Analysis of CRISPR Platforms
Performance Metrics Across Platforms
Table 1: Comprehensive comparison of advanced CRISPR screening platforms
| Platform Feature | Conventional CRISPR Screening | Multiplexed CRISPR Systems | CRISPR-StAR (Advanced ITMU) |
|---|---|---|---|
| Screening Context | Primarily in vitro | In vitro and some in vivo applications | Optimized for complex in vivo models (organoids, tumors) [30] |
| Genetic Targeting | Single gene knockout | Multiple gene knockouts; large deletions; structural variations [31] | Genome-wide with internal controls [30] |
| Internal Control System | Separate control population | Limited or no internal controls | Intrinsic single-cell-derived controls via UMIs [30] |
| Handling of Biological Noise | Limited, requires large cell numbers per sgRNA (500-1,000 cells) [30] | Moderate, still affected by heterogeneity | Excellent, overcomes heterogeneity and genetic drift [30] |
| Resolution in Complex Models | Low, excessive noise in vivo [30] | Moderate, improved but limited | High, maintains accuracy even with low sgRNA coverage [30] |
| Experimental Reproducibility | Variable (R = 0.07 at low coverage) [30] | Moderate | High (R > 0.68 at all coverages) [30] |
| Therapeutic Applicability | Limited by noise and specificity issues | Promising but requires validation | High, identifies in-vivo-specific dependencies [30] |
Technical Specifications and Capabilities
Table 2: Technical specifications and editing capabilities of advanced CRISPR systems
| Technical Parameter | CRISPR-Cas9 Base Editing | Prime Editing | Multiplexed Epigenetic Editing | CRISPR-StAR |
|---|---|---|---|---|
| Editing Type | Single-nucleotide changes without DSBs [32] | Precise insertions, deletions, and all base-to-base conversions [33] | Simultaneous gene activation/repression [31] | Conditional sgRNA activation with internal controls [30] |
| Efficiency | Varies by target site | ~20% insertion efficiency demonstrated [33] | Efficient multi-gene regulation [31] | 55-45% active/inactive sgRNA ratio optimized [30] |
| Specificity | Reduced off-target risks compared to nuclease editing [32] | 82% genome-wide specificity reported [33] | Target-specific but requires optimization | High, internally controlled for cell-intrinsic factors [30] |
| Key Innovation | Avoids double-strand breaks [32] | Reverse transcriptase-template editing [33] | dCas9-based transcriptional control [31] | Cre-inducible sgRNA with stochastic outcomes [30] |
| Therapeutic Evidence | VERVE-101/102 for PCSK9 inactivation [34] | Preclinical development | Research phase | Identified in-vivo-specific melanoma dependencies [30] |
Experimental Protocols for Advanced CRISPR Systems
CRISPR-StAR Implementation Workflow
The CRISPR-StAR (Stochastic Activation by Recombination) protocol represents a significant advancement for genetic screening in complex models. The methodology employs a Cre-inducible sgRNA expression system combined with single-cell barcoding using Unique Molecular Identifiers (UMIs) to generate internal controls at the single-cell level [30].
Step-by-Step Protocol:
Library Cloning: Clone sgRNA library (5,870 sgRNAs targeting 1,245 genes in demonstrated study) into the CRISPR-StAR backbone containing intercalated lox5171 and loxP sites [30].
Cell Engineering: Transduce target cells (e.g., mouse melanoma cells for in vivo screening) expressing Cas9 and Cre::ERT2 at high representation (>1,000 cells per sgRNA) [30].
Selection and Bottlenecking: Apply selection markers, then subject cells to artificial bottlenecks via limiting dilution to simulate in vivo engraftment conditions (1-1,024 cells per sgRNA) [30].
Clone Expansion: Re-expand cells to >1,000 cells per sgRNA to establish single-cell-derived clones tracked by UMIs [30].
Induction: Administer 4-OH tamoxifen (day 0) to induce Cre::ERT2-mediated recombination, generating either active sgRNAs (stop cassette excision) or inactive controls (tracr RNA excision) in a mutually exclusive manner [30].
In Vivo Engraftment: Inject induced cells into appropriate animal models (e.g., immunocompromised or immune-intact mice based on experimental needs) [30].
Harvest and Analysis: Harvest tumors/tissues after appropriate duration (14 days in proof-of-concept study), sequence UMIs and sgRNAs, and compare representation of active sgRNAs to internal UMI-matched inactive controls [30].
Critical Optimization Steps:
- Vector design refinement to achieve balanced 55:45 active:inactive sgRNA ratio
- UMI design ensuring minimal amplification bias between recombination outcomes
- Adjustment of tamoxifen concentration and timing for optimal recombination efficiency [30]
Multiplexed CRISPR Screening Protocol
Dual-Target Editing for Large Deletions and Structural Variations:
gRNA Design: Design paired gRNAs targeting flanking regions of target genomic loci for large deletions or specific orientations for inversions/duplications [31].
Vector Assembly: Utilize Golden Gate assembly or "PCR-on-ligation" methods for modular assembly of multiple gRNAs (up to 10 demonstrated) in single vectors [31].
Delivery: Package multiplexed gRNA arrays into lentiviral vectors with optimized promoters (human U6 and mouse U6) to prevent homologous recombination [31].
Screening: Transduce target cells at appropriate MOI to ensure single-copy integration, select with appropriate antibiotics, and harvest cells at timepoints appropriate for phenotypic readouts [31].
Analysis: Sequence genomic DNA to verify intended edits (deletions, inversions, translocations) and perform functional assays to assess phenotypic consequences [31].
Visualization of Advanced CRISPR Platforms
CRISPR-StAR Workflow and Mechanism
CRISPR-StAR Screening Workflow
Multiplexed CRISPR Engineering Applications
Multiplexed CRISPR Applications
Research Reagent Solutions for Advanced CRISPR Platforms
Table 3: Essential research reagents for implementing advanced CRISPR platforms
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Inducible Systems | Cre::ERT2; lox5171/loxP vectors [30] | Conditional sgRNA activation; internal control generation | Balanced recombination ratios (55:45 active:inactive) critical [30] |
| Delivery Vehicles | Lipid Nanoparticles (LNPs); AAVs; Lentiviral Vectors [35] | In vivo delivery; tissue-specific targeting | LNP liver tropism; AAV cargo size limitations (4.7kb) [36] [35] |
| Screening Libraries | CDKO libraries; genome-wide StAR libraries [31] [30] | Multiplexed screening; synthetic lethality studies | UMI barcoding essential for clonal tracking [30] |
| Editing Enhancers | Alt-R HDR Enhancer Protein [33] | Improve HDR efficiency in difficult cells (iPSCs, HSPCs) | 2-fold HDR improvement in hard-to-edit cells [33] |
| Specialized Nucleases | Cas12Max; high-fidelity Cas variants [33] [34] | Expanded targeting; improved specificity | Compact size for AAV packaging (hfCas12Max = 1080aa) [34] |
| Detection/Optimization | In situ sequencing; SORT nanoparticles [33] [35] | Spatial editing assessment; organ-specific targeting | Uniform hepatocyte editing across liver zones verified [33] |
Discussion and Future Perspectives
The evolution of CRISPR-based platforms from simple gene editing tools to sophisticated ITMU systems represents a paradigm shift in genetic research and therapeutic development. The data clearly demonstrates that advanced platforms like CRISPR-StAR overcome fundamental limitations of conventional screening methods, particularly in the context of complex in vivo models where biological heterogeneity has traditionally introduced excessive noise [30]. The ability to maintain high-resolution genetic screening capabilities in physiologically relevant environments opens new avenues for identifying therapeutic targets that would remain undetectable using traditional approaches.
The integration of multiplexed editing capabilities with increasingly sophisticated delivery systems creates a powerful foundation for addressing complex polygenic diseases and understanding intricate genetic networks [31]. Future developments will likely focus on enhancing the specificity and tissue targeting of these systems, particularly through engineered LNPs with selective organ targeting (SORT) capabilities and improved viral vectors that overcome current cargo limitations [35]. Additionally, the convergence of CRISPR technologies with single-cell analytics and spatial genomics promises to further refine our understanding of genetic function in native cellular contexts.
The therapeutic translation of these advanced platforms is already evident in clinical trials for conditions ranging from hereditary transthyretin amyloidosis to hypercholesterolemia, with early results demonstrating both the efficacy and safety of these approaches [36] [34]. As the field continues to evolve, the distinction between in vivo and in vitro platforms will likely blur further, with hybrid systems that leverage the controlled aspects of in vitro manipulation while maintaining the physiological relevance of in vivo contexts. This technological convergence positions CRISPR-based ITMU platforms as central tools in the future of precision medicine and functional genomics.
The pursuit of sustainable industrial processes has positioned enzyme engineering as a cornerstone of modern biotechnology, particularly in the development of efficient biofuel production pathways. Directed evolution (DE), a method that mimics natural selection in a laboratory setting, has emerged as one of the most powerful tools for optimizing enzymes, bypassing the need for complete structural knowledge to achieve user-defined goals [10]. This approach is indispensable for tailoring natural enzymes, which often lack the robustness, specificity, or activity required for industrial applications such as the conversion of raw biomass into biofuels like ethanol, biodiesel, and biogas [37] [38]. The core cycle of directed evolution involves iterative rounds of diversification (creating genetic variety), selection or screening (isolating improved variants), and amplification [10]. This process can be performed either within living cells (in vivo) or in cell-free systems (in vitro), with the choice of platform profoundly impacting the scale, throughput, and applicability of the engineering campaign. This article provides a comparative analysis of these two platforms, framing the discussion within the context of optimizing enzymes for the demanding environment of biofuel synthesis.
Platform Comparison: In Vivo vs. In Vitro Directed Evolution
The choice between in vivo and in vitro directed evolution involves a series of strategic trade-offs, balancing the authenticity of the cellular environment against the sheer scale and control of cell-free systems.
Table 1: Core Characteristics of Directed Evolution Platforms
| Feature | In Vivo Evolution | In Vitro Evolution |
|---|---|---|
| Cellular Environment | Uses living organisms (e.g., bacteria, yeast) [2]. | Performed in cell-free systems or emulsions [10]. |
| Genotype-Phenotype Link | Achieved through cellular compartmentalization [10]. | Requires covalent linkage (e.g., mRNA display) or compartmentalization in droplets [2] [10]. |
| Library Size | Limited by host cell transformation efficiency [2]. | Can be extremely large (up to 1015 variants) [10]. |
| Throughput | High when coupled with cell survival-based selection [10]. | Very high, compatible with pure in vitro selection methods [2]. |
| Environmental Relevance | Tests enzymes in a realistic cellular context with folding helpers and post-translational modifications [2]. | Lacks the full complexity of a living cell. |
| Selection/Screening Flexibility | Limited by cellular permeability and toxicity [10]. | Highly versatile; conditions can be freely adjusted [2]. |
| Suitability for Toxic Proteins | Poor, as toxicity can kill the host cell [2]. | Excellent, as no living cells are involved [2]. |
Workflow and Key Methodologies
The fundamental workflow of directed evolution is universal, but the specific techniques for diversification and screening differ between platforms. The logical flow of a typical directed evolution campaign, highlighting the parallel steps for each platform, is illustrated below.
Diagram 1: Generalized workflow for in vivo and in vitro directed evolution. The process is iterative, with the best variant from one round serving as the parent for the next.
Diversification Strategies
- In Vivo Diversification: Modern
in vivoapproaches often use engineered systems to target mutations to specific genes. Mutator strains of E. coli (e.g., XL1-Red), which are deficient in DNA repair, increase the global mutation rate [2]. More precise methods include CRISPR-based systems like EvolvR, which uses a Cas9-nickase fused to an error-prone polymerase to introduce mutations at a specific genomic locus, and base-editing techniques that enable targeted point mutations [6]. Techniques like Multiplex Automated Genome Engineering (MAGE) allow for the simultaneous optimization of multiple genes in a pathway by incorporating pools of mutagenic oligonucleotides [2]. - In Vitro Diversification: These methods are performed on genetic material in a test tube. Error-prone PCR (epPCR) introduces random point mutations by reducing the fidelity of the DNA polymerase [5] [6]. DNA shuffling physically fragments and reassembles genes from homologous parents, mimicking natural recombination to create chimeric libraries [5] [10]. Site-saturation mutagenesis is a focused approach where one or a few specific residues are randomized to all possible amino acids, often guided by structural knowledge [5].
Screening and Selection Methodologies
- In Vivo Screening: A major advantage of
in vivosystems is the ability to use growth-coupled selection, where enzyme activity is directly linked to cell survival, allowing for the screening of immense libraries with minimal effort [10]. When selection is not feasible, microtiter plate-based assays are common, where individual clones are cultured and their enzymatic activity measured using colorimetric or fluorimetric substrates [5]. - In Vitro Screening: Cell-free systems enable highly versatile screening. Display technologies, such as mRNA and ribosome display, create a physical link between the protein variant (phenotype) and its encoding mRNA (genotype), allowing for efficient affinity-based selection from libraries of billions [2] [10]. Fluorescence-Activated Cell Sorting (FACS) can be used with in vitro compartmentalization, where single genes and their expressed proteins are isolated in water-in-oil emulsion droplets, enabling high-throughput sorting based on fluorescent reporters of enzymatic activity [5].
Experimental Spotlight: Base-Editing-Mediated In Vivo Evolution of a Degron System
A 2025 study published in Nature Communications provides a compelling example of a sophisticated in vivo directed evolution campaign, showcasing the integration of CRISPR technology and base editors to solve a specific protein instability problem [39].
Experimental Objective and Rationale
The researchers aimed to improve the auxin-inducible degron (AID) 2.0 system, a tool for targeted protein degradation. While the OsTIR1(F74G)-based system was efficient, it suffered from limitations including high basal degradation (leakiness) and slow recovery of target proteins after removing the inducing ligand [39]. To overcome this, they employed a directed evolution approach to create superior OsTIR1 variants.
Detailed Protocol and Workflow
The experimental process combined several advanced in vivo techniques, as outlined below.
Diagram 2: Workflow for the base-editing-mediated directed evolution of the OsTIR1 degron system in human induced pluripotent stem cells (hiPSCs) [39].
- Library Diversification via Base Editing: The team used a custom-designed sgRNA library to target all possible regions of the OsTIR1 gene in human induced pluripotent stem cells (hiPSCs). They employed both cytosine base editors (CBEs) and adenine base editors (ABEs) to introduce a wide spectrum of point mutations across the gene, creating a comprehensive mutant library in a live-cell setting [39].
- Functional Selection and Screening: The mutant cell population was subjected to several rounds of functional selection. First, selection pressure was applied to enrich variants with minimal basal degradation (i.e., low leakiness in the absence of the ligand). Subsequently, Fluorescence-Activated Cell Sorting (FACS) was used as a high-throughput screen to isolate cells that exhibited faster recovery of a target protein after the inducing ligand was washed out [39].
- Validation: Individual clones were isolated, and the OsTIR1 gene was sequenced. The performance of leading candidates, such as the S210A variant, was rigorously validated using Western blot analysis to quantify degradation kinetics and recovery rates, confirming the superior properties of the newly evolved system, dubbed AID 2.1 [39].
Key Reagents and Research Solutions
Table 2: Essential Research Reagents from the OsTIR1 Directed Evolution Study
| Research Reagent | Function in the Experiment |
|---|---|
| Cytosine Base Editor (CBE) | Enzyme that catalyzes C•G to T•A base conversions, used for random mutagenesis [39]. |
| Adenine Base Editor (ABE) | Enzyme that catalyzes A•T to G•C base conversions, used for random mutagenesis [39]. |
| Custom sgRNA Library | A pool of guide RNAs designed to tile the entire OsTIR1 coding sequence, directing base editors to specific sites [39]. |
| Human Induced Pluripotent Stem Cells (hiPSCs) | The host cells for evolution; provide a human cellular context for OsTIR1 function [39]. |
| Fluorescence-Activated Cell Sorter (FACS) | High-throughput instrument used to screen and isolate individual cells based on fluorescent markers indicating protein recovery speed [39]. |
| Auxin Ligand (5-Ph-IAA) | The small molecule that induces the interaction between OsTIR1 and the degron-tagged target protein, triggering degradation [39]. |
Performance Data: Quantitative Comparison of Evolved Degrons
The success of the directed evolution campaign was quantified by comparing the performance of the evolved AID 2.1 system (featuring the OsTIR1-S210A variant) against the parent AID 2.0 system and other degradation technologies.
Table 3: Quantitative Performance Comparison of Degron Systems [39]
| Degron System | Basal Degradation (Leakiness) | Induced Degradation Efficiency (at 6h) | Recovery Rate (after ligand washout) | Impact on Cell Proliferation |
|---|---|---|---|---|
| AID 2.0 (OsTIR1-F74G) | High (Target-specific) | ~90-95% (Very High) | Slow | No significant impact |
| AID 2.1 (OsTIR1-S210A) | Significantly Reduced | ~90-95% (Maintained) | Faster | No significant impact |
| dTAG | Moderate | High | Moderate | Substantially reduced |
| HaloPROTAC | Low | Slow kinetics | Moderate | Substantially reduced |
| IKZF3 | Moderate | High | Moderate | Substantially reduced |
The data demonstrates that the in vivo directed evolution effort successfully generated an improved enzyme. The AID 2.1 variant retained the high induced degradation efficiency of its parent while addressing its key weaknesses: it exhibited minimal basal degradation and a faster recovery rate, creating a more precise and controllable tool for researchers [39].
The directed evolution of the OsTIR1 degron system exemplifies the power of modern in vivo platforms, particularly when enhanced by CRISPR-based diversification and high-throughput screening. The choice between in vivo and in vitro evolution is not a matter of which is universally superior, but which is more appropriate for the specific enzyme and desired function. For optimizing enzymes in biofuel pathways, in vivo evolution is crucial for ensuring functionality in a production host, while in vitro evolution can be unmatched for exploring radically non-natural chemistries or toxic reactions.
The future of directed evolution lies in the integration of both platforms with emerging technologies. Machine learning (ML) models are increasingly being used to analyze sequence-activity relationships and predict beneficial mutations, guiding library design to explore the most promising regions of sequence space [13]. Furthermore, the integration of laboratory automation enables the execution of complex, iterative evolution cycles with minimal human intervention, creating closed-loop systems that can rapidly converge on optimal enzyme variants [13]. As these tools mature, they will dramatically accelerate the engineering of robust biocatalysts, paving the way for more efficient and economically viable biofuel production processes.
In the rapidly advancing field of biotherapeutics, optimizing proteins and antibodies is a critical step for enhancing clinical efficacy and safety. The global therapeutic protein market, valued at hundreds of billions of dollars, is experiencing robust growth, driven by the increasing prevalence of chronic diseases and demands for targeted therapies [40] [41]. This growth is underpinned by relentless innovation in protein engineering technologies, particularly directed evolution platforms that enable researchers to enhance key drug properties such as binding affinity, specificity, and stability.
Directed evolution mimics natural selection in laboratory settings to generate biomolecules with improved or novel functions. These approaches are broadly categorized into in vitro (conducted in cell-free systems) and in vivo (performed within living cells) platforms [2]. While in vitro methods like phage display have historically dominated, recent advances in CRISPR-based genome editing and deep learning are accelerating in vivo techniques, offering distinct pathways for optimizing therapeutic candidates [6]. This guide provides an objective comparison of these platforms, focusing on their operational principles, experimental outputs, and applicability to therapeutic protein and antibody optimization.
Platform Comparison: In Vivo vs. In Vitro Directed Evolution
The choice between in vivo and in vitro directed evolution platforms depends heavily on project goals, required throughput, and available resources. The table below summarizes the core characteristics of each approach.
Table 1: Core Characteristics of In Vivo and In Vitro Directed Evolution Platforms
| Feature | In Vivo Platforms | In Vitro Platforms |
|---|---|---|
| Cellular Environment | Living cells (e.g., bacteria, yeast, mammalian cells) [2] | Cell-free systems (e.g., ribosome display, mRNA display) [2] |
| Key Strength | Ideal for complex phenotypes (e.g., metabolic pathways, cellular fitness); preserves native folding and post-translational modifications [2] | Vast library sizes (up to 10^15 variants); suitable for toxic proteins; direct control over selection conditions [2] |
| Throughput & Scalability | Library size constrained by transformation efficiency [2] | Extremely high throughput and scalability [2] |
| Typical Mutagenesis Methods | CRISPR-based editors (e.g., base editors, EvolvR) [6], mutator strains [2] | Error-prone PCR, DNA shuffling, site-saturation mutagenesis [6] |
| Best Suited For | Optimizing functions within a physiological context; pathway engineering; essential gene studies [39] [2] | Rapid affinity maturation; optimizing isolated protein domains; engineering stable proteins [2] [42] |
Recent studies highlight a trend toward hybrid and advanced strategies. For instance, base-editing-mediated directed evolution is an advanced in vivo method that uses CRISPR-Cas systems fused to deaminase enzymes to directly convert one base to another in the host's genome without causing double-strand breaks, enabling precise and efficient library generation [39] [6]. Another emerging approach is deep learning-guided evolution, which uses machine learning models trained on data from mutant libraries (often ~1,000 variants) to predict beneficial mutations, dramatically accelerating the optimization cycle [11].
Performance Benchmarking: Key Experimental Data
To objectively evaluate platform performance, researchers compare key metrics such as improvement in activity, binding affinity, and efficiency. The following table synthesizes experimental data from recent studies, providing a benchmark for what different platforms can achieve.
Table 2: Performance Benchmarking of Directed Evolution Platforms
| Platform / Technology | Target Protein | Key Improvement | Experimental Data / Outcome | Source / Context |
|---|---|---|---|---|
| DeepDE (AI-guided in vitro) | Green Fluorescent Protein (GFP) | Fluorescence Activity | 74.3-fold increase in activity achieved over 4 evolution rounds [11] | Applied triple mutants and a compact library of ~1,000 mutants for training [11] |
| Base-Editing (in vivo) | OsTIR1 (Auxin-inducible degron) | Degradation Efficiency | Reduced basal degradation; faster protein recovery after washout [39] | Generated gain-of-function variant (S210A) via base-editing and screening [39] |
| CRISPR-Directed Evolution (in vivo) | Antibodies in S. cerevisiae | Binding Affinity & Diversity | Rapid antibody enhancement using an improved, diversifying CRISPR base editor [6] | Platform enabled simultaneous cytosine and adenine base editing [6] |
| In Vitro Display Methods | Therapeutic Antibodies | Binding Affinity (KD) | High-affinity antibodies enabling lower dosing, better efficacy, and reduced side effects [42] | Standard industry practice for affinity maturation [42] |
Detailed Experimental Protocols
To ensure reproducibility, this section outlines the core methodologies for two high-performing platforms: a modern in vivo approach (Base-Editing-Mediated Evolution) and an advanced in vitro approach (Deep Learning-Guided Evolution).
Protocol 1: Base-Editing-Mediated Directed Evolution (In Vivo)
This protocol was used to evolve a superior auxin-inducible degron (AID 2.1) in human induced pluripotent stem cells (hiPSCs) [39].
Key Research Reagents:
- Cytosine Base Editor (CBE) or Adenine Base Editor (ABE): For introducing point mutations (e.g., C•G to T•A or A•T to G•C) without double-strand breaks [39].
- Custom sgRNA Library: A library of sgRNAs designed to target all possible coding regions of the gene of interest (e.g., OsTIR1) [39].
- Cell Line: hiPSCs (e.g., KOLF2.2J) harboring the system to be optimized (e.g., OsTIR1(F74G) knocked into the AAVS1 locus) [39].
- Selection Agents: Ligands or conditions for applying functional pressure (e.g., auxin for AID systems).
Methodology:
- Library Delivery: Co-transfect the hiPSCs with the plasmids encoding the base editor and the custom sgRNA library.
- Mutagenesis: Allow the base editors to introduce a diverse set of point mutations across the target gene pool in the cell population.
- Functional Selection: Apply selective pressure (e.g., treat with auxin) to enrich for cells containing OsTIR1 variants with desired gain-of-function phenotypes, such as reduced basal degradation.
- Screening & Isolation: After several rounds of selection, screen the enriched cell population for specific performance metrics (e.g., protein recovery rate after ligand washout via western blot). Isulate clonal cell lines.
- Sequence Validation: Sequence the target gene in superior clones to identify the beneficial mutations (e.g., the S210A mutation in OsTIR1) [39].
Protocol 2: Deep Learning-Guided Directed Evolution (In Vitro)
The DeepDE protocol was used to significantly enhance the activity of GFP, surpassing the benchmark superfolder GFP [11].
Key Research Reagents:
- DNA Library: A mutant library of the target gene (e.g., GFP), generated via methods like error-prone PCR.
- Expression System: A cell-free in vitro transcription/translation system or a microbial host (e.g., E. coli) for protein expression.
- High-Throughput Screening (HTS) Platform: Flow cytometry or microfluidic droplet systems for assaying thousands of variants.
- Deep Learning Model: A custom algorithm (e.g., DeepDE) trained on sequence-activity data.
Methodology:
- Initial Library Construction: Generate a initial mutant library of the target protein (e.g., ~1,000 triple mutants of GFP) [11].
- Expression & Screening: Express the library and measure the activity of each variant using a HTS method.
- Model Training: Train the deep learning model on the collected dataset, linking protein sequence to functional output.
- In Silico Prediction: Use the trained model to predict a subsequent set of mutations (e.g., new triple mutants) with high predicted activity.
- Iterative Rounds: Synthesize and test the model-predicted variants. Use the new data to retrain and refine the model for further rounds of evolution. Repeat until the performance target is met [11].
The Scientist's Toolkit: Essential Research Reagents
Successful execution of directed evolution campaigns relies on a suite of specialized reagents and platforms.
Table 3: Key Research Reagent Solutions for Directed Evolution
| Reagent / Solution | Core Function | Application Context |
|---|---|---|
| CRISPR Base Editors (CBE, ABE) | Enables precise, single-nucleotide mutations in a genome without double-strand breaks [39] [6]. | In vivo directed evolution for optimizing protein function in its native genomic context [39]. |
| In Vitro Display Systems (Phage, Yeast) | Links a protein's phenotype (e.g., binding) to its genotype by displaying it on the surface of a virus or cell [42]. | In vitro affinity maturation of antibodies and other binding proteins [42]. |
| Mutator Strains (e.g., E. coli XL1-Red) | Bacterial strains deficient in DNA repair, leading to elevated random mutation rates during replication [2]. | In vivo random mutagenesis of plasmid-borne genes in a prokaryotic host [2]. |
| Deep Learning Software (e.g., DeepDE) | Algorithm that predicts protein sequences with enhanced properties from limited experimental data [11]. | Guiding both in vivo and in vitro evolution by prioritizing variants for testing, drastically reducing screening burden [11]. |
| Cell-free Transcription/Translation Systems | Enables protein synthesis without the use of living cells [2]. | In vitro evolution methods like ribosome and mRNA display [2]. |
The landscape of therapeutic protein and antibody optimization is being reshaped by powerful directed evolution platforms. In vitro methods remain the gold standard for their unparalleled library diversity and straightforward selection for binding affinity. In contrast, modern in vivo platforms, supercharged by CRISPR and base-editing technologies, excel at solving complex optimization challenges that require a physiological context, such as improving degron systems or engineering metabolic pathways [39] [6].
The future of the field lies in the intelligent integration of these approaches. Combining the high-throughput capacity of in vitro screening with the physiological relevance of in vivo validation creates a powerful iterative cycle. Furthermore, the incorporation of deep learning acts as a force multiplier for both platforms, using data to navigate the vast sequence space more efficiently and effectively than ever before [11]. The choice of platform is not a binary one; rather, the most successful research and development pipelines will be those that strategically leverage the unique strengths of each method to achieve bespoke optimization goals.
Strategic Selection and Overcoming Platform-Specific Challenges
Directed evolution is a powerful cornerstone of modern biotechnology, enabling researchers to engineer proteins, pathways, and whole cells for applications ranging from drug discovery to sustainable bioproduction. A critical initial choice in any directed evolution campaign is the platform: in vivo, within a living cell, or in vitro, in a cell-free environment. This guide provides an objective, data-driven framework to inform this fundamental decision, helping you select the optimal path for your specific research goals.
Core Concepts: Defining the Platforms
In vivo directed evolution performs both the generation of genetic diversity and the selection of improved variants within a living host organism, such as bacteria or yeast [2]. The entire process leverages the cellular machinery and takes place in a natural biological context.
In vitro directed evolution conducts diversification and selection outside a living organism [2]. Key examples include mRNA and ribosome display, where the gene library is translated in a test tube and selected for desired properties like binding affinity [2].
The table below summarizes the fundamental characteristics and typical applications of each platform.
| Feature | In Vivo Directed Evolution | In Vitro Directed Evolution |
|---|---|---|
| Environment | Living host cells (e.g., E. coli, yeast) [2] | Cell-free system (e.g., test tube) [2] |
| Diversity Generation | Cellular mutator strains, hypermutator systems, CRISPR-based editing in cells [2] [43] [6] | Error-prone PCR, DNA shuffling, and other PCR-based techniques [2] [44] |
| Selection Context | Native cellular environment with folding, post-translational modifications, and metabolic pathways [2] | Highly controlled, simplified environment [2] |
| Typical Applications | Engineering metabolic pathways, improving protein stability & function in a cellular context, whole-cell biocatalysts [2] [44] [43] | Optimizing binding affinity (antibodies, aptamers), evolving proteins toxic to cells, achieving extremely large library sizes [2] [44] |
Comparative Analysis: A Data-Driven Breakdown
Choosing a platform involves weighing specific performance metrics and practical constraints. The following tables provide a detailed, side-by-side comparison to guide your assessment.
Table 1: Performance and Operational Characteristics
| Parameter | In Vivo Platform | In Vitro Platform | Experimental Basis & Context |
|---|---|---|---|
| Library Size | Limited by host transformation efficiency (~10^8-10^9 for bacteria) [2] | Vast, not limited by transformation (>10^13) [2] | In vivo size is a biological bottleneck; in vitro is a physical-chemical bottleneck. |
| Selection Throughput | High when coupled to growth/fluorescence (FACS) [43] | High in display techniques [44] | Both support high-throughput screening when selection is coupled to a detectable output. |
| Mutation Control | Moderate to High (with targeted systems like CRISPR) [15] [6] | High (direct control over gene library) [2] | New CRISPR tools (EvolvR, MAGE) improve in vivo targeting [15] [6]. |
| Functional Context | High biological relevance; native folding, PTMs, and metabolic integration [2] | Low biological relevance; lacks cellular environment [2] | In vivo is superior for selecting functions that depend on cellular metabolism or complex interactions. |
| Automation & Speed | Amenable to automated biofoundries for continuous evolution [43] | Individual rounds can be faster, but requires iterative steps [2] | Automated in vivo workflows can run continuously for weeks with minimal intervention [43]. |
Table 2: Practical and Strategic Considerations
| Consideration | In Vivo Platform | In Vitro Platform |
|---|---|---|
| Host/System Choice | Critical; impacts PTMs, toxicity, and selection design [2] [15] | Flexible; choice is based on translation efficiency (e.g., rabbit reticulocyte lysate) |
| Handling Toxic Proteins | Challenging; can kill the host cell [2] | Ideal; no host viability concerns [2] |
| Technical Complexity | Requires expertise in molecular biology and microbiology | Requires expertise in biochemistry and in vitro techniques |
| Resource Requirements | Requires cell culture facilities and maintenance | Requires purified components and translation systems |
| Best For |
Experimental Protocols and Workflows
To ground this comparison in practical laboratory work, here are detailed methodologies for representative campaigns in each platform.
Protocol 1: In Vivo Continuous Evolution Using a Hypermutator System
This protocol uses engineered host strains to accelerate evolution within cells, ideal for optimizing biosynthetic pathways or cellular functions [2] [43].
- Library Construction & Transformation: Clone the target gene(s) into a plasmid. For multi-gene pathways, use a multiplexed system like MAGE [2]. Transform the library into a specialized mutator strain (e.g., E. coli XL1-Red, deficient in DNA repair) or a strain expressing a CRISPR-based hypermutator like EvolvR [2] [6].
- Growth-Coupled Selection: Culture the transformed library in conditions where the desired activity (e.g., production of a target compound) is coupled to cellular growth or survival. For example, the target compound could complement an auxotrophy or provide resistance to an antibiotic [43].
- Continuous Cultivation & Monitoring: Propagate the culture in bioreactors or multi-well plates for multiple generations, allowing beneficial mutations to accumulate and enrich. Automated biofoundries can be used to maintain optimal evolution conditions for extended periods [43].
- Variant Screening & Isolation: After several cycles, plate the enriched culture on solid media. Isolate single colonies and screen them using assays (e.g., HPLC for product yield, or fluorescence-based functional assays) to identify top performers [43].
- Sequence & Validation: Sequence the evolved gene(s) from the best clones and reconstruct the variant in a fresh host to validate the improved phenotype.
Protocol 2: In Vitro Evolution using mRNA Display
This protocol is a pure in vitro method excellent for evolving high-affinity binders (peptides, antibodies) without cellular constraints [2].
- Library Generation: Create a DNA library of your protein of interest using error-prone PCR or gene synthesis. The library must be flanked by the necessary sequences for in vitro transcription and a puromycin linkage site [2].
- In Vitro Transcription & Translation: Transcribe the DNA library into mRNA. Incubate the mRNA in a cell-free translation system (e.g., wheat germ or rabbit reticulocyte extract) that contains puromycin. Puromycin covalently links the translated protein to its own encoding mRNA molecule, creating a genotype-phenotype linkage [2].
- Affinity Selection: Incubate the mRNA-protein fusions against an immobilized target of interest (e.g., a receptor or antigen). Wash away unbound and weakly bound fusions.
- Elution & Recovery: Elute the tightly bound fusions using denaturing conditions (e.g., low pH) or a competitive ligand.
- Reverse Transcription & Amplification: Reverse transcribe the recovered mRNA into cDNA. Amplify the cDNA using PCR to create a template for the next round of diversification and selection. Repeat steps 2-5 for 3-10 rounds to enrich for high-affinity binders [2].
Visualizing the Decision Workflow
The following diagram maps out the logical decision process for selecting between in vivo and in vitro platforms, incorporating key questions from the comparison tables.
The Scientist's Toolkit: Essential Research Reagents
Successful execution of directed evolution campaigns relies on specialized reagents and tools. The following table details key solutions for both platforms.
Table 3: Key Research Reagent Solutions
| Reagent / Tool | Function | Platform |
|---|---|---|
| Mutator Strains (e.g., E. coli XL1-Red) | Deficient in DNA repair pathways to increase random mutation rates in the host [2]. | In Vivo |
| CRISPR Base Editors (e.g., BE, ABE) | Enable precise, targeted point mutations (C•G to T•A or A•T to G•C) without double-strand breaks for focused library generation [39] [6]. | In Vivo |
| Broad Host-Range Mutagenesis Systems (e.g., ITMU) | Enable targeted in vivo mutagenesis across diverse bacterial and yeast hosts, expanding evolution beyond E. coli [15]. | In Vivo |
| Error-Prone PCR Kits | Use biased nucleotide concentrations or error-prone polymerases to introduce random mutations during gene amplification [44]. | In Vitro |
| Cell-Free Protein Synthesis Systems | Lysates (e.g., from E. coli, wheat germ) containing ribosomes and translation factors for in vitro transcription and translation [2]. | In Vitro |
| Puromycin Linkage Reagents | Critical for mRNA display; covalently links a translated protein to its encoding mRNA molecule [2]. | In Vitro |
The choice between in vivo and in vitro directed evolution is not a matter of which platform is superior, but which is optimal for your specific protein, pathway, and desired function. Use this framework as a starting point: if your goal requires a cellular context, is non-toxic, and benefits from growth coupling, an in vivo approach is robust and effective. If you need to evolve toxic proteins, explore ultra-deep libraries, or simply optimize binding affinity, an in vitro platform offers unparalleled control and scale. Emerging technologies that blend automation, machine learning, and hybrid strategies are continually blurring the lines, offering scientists an ever-expanding toolkit for engineering biology [43].
Directed evolution (DE) is a powerful protein engineering method that mimics natural evolution by employing iterative rounds of diversity generation and screening or selection to isolate biomolecules with enhanced traits [9]. When performed in vivo—within living cells—this process leverages the host's natural cellular machinery, enabling the selection of functionalities that depend on complex physiological contexts, such as specific signaling kinetics or drug resistance [45]. However, this approach is fraught with significant challenges, including host toxicity from expressed proteins or mutagenesis systems, limitations in transformation efficiency that restrict library diversity, and the emergence of cheater variants that exploit the selection system without contributing the desired function [46]. This guide objectively compares how modern in vivo platforms address these hurdles, providing a detailed analysis of their performance against traditional in vitro methods and other alternatives.
Comparative Platform Performance Analysis
The table below summarizes the quantitative performance and key characteristics of different directed evolution platforms in addressing core in vivo challenges.
Table 1: Platform Comparison for Addressing In Vivo Hurdles
| Platform / Feature | EvolvR in Mammalian Cells [45] | VEGAS / Viral Systems [45] | CRISPR-guided Deaminases [45] | Traditional In Vitro DE [9] |
|---|---|---|---|---|
| Primary Diversity Mechanism | CRISPR-guided error-prone DNA polymerase (EvolvR) | Orthogonal viral error-prone polymerases/replicases | CRISPR-guided nucleobase deaminases (e.g., C>T, A>G) | Error-prone PCR, DNA shuffling |
| Mutation Types | All 4 nucleotides; all 12 possible substitutions [45] | All 4 nucleotides (in principle) | Primarily transition mutations (C>T, A>G, G>A, T>C) [45] | All 4 nucleotides |
| Typical Mutation Window | At least 40 base pairs [45] | Dependent on viral genome | ~50 base pairs to thousands of base pairs [45] | Entire gene |
| Context for Selection | Native genomic locus in mammalian cells [45] | Within viral genomes; requires coupling to viral propagation [45] | Native genomic locus in mammalian cells [45] | Outside living organism; controlled lab setting [47] |
| Addresses Host Toxicity? | Enables study of toxic phenotypes under native regulation | Limited; viral infection can be cytotoxic and context is artificial | Potential for off-target effects, but enables genomic targeting | N/A - not in a living host [47] |
| Transformation Efficiency Bottleneck? | No; diversifies genes in native genome, bypassing transformation [45] | No; diversity generated in vivo via viral replication [45] | No; diversifies genomic loci in situ [45] | Yes; library delivery limited by host cell transformation [45] |
| Addresses Cheater Variants? | Not explicitly reported, but selection in native context reduces cheating opportunities | Prone to cheater variants that enhance viral propagation without desired function [45] | Not explicitly reported | N/A - selection is externally controlled [46] |
Experimental Protocols and Methodologies
Protocol for EvolvR-based Directed Evolution
The EvolvR system exemplifies a modern approach to overcoming in vivo hurdles. The following is a generalized protocol for implementing it in mammalian cells [45].
System Design and Cloning:
- Construct Assembly: Fuse a gene encoding a nickase-derived Cas9 (e.g., enCas9 with D10A and other mutations like K848A, K1003A, R1060A) to a gene encoding an error-prone E. coli DNA polymerase I (PolI3M or PolI5M). Include nuclear localization sequences (NLS) and a fluorescent reporter (e.g., mCherry) for tracking.
- gRNA Design: Design a 20nt gRNA to direct the EvolvR complex to a specific genomic locus. The use of a PAM-flexible Cas9 ortholog (e.g., recognizing NNG) increases the number of potential target sites.
Cell Transfection and Expression:
- Transfert the plasmid driving EvolvR expression, along with the gRNA plasmid, into the target mammalian cell line (e.g., HEK293, A375).
- Use a strong promoter to ensure high expression of the EvolvR construct.
Diversity Generation and Selection:
- The EvolvR complex generates a nick at the target site. The error-prone polymerase uses the nicked strand's 3' end as a primer for low-fidelity DNA synthesis, introducing substitutions within a window of at least 40 bp.
- Apply the selective pressure (e.g., an anticancer drug like trametinib for evolving resistant MAP2K1 variants) to the population of mutagenized cells.
- Culture cells for several generations to allow mutations to be "locked in" after cell division or upon evasion of mismatch repair.
Analysis and Hit Validation:
- After selection, harvest genomic DNA from surviving cells.
- Amplify and sequence the targeted locus to identify the mutations conferring the desired phenotype.
- Validate the function of specific hits by reintroducing the mutated gene into a clean background and re-testing under selective conditions.
Protocol for Identifying and Controlling Cheater Variants
Cheater variants are a fundamental challenge in microbial social evolution and in vivo selection systems. The following methodology outlines strategies for their control, as derived from microbial ecology [46].
Establishing the Model System:
- Use a well-characterized model for social cooperation and cheating, such as the fruiting body formation in Myxococcus xanthus or public good production in yeast.
- Define a "public good"—a resource like an enzyme or siderophore that is costly for an individual to produce but benefits the entire group.
Inducing and Detecting Cheaters:
- In a mixed population of cooperators and potential defectors, apply selection for the public good. Cheaters, which do not pay the cost of production but still benefit, will have a relative fitness advantage.
- Monitor the population dynamics using selective plates, fluorescent tags, or other markers to track the frequency of cooperator and cheater genotypes over time.
Implementing Control Strategies:
- Policing (Punishment): Engineer cooperator genotypes to produce a toxin to which only defectors are susceptible. Introduce a "green-beard gene" system where the toxin's production and immunity are genetically linked.
- Targeted Benefit (Kin Selection): Structure the population so that benefits of cooperation are preferentially directed toward relatives (other cooperators). This can be achieved by using a "germ-soma" division of labor or by ensuring that only cells expressing a specific cooperation signal can access the group benefit.
- Screening for Intrinsic Defector Inferiority: In a pure culture of cheaters, assess whether they can perform the selected function at all. Pure cheater groups are often at a disadvantage compared to pure cooperator groups.
Quantifying the "Cheating Load":
- Compare the fitness or productivity of pure cooperator groups with that of chimeric groups containing a mix of cooperators and cheaters.
- The difference in group-level benefits quantifies the "cheating load"—the cost imposed by cheaters on the social group [46].
Visualizing Experimental Systems and Strategies
EvolvR System Mechanism
The diagram below illustrates the mechanism of the EvolvR system for generating targeted genetic diversity in vivo.
Cheater Control Strategies
This diagram outlines the fundamental strategies for controlling cheater variants in a cooperative system.
The Scientist's Toolkit: Key Research Reagents
The table below details essential reagents and their functions for conducting directed evolution experiments, particularly those focused on addressing in vivo challenges.
Table 2: Essential Research Reagents for In Vivo Directed Evolution
| Reagent / Tool | Function / Application | Key Characteristics |
|---|---|---|
| EvolvR Construct (e.g., nCas9-PolI fusion) [45] | Targets mutagenesis to specific genomic loci in mammalian cells. | Generates all 12 substitution mutations; bypasses transformation bottlenecks. |
| PAM-flexible nCas9 (e.g., recognizing NNG) [45] | Increases the number of targetable genomic sites for EvolvR. | Enhances flexibility in gRNA design and broadens the scope of targetable genes. |
| Error-Prone Polymerase I (PolI3M/5M) [45] | The catalytic engine for introducing mutations during in vivo DNA synthesis. | Contains specific point mutations (e.g., D424A, I709N, A759R) to increase error rate. |
| gRNA Libraries | Guides mutagenic machinery to specific DNA sequences. | 20nt guide RNA; design impacts mutagenesis efficiency and window [45]. |
| Selective Agents (e.g., Trametinib) [45] | Applies selective pressure to enrich for desired phenotypes (e.g., drug resistance). | Critical for distinguishing functional variants from non-functional or cheater variants. |
| Fluorescent Reporters (e.g., BFP) [45] | Enables rapid and sensitive measurement of editing frequency and variant function. | Allows for FACS-based screening and enrichment of mutated cell populations. |
| Matrigel / ECM | Provides a 3D extracellular matrix for complex in vitro models (CIVMs) like organoids [48]. | Better mimics the in vivo tissue microenvironment for more physiologically relevant screening. |
In the field of directed evolution, researchers face a fundamental trade-off: in vitro systems offer unparalleled control and throughput for protein engineering, while in vivo systems provide the native cellular context essential for proper protein folding, modification, and function. This comparison guide objectively examines this critical divide, focusing specifically on how the absence of authentic cellular environments and post-translational modifications (PTMs) in test tube systems limits their application for optimizing therapeutic proteins. As protein-based therapeutics now constitute approximately 30% of all new US Food and Drug Administration (FDA) approved drugs, addressing these limitations has become increasingly urgent for drug development pipelines [49].
The fundamental challenge stems from the artificial nature of in vitro environments, which lack the complex molecular machinery found within living cells. This machinery is responsible for critical biochemical processes including protein folding, quality control, and the installation of PTMs—chemical modifications that occur after protein synthesis and profoundly influence stability, activity, and molecular interactions [49] [50]. For researchers selecting between directed evolution platforms, understanding the scope and solutions for these limitations is essential for developing biologically relevant therapeutics.
Comparative Analysis of Evolution Platforms
The table below summarizes the core differences between in vivo and in vitro directed evolution platforms, with particular emphasis on their handling of cellular context and PTMs.
Table 1: Platform Comparison for Cellular Context and PTM Handling
| Feature | In Vivo Evolution Systems | Traditional In Vitro Systems | Advanced In Vitro Solutions |
|---|---|---|---|
| PTM Capability | Native, authentic PTMs enabled by cellular machinery [2] | Limited to no native PTMs [49] | Engineered pathways for specific PTMs (e.g., glycosylation, cyclization) [49] |
| Cellular Environment | Full physiological context (folding chaperones, ion concentrations, pH gradients) [2] | Simplified buffer system, lacking cellular complexity [2] | Supplementation with specific machinery (e.g., microsomes, purified enzymes) [49] |
| Throughput & Control | Lower throughput, limited by transformation efficiency and cell growth [2] | Very high throughput, no transformation required [2] [49] | High throughput amenable to 384- or 1,536-well plate formats [49] |
| Selection Pressure | Suitable for complex phenotypes (e.g., metabolic engineering, fitness) [2] | Best for simple, bind-and-elute selection (e.g., affinity maturation) [2] | Expanding to more complex functions via coupled assays [49] |
| Key Limitation | Difficult to target mutagenesis specifically; host cell damage concerns [2] | Lack of PTMs and cellular context limits biological relevance [2] [49] | Engineering pathways is complex; may not recapitulate full PTM complexity [49] |
Experimental Workflows for PTM Engineering
To overcome the limitation of PTM absence in vitro, researchers have developed sophisticated cell-free workflows that incorporate specific modification machinery. The following diagram and protocol detail one such advanced methodology.
Diagram Title: High-Throughput PTM Engineering Workflow
Detailed Protocol: Cell-Free PTM Characterization with AlphaLISA
This protocol enables high-throughput characterization and engineering of PTMs by coupling cell-free gene expression with a bead-based detection assay [49].
Table 2: Key Research Reagent Solutions for PTM Workflow
| Reagent / Material | Function in Experiment |
|---|---|
| PUREfrex CFE System | Provides transcription/translation machinery for protein synthesis without living cells [49]. |
| DNA Template | Encodes the target protein/peptide and/or PTM enzyme; allows rapid variant testing [49]. |
| AlphaLISA Beads | Anti-FLAG donor and anti-MBP acceptor beads enable proximity-based signal detection [49]. |
| sFLAG-tagged Peptide | Allows universal detection of expressed peptide substrates in the assay [49]. |
| MBP-tagged RRE/Enzyme | Maltose-binding protein fusion enhances soluble expression and enables detection [49]. |
Procedure:
- Template Preparation: Design DNA templates encoding your protein or peptide substrate of interest with an N-terminal sFLAG tag. For the modifying enzyme (e.g., Oligosaccharyltransferase for glycosylation or RRE for RiPPs), use an MBP-tagged construct [49].
- Cell-Free Expression: Set up individual PUREfrex reactions for the substrate and the enzyme. Express them separately in small volumes (1-5 µL) for 1-2 hours at 37°C [49].
- Reaction Mixing: Combine the substrate-expressing and enzyme-expressing cell-free reactions in a 384-well or 1,536-well plate [49].
- PTM Reaction Incubation: Allow the mixture to incubate to enable the enzymatic modification (e.g., glycosylation or RRE-binding). Incubation time may vary (30 minutes to 2 hours) based on the specific enzyme kinetics [49].
- AlphaLISA Detection: Add the anti-FLAG donor beads and anti-MBP acceptor beads to the reaction mixture. After incubation in the dark (typically 1-2 hours), measure the chemiluminescent signal. The signal is generated only when the tagged substrate and enzyme interact closely, bringing the beads into proximity [49].
- Data Analysis: Normalize the signal output relative to positive and negative controls. The signal intensity correlates with the efficiency of the PTM installation or molecular interaction [49].
In Vivo Systems: Harnessing Natural Cellular Machinery
In vivo directed evolution leverages the full complexity of living cells, utilizing natural cellular processes to generate and select functional proteins. The following diagram illustrates the core principle of linking genotype to phenotype in a cellular environment.
Diagram Title: In Vivo Directed Evolution Cycle
Key Methodologies and Supporting Data
In vivo platforms employ various strategies to increase mutation rates and select for desired functions.
Table 3: In Vivo Mutator Systems and Applications
| System / Organism | Mutagenesis Mechanism | Key Application & Result |
|---|---|---|
| E. coli XL1-Red | DNA repair-deficient (mutD, mutS, mutT); mutation rate: ~1/2000 bp [2] | Shifted pH optimum of L. gasseri beta-glucuronidase to neutral pH [2]. |
| Error-Prone Pol I E. coli | Targeted plasmid mutagenesis via fidelity-mutated DNA Pol I; 80,000-fold increase [2] | Evolved TEM-1 β-lactamase for 150-fold increased resistance to aztreonam [2]. |
| MAGE (E. coli EcNR2) | Oligonucleotide incorporation via λ-Red β protein; targets multiple genes [2] | Optimized DXP pathway for 5-fold increased lycopene production [2]. |
Emerging Solutions and Future Outlook
Integrating Artificial Intelligence and Machine Learning
The integration of AI and ML with advanced in vitro models is a growing trend to overcome limitation. These algorithms analyze complex, high-dimensional data (e.g., from transcriptomics or phenotypic screens) to identify patterns of efficacy and toxicity that might be missed by conventional analysis, thereby enhancing the predictive power of in vitro systems [51]. For instance, deep learning models have been successfully used to predict PTM crosstalk on complex proteins like Hsp90, offering a highly efficient and rapid approach to deciphering how multiple modifications interact to regulate protein function [52].
Compartmentalization to Mitigate Parasitism
A significant challenge in evolving self-replicating systems in vitro is the emergence of parasitic sequences that replicate but do not contribute to the system's function. Research on translation-coupled RNA replication systems demonstrates that molecular parasites readily evolve and can lead to population collapse [53]. A proposed solution is compartmentalization within cell-like structures, which physically links a genotype (RNA) to its phenotype (translated replicase), protecting functional replicators and enabling sustainable evolution [53]. This principle is crucial for efforts to evolve complex molecular systems toward higher functionality.
Bio-Inspired In Vitro Evolution
Learning from natural immune systems provides a strategic path for in vitro antibody evolution. Analysis of large-scale human antibody repertoire data shows that in vivo evolution follows germline gene-defined paths, with substitutions occurring not only in complementarity determining regions but also in framework and core regions [54]. Mimicking these natural evolutionary trajectories during in vitro antibody optimization can guide library design, potentially leading to antibodies with superior affinity and developability, while also minimizing immunogenicity [54].
The divergence between in vivo and in vitro directed evolution platforms fundamentally centers on their reconciliation of throughput with biological relevance. While in vivo systems natively offer the complex cellular context and PTM machinery essential for many therapeutic proteins, advanced in vitro solutions are rapidly closing this gap. The development of high-throughput cell-free workflows incorporating specific PTM pathways, coupled with emerging technologies like AI-driven prediction and bio-inspired library design, provides researchers with an expanding toolkit. The optimal platform choice is not absolute but depends on the specific protein target, the desired properties for optimization, and the required biological fidelity. As these technologies mature and undergo rigorous validation, they hold the promise of delivering more effective and manufacturable protein therapeutics with reduced reliance on animal models.
In the field of directed evolution, the quality and breadth of the mutant library often determine the success of entire campaigns aimed at improving protein function, metabolic pathways, or entire genomes. Library generation encompasses the methodologies for creating genetic diversity, while mutational coverage refers to the effective sampling of this diversity to identify beneficial variants. The fundamental challenge lies in balancing the creation of sufficient diversity with practical screening capabilities, as the sequence space for even a modest-sized protein exceeds what can be experimentally screened.
This guide systematically compares the library generation strategies and diversity coverage of contemporary in vivo and in vitro directed evolution platforms, providing researchers with objective performance data and methodological details to inform their experimental design. We focus specifically on technologies reported in 2024-2025, representing the current state of the art in this rapidly advancing field.
Comparative Analysis of Platform Performance
The table below summarizes key performance metrics and characteristics of modern directed evolution platforms, highlighting their approaches to library generation and resulting mutational diversity.
Table 1: Performance Comparison of Directed Evolution Platforms
| Platform/Technology | Mutation Mechanism | Theoretical Diversity | Mutation Rate/Frequency | Key Applications Demonstrated |
|---|---|---|---|---|
| Base-editing-mediated evolution [39] | Cytosine/adenine base editors | Target-specific, limited to C→T, A→G | Not quantified | OsTIR1 evolution for improved degron system |
| Barcoded eVLP evolution [12] | Capsid protein mutagenesis | Limited only by barcode diversity | Not quantified | Improved eVLP production and transduction efficiency |
| PROTEUS (VLV-based) [20] | Error-prone RNA polymerase + ADAR | Entire transgene mutagenesis | ~2.6 mutations/10^5 cells (with ADAR bias) | Tetracycline transactivator evolution |
| Bacterial mutator strains [2] | DNA repair deficiencies | Genome-wide mutagenesis | ~1 mutation/2,000 bp (XL1-Red) | Esterase, β-glucuronidase evolution |
| DeepDE (AI-guided) [11] | Focused triple mutants | Targeted exploration of sequence space | 74.3-fold GFP improvement in 4 rounds | GFP optimization |
| CRISPR-directed evolution [6] | CRISPR-guided nucleases + DNA repair | Target-specific diversity | Varies by specific method | Enzyme engineering, metabolic pathways |
Table 2: Operational Characteristics and Implementation Requirements
| Platform | Screening Throughput | Selection Principle | Implementation Complexity | Best Suited For |
|---|---|---|---|---|
| Base-editing evolution [39] [55] | Medium to high | Functional screening | Medium | Target-specific protein optimization |
| Barcoded eVLP [12] | Very high | Barcode enrichment | High | Viral vector and delivery system optimization |
| PROTEUS [20] | High | Circuit-coupled replication advantage | High | Mammalian protein optimization |
| Bacterial mutator [2] | Low to medium | Growth-based selection | Low | Whole-cell or pathway optimization |
| DeepDE [11] | Medium (~1,000 variants) | AI-predicted fitness | High (requires ML expertise) | Protein activity optimization |
| CRISPR-directed evolution [6] | Medium to high | Growth or reporter-based | Medium | Pathway and genome-scale evolution |
Experimental Protocols for Key Platforms
Base-Editing-Mediated Directed Evolution
This approach utilizes cytosine and adenine base editors to create targeted diversity in protein-coding sequences, as demonstrated in the evolution of OsTIR1 for superior auxin-inducible degron technology [39].
Detailed Protocol:
- Library Design: Design a custom sgRNA library targeting all possible regions of interest in the gene (e.g., OsTIR1) for comprehensive coverage.
- Base Editor Delivery: Co-transfect cells with both cytosine (BE) and adenine (ABE) base editors along with the sgRNA library.
- Hypermutation: Allow base editors to introduce C→T and A→G mutations across the target regions through in vivo hypermutation.
- Functional Screening: Subject the mutated population to several rounds of functional selection based on desired characteristics (e.g., reduced basal degradation, faster recovery).
- Variant Isolation: Screen for and isolate individual gain-of-function variants (e.g., OsTIR1 S210A) that exhibit improved properties.
- Validation: Characterize superior variants in the final system (e.g., AID 2.1 degron system) and validate performance metrics.
Critical Considerations:
- This method is limited to transition mutations (C→T, A→G), which covers approximately 34% of all possible amino acid changes [39].
- Mutation efficiency varies depending on base editor performance and PAM compatibility.
- The approach enables focused exploration of mutational space around beneficial regions.
Barcoded eVLP Directed Evolution
This innovative system enables directed evolution of engineered virus-like particles by linking eVLP variant identity to barcoded sgRNAs packaged within the particles [12].
Detailed Protocol:
- Library Construction: Generate a library of eVLP production vectors, each expressing both an eVLP variant (e.g., capsid mutant) and a uniquely barcoded sgRNA.
- Producer Cell Transfection: Introduce barcoded vectors into producer cells under single-vector conditions to ensure each cell produces only one eVLP variant–barcode combination.
- eVLP Production: Harvest barcoded eVLPs from producer cells, with each particle packaging sgRNAs containing the unique barcode corresponding to its variant.
- Selection Pressure: Subject the barcoded eVLP library to selections for desired properties (e.g., improved production, enhanced transduction efficiency).
- Variant Identification: Sequence sgRNAs from post-selection populations to identify enriched barcodes, revealing eVLP variants with improved properties.
- Variant Combination: Combine beneficial mutations to generate superior eVLP generations (e.g., v5 eVLPs with 2-4-fold increased delivery potency).
Critical Considerations:
- Barcode diversity determines the maximum library size that can be effectively screened.
- The system requires careful optimization to ensure barcode retention and accurate representation.
- This method is particularly powerful for evolving delivery vehicles and viral vectors.
PROTEUS Platform for Mammalian Directed Evolution
PROTEUS (PROTein Evolution Using Selection) uses chimeric virus-like vesicles (VLVs) to enable extended mammalian directed evolution campaigns [20].
Detailed Protocol:
- System Setup: Establish the pSFV-DE replicon construct containing the target transgene in the attenuated SFV backbone.
- VLV Production: Transfert BHK-21 producer cells with the replicon vector and pCMV_VSVG envelope protein vector.
- Evolution Cycles: Propagate VLVs through multiple rounds of transduction in naive cells transfected to express VSVG.
- Diversification: Leverage the error-prone RNA-dependent RNA polymerase and ADAR activity to accumulate mutations (~2.6 mutations/10^5 cells).
- Selection Pressure: Apply circuit-coupled selection where transgene activity drives VSVG production and thus VLV propagation advantage.
- Variant Recovery: Sequence surviving replicons after multiple evolution rounds to identify beneficial mutations.
Critical Considerations:
- The system shows strong A-to-G and U-to-C mutational bias due to ADAR activity.
- ADAR/ADARB1 knockout reduces bias but decreases mutation rate 3-fold.
- Progressive transgene truncation occurs without selective pressure, requiring tight coupling between transgene activity and replication fitness.
Workflow Visualization
The following diagram illustrates the core workflow differences between general in vivo and in vitro directed evolution approaches, highlighting their distinct pathways for library generation and variant selection.
Workflow comparison of in vivo versus in vitro directed evolution platforms
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of directed evolution campaigns requires specific molecular tools and reagents. The table below details essential components for establishing these platforms.
Table 3: Essential Research Reagents for Directed Evolution Platforms
| Reagent Category | Specific Examples | Function/Purpose | Platform Applications |
|---|---|---|---|
| Base Editors | Cytosine base editor (BE), Adenine base editor (ABE8e) [39] [55] | Introduce C→T and A→G mutations | Base-editing evolution, continuous evolution platforms |
| CRISPR Systems | Cas9, Cas12a, guide RNA libraries [6] | Targeted DNA cleavage or modulation | CRISPR-directed evolution, library introduction |
| Mutagenic Enzymes | MutaT7, rApo1, PmCDA1, TadA-8e [55] | In vivo mutagenesis during transcription or replication | Bacterial and mammalian continuous evolution |
| Barcoding Systems | Tetraloop-barcoded sgRNAs [12] | Unique variant identification in pooled screens | Barcoded eVLP evolution, high-throughput screening |
| Biosensors | β-alanine-responsive biosensor [55] | Link product concentration to selectable phenotype | Growth-coupled selection, metabolic engineering |
| Error-Prone Polymerases | RNA-dependent RNA polymerase [20] | Generate diversity during replication | PROTEUS platform, viral vector evolution |
| Selection Circuits | Tetracycline-responsive circuits [20] | Couple protein function to replication advantage | Mammalian directed evolution |
| Delivery Vehicles | Engineered VLPs (eVLPs) [12] | Deliver editing components or serve as evolution target | Delivery and optimization of macromolecular cargo |
The choice between in vivo and in vitro directed evolution platforms involves significant trade-offs between library diversity, biological relevance, and practical implementability. In vivo platforms like PROTEUS, base-editing evolution, and barcoded eVLP systems offer the advantage of cellular context, including proper protein folding, post-translational modifications, and functional activity within complex cellular environments. However, they typically generate smaller library sizes and are limited by transformation efficiency and cellular viability constraints.
In contrast, in vitro methods like mRNA display and ribosome display can create vastly larger libraries (10^13-10^15 members) unrestricted by cellular transformation, enabling more comprehensive exploration of sequence space. The trade-off is the absence of cellular context, which can be critical for proteins whose function depends on specific cellular environments or complex interactions.
Recent advances, particularly the integration of base editing, barcoding strategies, and machine learning guidance, are blurring the traditional boundaries between these approaches. Platforms like DeepDE demonstrate how limited but intelligent screening can dramatically enhance evolutionary outcomes, while barcoded eVLP systems enable evolution of delivery vehicles themselves. The optimal choice depends critically on the specific protein or system being evolved, the desired properties, and the available screening capacity.
For researchers designing directed evolution campaigns, we recommend carefully considering the mutational coverage required, the importance of cellular context for the target protein, and the available high-throughput screening methods. Hybrid approaches that leverage the strengths of both in vivo and in vitro methods often provide the most powerful solutions for challenging protein engineering problems.
In the quest to engineer proteins, pathways, and entire genomes with enhanced functions, directed evolution has emerged as a cornerstone of modern biotechnology, deliberately harnessing the principles of natural evolution in laboratory settings to tailor biological systems for human-defined applications [1]. This process operates through an iterative algorithm of diversification and selection, where libraries of genetic variants are created and then screened for improved properties [1]. The ultimate success of any directed evolution campaign hinges on a critical step: efficiently linking a variant's genetic code (genotype) to its observable functional output (phenotype) [1]. This genotype-to-phenotype bridge is the domain of high-throughput screening (HTS), a field that has become the pivotal bottleneck determining the pace and success of biological engineering.
The strategic importance of HTS is amplified by the ongoing debate between two fundamental platforms for conducting directed evolution: in vivo systems, where both diversification and selection occur within living cells, and in vitro systems, where these processes are performed in a cell-free environment [2]. In vivo systems benefit from a natural cellular context, including proper protein folding, post-translational modifications, and integration into complex metabolic pathways, which can be difficult to reproduce artificially [2]. Conversely, in vitro systems can access a vastly larger sequence space, as they are not constrained by transformation efficiency or host cell viability, and can handle proteins that are toxic or unstable in cells [2]. This guide provides a comparative analysis of these platforms, focusing on how advanced HTS technologies are enabling researchers to navigate this strategic trade-off and accelerating the discovery of novel biomolecules.
Platform Comparison: In Vivo vs. In Vitro Directed Evolution
The choice between in vivo and in vitro directed evolution involves a series of strategic trade-offs that directly impact the efficiency of bridging the genotype-phenotype gap. The table below summarizes the core characteristics of each platform.
Table 1: Core Characteristics of In Vivo and In Vitro Directed Evolution Platforms
| Feature | In Vivo Platforms | In Vitro Platforms |
|---|---|---|
| Cellular Environment | Realistic, with folding, modifications, and complex interactions [2] | Artificial, lacking many native cellular processes [2] |
| Library Size & Diversity | Limited by host transformation efficiency [2] | Extremely large (e.g., >1012), not limited by transformation [2] |
| Throughput of Screening | High, but can be limited by culturing and assay setup [56] | Can be ultra-high-throughput, especially with droplet microfluidics [57] |
| Handling Toxic/Unstable Proteins | Challenging, can affect host viability [2] | Ideal, as the protein is isolated from cellular viability [2] |
| Typical HTS Methods | FACS, growth-based selections, microtiter plate assays [1] [56] | mRNA/ribosome display, droplet microfluidics [2] [57] |
| Automation & Miniaturization | Possible with advanced tools like the Digital Colony Picker [57] | Inherently more amenable to miniaturization and automation [57] |
Recent technological advancements are blurring the lines between these platforms and addressing their inherent limitations. For instance, the Digital Colony Picker (DCP) is an AI-powered platform that enhances in vivo screening by using a microfluidic chip with 16,000 picoliter-scale microchambers. This system dynamically monitors single-cell morphology, proliferation, and metabolic activities, enabling AI-driven identification and contact-free export of clones with desired phenotypes [57]. This represents a significant leap over traditional colony-picking methods, which rely on macroscopic observations and lack the resolution to detect subtle phenotypic advantages [57].
Case Studies and Experimental Data
Case Study 1: Evolving an Improved Degron System In Vivo
A systematic comparison of inducible protein degradation systems identified the auxin-inducible degron (AID) as a powerful tool for studying gene function. However, the high efficiency of the OsTIR1-based AID 2.0 system came with limitations, including significant basal degradation (leakiness) and slow recovery of the target protein after removing the inducing ligand [39].
Experimental Protocol: To overcome these limitations, researchers employed a directed evolution strategy entirely within human induced pluripotent stem cells (hiPSCs).
- Diversification: A custom-designed sgRNA library was used with cytosine and adenine base editors (BEs) to perform comprehensive mutational scanning of the OsTIR1 gene. This base-editing-mediated mutagenesis created a vast library of OsTIR1 variants within the native cellular environment [39].
- Selection & Screening: The mutant cell population underwent several rounds of functional selection and screening. The screening was designed for multiple phenotypes: efficient inducible degradation, minimal basal degradation, and rapid recovery after ligand washout [39].
- Output: This in vivo campaign successfully yielded gain-of-function OsTIR1 variants, most notably the S210A mutant. The resulting improved system, termed AID 2.1, maintained effective target protein depletion while exhibiting substantially reduced basal degradation and faster recovery kinetics [39].
Performance Data: The quantitative outcomes of this evolution campaign are summarized in the table below.
Table 2: Quantitative Performance Comparison of Evolved Degron Systems [39]
| System | Inducible Degradation Efficiency | Basal Degradation (Leakiness) | Recovery Rate after Washout |
|---|---|---|---|
| AID 2.0 (Parent) | High (Baseline) | High (Baseline) | Slow (Baseline) |
| AID 2.1 (Evolved S210A) | Maintained high efficiency | Significantly reduced | Faster |
Case Study 2: In Vivo Evolution of Aminoacyl-tRNA Synthetases
Genetic code expansion (GCE) relies on engineered aminoacyl-tRNA synthetase (aaRS) enzymes to incorporate non-canonical amino acids (ncAAs) into proteins. A major bottleneck has been the labor-intensive process of evolving efficient and specific aaRSs [56].
- Experimental Protocol: An advanced in vivo platform called OrthoRep was used to overcome this bottleneck.
- Platform: In the yeast S. cerevisiae, OrthoRep is an orthogonal error-prone DNA replication system that continuously mutates genes of interest encoded on a special plasmid at a rate of ~10-5 substitutions per base, while leaving the host genome untouched [56].
- Screening: The selection for improved aaRSs was performed using a ratiometric dual-fluorescence reporter (RXG). An amber stop codon was placed between genes for RFP and GFP. The critical readout was the relative readthrough efficiency (RRE), calculated as the ratio of GFP/RFP fluorescence in the presence of the ncAA versus its absence. This provided a sensitive, quantitative measure of ncAA-dependent translation efficiency [56].
- Output: This platform successfully evolved aaRSs for 13 different ncAAs. Some evolved systems achieved ncAA incorporation efficiencies that matched the efficiency of natural translation at sense codons, a significant milestone in the field. The campaign also serendipitously discovered an aaRS that had evolved to autoregulate its own expression based on ncAA availability, minimizing leakiness in the absence of the ncAA [56].
The Scientist's Toolkit: Essential Research Reagents and Solutions
The successful implementation of high-throughput screening protocols relies on a suite of specialized reagents and tools. The following table details key solutions used in the featured experiments.
Table 3: Key Research Reagent Solutions for High-Throughput Screening
| Reagent / Solution | Function / Explanation |
|---|---|
| Base Editors (BEs) | CRISPR-based tools that enable precise, programmable conversion of one DNA base into another (e.g., C to T or A to G) without causing double-strand breaks, used for focused library generation [39] [6]. |
| Error-Prone OrthoRep System | An orthogonal DNA polymerase in yeast that replicates a specific plasmid with high error rates, enabling continuous in vivo mutagenesis of target genes over many generations [56]. |
| Ratiometric Fluorescence Reporter (RXG) | A dual-fluorescent protein reporter used to quantify the efficiency of stop-codon readthrough or splicing, normalizing for cell-to-cell variation in expression and enabling highly sensitive phenotypic screening [56]. |
| Microfluidic Chips (DCP) | Chips containing thousands of addressable picoliter-scale chambers for isolating and culturing single cells, allowing for dynamic, high-resolution phenotypic monitoring and sorting [57]. |
| Ligand-Inducible Degrons (e.g., dTAG, AID) | Small protein tags that can be fused to a target protein, inducing its rapid degradation upon addition of a specific small-molecule ligand, useful for probing gene function [39]. |
Workflow Visualization and Technical Diagrams
The following diagrams illustrate the core workflows and logical relationships of the key technologies discussed in this guide.
Directed Evolution Core Cycle
In Vivo Base Editing Workflow
Digital Colony Picker Screening
The bridge between genotype and phenotype is no longer a formidable chasm but an actively engineered pathway, thanks to advanced high-throughput screening technologies. The strategic choice between in vivo and in vitro directed evolution platforms is increasingly not a binary one but a synergistic combination. As demonstrated by platforms like OrthoRep for continuous in vivo evolution and the Digital Colony Picker for AI-powered phenotypic screening, the future lies in integrated systems that leverage the strengths of both approaches. These systems offer greater scalability, precision, and depth in exploring functional sequence space, thereby accelerating the development of novel enzymes, biosynthetic pathways, and therapeutic agents. For researchers and drug development professionals, mastering these tools and understanding their comparative applications is essential for leading the next wave of innovation in biotechnology.
Data-Driven Analysis: Performance, Validation, and Future Trends
Directed evolution stands as a cornerstone of modern protein engineering, enabling the development of biomolecules with enhanced or entirely novel functions. The core process involves iterative cycles of diversity generation and screening to emulate natural evolution on a laboratory timescale. A fundamental distinction in this field lies in the choice between in vitro and in vivo platforms, each with distinct operational paradigms and performance characteristics. In vitro systems conduct diversification and selection outside living cells, while in vivo systems perform these functions within a cellular host. This guide provides a head-to-head comparison of these platforms, summarizing key performance metrics and experimental data to inform researchers in selecting the optimal system for their specific protein engineering challenges.
Performance Metrics Comparison: In Vivo vs. In Vitro Platforms
The table below summarizes the core performance metrics of in vivo and in vitro directed evolution platforms, highlighting their respective advantages and limitations.
Table 1: Key Performance Metrics of Directed Evolution Platforms
| Performance Metric | In Vivo Platforms | In Vitro Platforms |
|---|---|---|
| Library Size & Diversity | Limited by host transformation efficiency (typically ≤10^9 variants) [2] [5] | Vastly higher; not limited by transformation (can reach >10^13 variants) [2] [5] |
| Mutation Generation | Continuous, targeted mutagenesis during host cell division [2] [14] | Discrete, performed in vitro (e.g., error-prone PCR, DNA shuffling) [2] [5] |
| Cellular Context | Native folding, post-translational modifications, and complex interactions [2] [3] | Absent; may not reflect true in vivo protein behavior [2] |
| Throughput & Screening | Coupled to cellular fitness or FACS; ultrahigh-throughput possible with biosensors [5] [14] | Pure in vitro selection (e.g., ribosome display) or laborious individual screening [2] [5] |
| Automation & Labor | Potential for continuous evolution with minimal intervention (e.g., PACE, OrthoRep) [14] | Iterative, labor-intensive cycles of in vitro steps required [2] [14] |
| Target Applicability | Ideal for optimizing function within metabolic pathways or requiring cellular components [2] [14] | Superior for toxic proteins, simple affinity selection, or sequences unstable in cells [2] |
Experimental Protocols for Key Platforms
PROTEUS: A Mammalian In Vivo Directed Evolution Platform
The PROTEUS platform uses chimeric virus-like vesicles (VLVs) to enable directed evolution in mammalian cells, preserving the native cellular environment for proteins with complex modifications or interactions [3].
- Workflow: The platform is based on a two-component system. A modified Semliki Forest Virus replicon encodes the non-structural proteins and the target transgene. The infectivity of the VLVs is contingent on host cell expression of the VSVG coat protein, which is placed under the control of a circuit activated by the target transgene's activity [3].
- Diversification: The error-prone RNA-dependent RNA polymerase (RdRp) of the alphavirus backbone naturally introduces mutations during replication at a rate of approximately 2.6 mutations per 100,000 nucleotides per replication cycle, providing continuous diversity generation [3].
- Selection: Cells expressing improved transgene variants activate the VSVG expression circuit more strongly, producing more infectious VLVs that outcompete others. In a proof-of-concept, circuit-activating VLVs outcompeted neutral controls at dilutions up to 1:1000 within three rounds [3].
The following diagram illustrates the PROTEUS platform's workflow for evolving proteins within mammalian cells.
Temperature-Controlled In Vivo Evolution in E. coli
This platform in E. coli uses a thermal-responsive system to regulate mutagenesis, combining an engineered error-prone DNA polymerase I and a genomic MutS defect for efficient mutation fixation [14].
- Genetic Construction: A two-plasmid system is used. A low-copy mutator plasmid carries an evolved, thermo-sensitive repressor (cI857*) controlling the expression of an error-prone DNA Pol I variant. A separate high-copy target plasmid with a ColE1 origin carries the gene of interest [14].
- Diversification Protocol: Cultures are grown at a permissive temperature (30°C). A temperature upshift to 37-42°C induces the expression of the error-prone Pol I. Concurrently, a temperature-sensitive defect in the genomic MutS mismatch repair protein is exploited to enhance the fixation of mutations introduced by Pol I on the target plasmid [14].
- Performance: This system achieved an approximately 600-fold increase in the targeted mutation rate compared to baseline, enabling rapid evolution [14].
- Screening Integration: The platform can be coupled with ultrahigh-throughput screening. For example, evolved α-amylase libraries can be screened using microfluidic droplets, while biosynthetic pathways can be coupled with biosensors and selected via fluorescence-activated cell sorting (FACS) [14].
In Vitro Ribosome Display
Ribosome display is a pure in vitro selection technique that directly links genotype to phenotype without using living cells [2].
- Library Construction: A DNA library is first generated via methods like error-prone PCR. This library is then transcribed and translated in vitro to create a pool of proteins. A key feature is the absence of a stop codon in the DNA construct, which prevents the release of the nascent protein from the ribosome and the mRNA, forming stable mRNA-protein-ribosome complexes [2].
- Selection Process: The complex pool is incubated with an immobilized target ligand. Non-binding complexes are washed away. The mRNA from the bound complexes is then recovered, typically by dissociating the ribosome complex. This mRNA encodes the proteins with the highest affinity for the target [2].
- Amplification & Iteration: The recovered mRNA is reverse transcribed into cDNA, which is then amplified by PCR. The amplified DNA serves as the template for the next round of transcription, translation, and selection. This cycle is typically repeated 3-10 times to enrich high-affinity binders [2] [5].
The diagram below outlines the iterative process of ribosome display for in vitro protein selection.
The Scientist's Toolkit: Essential Research Reagents
Successful directed evolution campaigns rely on a suite of specialized reagents and tools. The following table details key solutions for setting up directed evolution experiments.
Table 2: Key Research Reagent Solutions for Directed Evolution
| Research Reagent | Function in Directed Evolution | Example Application / Note |
|---|---|---|
| Error-Prone DNA Polymerase I (Pol I*) | In vivo mutagenesis agent for target plasmids with ColE1 origin [14]. | Engineered variant (D424A, I709N, A759R) with high error rate. Expression is often controlled by a thermo-sensitive promoter [14]. |
| Mutator Strains (e.g., E. coli XL1-Red) | In vivo mutagenesis via defects in DNA repair pathways (e.g., mutD, mutS, mutT) [2]. | Provides a mutation frequency of ~1/2000 bp; simple to use but mutagenesis is genome-wide and not target-specific [2]. |
| Thermo-Sensitive Repressor (cI857*) | Regulates mutagenesis in vivo; represses mutator gene expression at low temperatures [14]. | An evolved variant (ΔT57, A400T, T418A) shows reduced leakage and stronger induction, improving system control [14]. |
| Barcoded Guide RNAs (sgRNAs) | Encodes the identity of individual eVLP variants during directed evolution of delivery vehicles [12]. | A 15-bp barcode in the sgRNA tetraloop enables tracking and enrichment analysis of eVLP variants without packaged viral genomes [12]. |
| Transcription Factor-Based Biosensors | In vivo reporters that link metabolite concentration to fluorescent signal for ultrahigh-throughput screening [14]. | Enables FACS-based selection for improved metabolic pathway flux, as demonstrated in the evolution of a resveratrol pathway [14]. |
| Microfluidic Droplet Systems | Ultrahigh-throughput screening by compartmentalizing single cells and assays in picoliter droplets [14]. | Used to screen for improved α-amylase activity, identifying a mutant with a 48.3% improvement [14]. |
The choice between in vivo and in vitro directed evolution platforms is not a matter of superiority but of strategic alignment with the project's goals. In vivo platforms excel when the target function is complex, dependent on cellular machinery, or can be linked to cellular fitness, benefiting from continuous evolution formats that reduce labor. In vitro platforms are indispensable for evolving proteins toxic to cells, for achieving the largest possible library sizes, or for selections based primarily on binding affinity. Emerging trends, including the integration of ultrahigh-throughput screening, machine learning, and computational metrics like COMPSS [58], are blurring the lines between these platforms. By leveraging the quantitative data and experimental details in this guide, researchers can make an informed decision, selecting and optimizing the directed evolution platform that most efficiently navigates the fitness landscape toward their desired biomolecular function.
This guide provides an objective comparison of in vivo and in vitro directed evolution platforms, contextualized within advanced enzyme engineering research. We analyze their performance based on throughput, control, and efficiency metrics, using the engineering of hydrocarbon-producing enzymes like the cytochrome P450 fatty acid decarboxylase, OleTJE, as a representative case [9]. Supporting experimental data is synthesized into comparative tables to guide platform selection for research and development applications.
Directed evolution (DE) mimics natural selection in the laboratory to generate biomolecules with enhanced or novel properties. The process involves iterative cycles of diversity generation and screening or selection for improved variants [5]. The choice between in vivo (within living cells) and in vitro (in a cell-free system) platforms is fundamental, influencing the scale, scope, and outcome of an enzyme engineering campaign.
For engineering hydrocarbon-producing enzymes like alkane/alkene synthases, the challenge is particularly acute. The target molecules are often insoluble, gaseous, or chemically inert, making their detection and dynamic coupling to cellular fitness difficult [9]. This case study dissects how different evolution platforms address these challenges, providing a framework for selecting the optimal strategy.
Platform Comparison: In Vivo vs. In Vitro Directed Evolution
The table below summarizes the core characteristics of in vivo and in vitro directed evolution platforms.
Table 1: Comparative Analysis of In Vivo and In Vitro Directed Evolution Platforms
| Feature | In Vivo Directed Evolution | In Vitro Directed Evolution |
|---|---|---|
| Core Principle | Mutagenesis and selection occur within living cells [2]. | Mutagenesis and selection are performed in a cell-free environment [2]. |
| Diversity Generation | - Error-prone replication- CRISPR-based mutators [6]- Bacterial mutator strains (e.g., XL1-Red) [2] | - Error-prone PCR (epPCR)- DNA shuffling- Site-saturation mutagenesis [5] [6] |
| Typical Throughput | Very High (up to >10^9 with FACS/droplets) [14] | High (10^7 - 10^13 with ribosome/mRNA display) [2] |
| Key Advantage | - Direct selection for complex cellular functions- Realistic cellular environment (folding, modifications) [2] | - No transformation efficiency bottleneck- Access to toxic or unstable proteins [2] |
| Key Limitation | - Cellular fitness not always linked to desired trait- Mutagenesis not always target-specific [9] | - Poorly suited for optimizing complex metabolic pathways- Lack of native cellular environment [2] |
| Ideal Use Case | - Evolving metabolic pathways- Improving enzyme solubility/function in vivo [14] | - Engineering binding affinity (antibodies, receptors)- Evolving proteins toxic to cells [2] |
Case Study: Engineering a Hydrocarbon-Producing Enzyme
The Challenge of Hydrocarbon-Producing Enzymes
Enzymes like OleTJE, a cytochrome P450 that decarboxylates fatty acids to produce alkenes, are promising biocatalysts for "drop-in" biofuel production [9]. However, their native activity, stability, and specificity are often insufficient for industrial application. A primary obstacle in their directed evolution is the lack of a high-throughput, growth-coupled selection method. Hydrocarbon products do not inherently provide a selective advantage to a host cell, necessitating sophisticated screening or selection strategies [9].
Experimental Data from Platform Implementations
Different research approaches have employed various platforms to overcome these challenges. The following table summarizes quantitative outcomes from representative methodologies.
Table 2: Experimental Outcomes from Different Directed Evolution Approaches
| Evolution Platform / Technique | Target Enzyme/System | Key Experimental Outcome | Reference |
|---|---|---|---|
| In Vivo Continuous Evolution (Thermal-Responsive Pol I*) | α-Amylase | After iterative enrichment via microfluidic droplet screening, a mutant with a 48.3% improvement in activity was identified [14]. | [14] |
| In Vivo Base-Editing-Mediated Evolution | OsTIR1 (Auxin-inducible degron) | Directed evolution using cytosine and adenine base editors generated gain-of-function variants (e.g., S210A), leading to the improved AID 2.1 system [39]. | [39] |
| In Vitro Machine Learning-Assisted DE (MLDE) | Various (GB1, Dihydrofolate reductase, etc.) | On challenging, epistatic fitness landscapes, MLDE strategies consistently identified high-fitness variants more efficiently than typical directed evolution [59]. | [59] |
| Semi-Rational Design (Incremental Challenge) | Cytochrome P450 Fatty Acid Hydroxylase | The enzyme was progressively evolved into a highly efficient propane hydroxylase, an activity absent in the native enzyme [60]. | [60] |
Detailed Experimental Protocols
Protocol 1: In Vivo Continuous Evolution with Ultrahigh-Throughput Screening
This protocol, adapted from [14], is effective for evolving enzyme activity where no direct growth selection exists.
- System Construction: A two-plasmid system is established in E. coli.
- Mutator Plasmid (pSC101): A low-copy plasmid carrying a gene for an error-prone DNA polymerase I (Pol I) under the control of a thermo-inducible promoter (λPR–cI857).
- Target Plasmid (pET28a): A high-copy ColE1-based plasmid carrying the gene of interest (e.g., a hydrocarbon-producing enzyme).
- In Vivo Mutagenesis: Culture temperature is shifted to 37-42°C to induce expression of Pol I*. This enzyme preferentially replicates the target plasmid, introducing random mutations during replication [14].
- Library Generation: Cells are cultured for multiple generations to accumulate mutations in the target gene.
- Ultrahigh-Throughput Screening:
- For Secreted Enzymes (e.g., α-amylase): Single cells are encapsulated in microfluidic droplets with a fluorescent substrate. Active variants produce a fluorescent signal, enabling sorting of the entire droplet [14].
- For Metabolites (e.g., resveratrol): An in vivo transcription factor-based biosensor is used. Metabolite production triggers expression of a fluorescent protein, and high-producing cells are isolated using Fluorescence-Activated Cell Sorting (FACS) [14].
- Iteration: Enriched populations are subjected to further rounds of mutagenesis and screening.
The following diagram illustrates the core workflow of this in vivo method.
Protocol 2: CRISPR-Based In Vivo Directed Evolution
This protocol utilizes CRISPR-Cas systems for targeted diversity generation, as outlined in [6].
- gRNA Library Design: Design a library of guide RNAs (gRNAs) to target the specific genomic locus or gene of interest.
- Diversity Generation:
- DSB-Dependent Method: Co-express a Cas nuclease (e.g., Cas9) with the gRNA library. The resulting double-strand breaks are repaired via the error-prone Non-Homologous End Joining (NHEJ) pathway, introducing insertions and deletions (indels) at the target site [6].
- DSB-Independent Method (Base Editing): Use a catalytically impaired Cas protein fused to a deaminase (e.g., adenine or cytosine base editor). The complex is directed by the gRNA to introduce specific point mutations (C>T or A>G) within a defined window without creating double-strand breaks [39] [6].
- Selection & Screening: Apply selective pressure (e.g., presence of a toxic intermediate, limited nutrient) or employ a high-throughput screen (e.g., biosensor-based FACS) to identify improved variants [6].
- Variant Characterization: Sequence the evolved gene(s) and characterize the purified enzyme or strain for enhanced hydrocarbon production.
The diagram below contrasts these two primary CRISPR-based mechanisms.
The Scientist's Toolkit: Key Research Reagents and Solutions
Successful execution of directed evolution campaigns relies on specialized reagents and tools. The following table details essential solutions for setting up these platforms.
Table 3: Essential Research Reagents for Directed Evolution Platforms
| Reagent / Solution | Function | Example Application |
|---|---|---|
| Error-Prone PCR Kits | Introduces random point mutations across a gene during amplification in vitro [5]. | Creating diverse libraries for in vitro screening or display technologies. |
| CRISPR Base Editor Kits | Enables targeted point mutations at specific genomic loci without double-strand breaks [39] [6]. | Saturation mutagenesis of active site residues in vivo for hydrocarbon-producing enzymes. |
| Mutator Strains (e.g., E. coli XL1-Red) | Deficient in DNA repair pathways, leading to increased random mutation rates across the host genome and plasmids [2]. | Broad, untargeted in vivo evolution of plasmids carrying a target gene. |
| Microfluidic Droplet Generators | Encapsulates single cells and assay reagents in picoliter droplets for ultrahigh-throughput screening [14]. | Screening hydrolytic enzyme activity (e.g., α-amylase) using fluorescent substrates. |
| Transcription Factor-Based Biosensors | Links intracellular metabolite concentration to a reporter gene (e.g., GFP) output [14]. | FACS-based selection of high-producing strains in metabolic pathway engineering. |
The choice between in vivo and in vitro directed evolution is not a matter of superiority but of strategic alignment with project goals.
- For engineering metabolic pathways or optimizing enzyme function within the complex environment of a cell, in vivo platforms are indispensable. When coupled with biosensors or microfluidic screening, they provide a powerful route for evolving traits like hydrocarbon production [9] [14].
- For maximizing mutational diversity and screening library size for individual protein traits like binding affinity or stability, in vitro platforms remain the gold standard due to their freedom from cellular transformation [2].
- CRISPR-based methods represent a versatile hybrid, offering the target-specificity of rational design with the exploratory power of evolution directly in the genome [6].
- Machine Learning-assisted DE (MLDE) is emerging as a transformative force across both platforms, using computational models to predict high-fitness variants and navigate complex fitness landscapes more efficiently than traditional screening alone [59].
Researchers are increasingly adopting a hybridized approach, leveraging the strengths of multiple platforms to accelerate the engineering of robust biocatalysts for sustainable fuel and chemical production.
Directed evolution serves as a powerful tool in protein engineering, enabling the development of biomolecules with enhanced or novel functions by mimicking natural selection in a laboratory setting. [5] This process is primarily categorized into in vitro and in vivo approaches, each with distinct advantages and limitations. [2] The choice between these platforms significantly impacts the efficiency, depth, and practical outcomes of an evolution campaign. This case study objectively compares the performance of a novel in vivo directed evolution method against traditional in vitro techniques through the lens of a specific application: enhancing the organic acid tolerance and activity of β-glucosidase. [8] β-glucosidases are critical enzymes in industrial processes such as the bioconversion of lignocellulose to biofuels, but their efficiency is often hampered by inhibition from organic acids like formic acid generated during biomass pretreatment. [8] The comparative data and methodologies presented herein provide a framework for selecting appropriate evolution platforms for specific research goals.
Comparative Analysis of In Vivo vs. In Vitro Directed Evolution Platforms
Directed evolution strategies are broadly defined by where the crucial step of genetic diversification occurs. The following table summarizes the core distinctions between these platforms, which form the basis for the methodological comparison in this case study.
Table 1: Fundamental Comparison of In Vivo and In Vitro Directed Evolution Platforms
| Feature | In Vitro Directed Evolution | In Vivo Directed Evolution |
|---|---|---|
| Diversification Site | Outside a living cell (e.g., test tube) | Within a living host organism (e.g., yeast, bacteria) |
| Core Principle | Gene mutagenesis performed in vitro, followed by host transformation/transfection and screening. [2] | Mutagenesis and selection are performed simultaneously within the cellular environment. [2] |
| Typical Methods | Error-prone PCR, DNA shuffling, phage/mRNA/ribosome display. [2] [5] | Mutator strains (e.g., E. coli XL1-Red), orthogonal DNA replication systems (e.g., OrthoRep), prokaryotic in vivo evolution systems. [2] [14] [61] |
| Key Advantage | Can work with toxic or unstable protein sequences; library size not limited by transformation efficiency in pure systems. [2] | Occurs within a real-life cellular environment, accommodating complex factors like protein folding, post-translational modifications, and multi-protein interactions. [2] |
| Primary Limitation | Iterative cycles are laborious; screening in eukaryotic cells is complex; difficult to optimize complex metabolic pathways. [2] [5] | The analyzable library size can be restricted by host cell transformation efficiency; challenging to mutagenize target without cellular damage. [2] |
Experimental Comparison: SEP/DDS vs. Traditional Methods
The directed evolution of β-glucosidase for enhanced activity and organic acid tolerance provides a concrete example for comparing platform performance. The following experiment illustrates the application of a novel in vivo method against traditional in vitro techniques.
Experimental Objective and Rationale
The goal was to simultaneously improve the catalytic activity of Penicillium oxalicum 16 β-glucosidase (16BGL) and its tolerance to formic acid. [8] Organic acids like formic acid are potent inhibitors of enzymatic hydrolysis during lignocellulose processing, making this a critical industrial objective. Prior attempts using rational design (targeting surface charges and hydrogen bonds) and traditional directed evolution (error-prone PCR and DNA shuffling) failed to produce significant improvements, highlighting the limitations of these approaches for large, complex genes. [8]
Methodologies and Workflows
Novel In Vivo Approach: Segmental Error-Prone PCR (SEP) & Directed DNA Shuffling (DDS)
This approach combines in vitro mutagenesis with in vivo assembly and selection in yeast. [8]
Detailed Protocol:
- Gene Segmentation: The large 16bgl gene (~2.5 kb) was divided into four smaller, overlapping fragments (F1-F4). [8]
- In Vitro Mutagenesis: Fragments F1, F2, and F4 were subjected to independent error-prone PCR to introduce random mutations. Fragment F3 was left unmutated as a control. [8]
- In Vivo Assembly: The mutagenized fragments were co-transformed with a linearized plasmid vector into Saccharomyces cerevisiae. The yeast's highly efficient homologous recombination machinery assembled the fragments into a full-length, mutagenized gene within the plasmid. [8]
- Library Creation: The assembled plasmids constituted the primary mutant library. [8]
- Directed DNA Shuffling (DDS): Mutated fragments from identified positive variants after screening were amplified and reassembled in yeast to cumulatively combine beneficial mutations. [8]
The workflow for this method is illustrated below.
Traditional In Vitro Approach: Error-Prone PCR & DNA Shuffling
This standard method relies entirely on in vitro steps. [8]
Detailed Protocol:
- Error-Prone PCR: The full-length 16bgl gene is subjected to error-prone PCR using conditions that promote nucleotide misincorporation. [5] [8]
- Ligation: The resulting mutagenized DNA is ligated into an expression plasmid vector. [2]
- Transformation: The ligated plasmids are transformed into a host organism (e.g., E. coli) for protein expression. This step is a major bottleneck due to limited transformation efficiency. [2]
- Screening: Transformed colonies are screened for improved traits. [2]
- DNA Shuffling: Beneficial mutants identified from the screen are digested with DNase I, and the fragments are reassembled in a primerless PCR to recombine mutations. This process is complex and prone to generating reverse mutations. [8]
Performance Data and Results
The SEP/DDS method demonstrated clear advantages over the traditional in vitro approach in this application, as quantified by the following experimental outcomes.
Table 2: Experimental Outcomes of β-Glucosidase Directed Evolution Campaigns
| Evolution Method | Key Mutagenesis Characteristics | Documented Outcome on 16BGL | Primary Advantage |
|---|---|---|---|
| Rational Design | Targeted mutagenesis of 9 specific surface residues. [8] | No significant improvement in activity or acid tolerance. [8] | Requires no screening; based on structural hypothesis. |
| Traditional In Vitro | Error-prone PCR on full-length gene; DNA shuffling. [8] | Failed to produce improved variants. [8] | Well-established, standardized protocols. |
| Novel In Vivo (SEP/DDS) | Even distribution of mutations; reduced reverse mutations; in vivo recombination of positive fragments. [8] | Successfully generated variants with simultaneously enhanced activity and formic acid tolerance. [8] | Overcomes limitations of large gene size; efficiently combines beneficial mutations. |
The Scientist's Toolkit: Key Reagents for Directed Evolution
Successful directed evolution, whether in vivo or in vitro, relies on a suite of specialized reagents and genetic tools.
Table 3: Essential Research Reagents for Directed Evolution Experiments
| Reagent / Tool | Function / Description | Example Application |
|---|---|---|
| Mutator Strains | Host organisms with defective DNA repair pathways to elevate mutation rates. [2] | E. coli XL1-Red strain (deficient in mutD, mutS, mutT) used to evolve esterases and β-glucuronidases. [2] |
| Error-Prone PCR | A PCR technique that utilizes conditions (e.g., unbalanced dNTPs, Mn²⁺) to introduce random point mutations. [5] [8] | Standard method for creating random mutagenesis libraries for gene diversification. [5] |
| Specialized Vectors | Plasmid constructs designed for specific hosts, containing replicons, promoters, and selection markers. [8] [14] | pYAT22 vector for constitutive secretion in S. cerevisiae; pET28a with ColE1 ori for targeted mutagenesis in E. coli. [8] [14] |
| Orthogonal Replication Systems | Engineered genetic systems that mutate a target gene at high rates without affecting the host genome. [61] | OrthoRep in S. cerevisiae mutates user-selected genes at ~10⁻⁵ substitutions per base pair. [61] |
| Microfluidic Droplet Screening | Ultrahigh-throughput technology that encapsulates single cells in picoliter droplets for assay. [14] | Enabled screening of an α-amylase library, identifying a mutant with 48.3% improved activity. [14] |
This case study demonstrates that the choice of directed evolution platform is pivotal to success. The novel in vivo SEP/DDS approach proved uniquely capable of engineering complex, multi-property enhancements in a large β-glucosidase gene where both rational design and traditional in vitro evolution had failed. [8] Its key innovation lies in segmenting the mutagenesis problem and leveraging cellular machinery for efficient assembly, thereby ensuring an even distribution of beneficial mutations and mitigating common issues like reverse mutations. [8]
The field continues to advance with the development of continuous directed evolution platforms like PACE (phage-assisted) and OrthoRep, which can dramatically accelerate evolution campaigns by combining continuous mutagenesis and selection in a single vessel. [14] [61] Furthermore, the integration of ultrahigh-throughput screening methods (e.g., microfluidics, FACS with biosensors) and machine learning is beginning to address the critical bottleneck of identifying improved variants from vast libraries. [14] For researchers aiming to engineer enzymes for demanding industrial environments, such as those requiring organic acid tolerance, modern in vivo and continuous evolution systems offer a powerful and increasingly accessible path forward.
Directed evolution stands as a powerful protein engineering methodology that harnesses natural evolutionary principles on an accelerated timescale, enabling researchers to rapidly select biomolecular variants with properties optimized for specific applications [5]. This field has diversified significantly since its early in vitro beginnings in the 1960s, expanding from altering simple binding sites to improving complex enzyme kinetic parameters, substrate specificity, and performance in industrial biocatalysis [5]. The fundamental process involves two critical steps: generating genetic diversity in a parental sequence (library generation) and isolating variants with desired traits (selection), with the primary distinction between approaches lying in whether these steps occur within living cells (in vivo) or in laboratory environments (in vitro) [2] [5].
The choice between in vivo and in vitro evolution platforms carries significant implications for research outcomes, efficiency, and applicability. While traditional in vitro methods have proven powerful for optimizing proteins, they often face limitations including host cell transformation efficiency restrictions and difficulties in reproducing complex intracellular environments [2]. In vivo systems address these challenges by performing diversification and selection within living cells, providing a natural environment affected by cellular parameters like ion concentrations, pH, folding mechanisms, and post-translational modifications that profoundly influence protein function [2]. This comparative analysis examines genomic evidence and experimental data from both platforms to elucidate their respective advantages, limitations, and optimal applications in modern biotechnology and drug development.
Methodological Approaches and Key Technologies
In Vivo Directed Evolution Systems
In vivo directed evolution systems leverage the complex cellular machinery of living organisms to generate diversity and select functional variants, simulating natural evolutionary processes within controlled laboratory settings. These systems utilize various mutational mechanisms operating within prokaryotic or eukaryotic cells:
Prokaryotic Mutator Strains: Engineered bacterial strains with enhanced mutation rates serve as foundational in vivo evolution platforms. The commercially available E. coli XL1-Red strain exemplifies this approach, featuring deficiencies in DNA repair genes (mutD, mutS, and mutT) that elevate mutation frequencies to approximately 1 base change per 2,000 nucleotides [2]. This system has successfully modified substrate specificity in Pseudomonas fluorescens esterase to hydrolyze sterically hindered 3-hydroxy esters—key components in epothilone synthesis—and shifted the pH activity optimum of Lactobacillus gasseri ADH beta-glucuronidase from acidic to neutral ranges for broader application across host organisms [2].
Specialized Enzymatic Systems: Beyond simple mutator strains, researchers have developed more targeted in vivo approaches. One innovative system exploits an engineered error-prone DNA polymerase I (Pol I) that preferentially mutagenizes specific plasmid regions, achieving mutation rates of 8.1 × 10⁻⁴ mutations per base pair—an 80,000-fold increase over natural levels [2]. This technology successfully evolved TEM-1 β-lactamase variants with 150-fold increased resistance to the antibiotic aztreonam [2]. Recent advancements incorporate tunable thermal-responsive systems; researchers developed a temperature-sensitive platform using engineered repressor cI857* to control error-prone Pol I expression, coupled with genomic MutS defects for mutation fixation, achieving a 600-fold increase in targeted mutation rates in E. coli [14].
Multiplex Automated Genome Engineering (MAGE): This high-throughput approach enables simultaneous optimization of multiple genes within complex biosynthetic pathways. Utilizing E. coli EcNR2 expressing bacteriophage λ-Red ssDNA-binding protein β, MAGE repeatedly incorporates mutant oligonucleotides into lagging DNA strands during replication [2]. Under optimal conditions, approximately 30% of cell populations accumulate targeted modifications each cycle, potentially generating billions of genetic variants. Applied to the 1-deoxy-d-xylulose-5-phosphate (DXP) pathway, MAGE rapidly isolated strains with fivefold increased lycopene production within just three days [2].
In Vitro Directed Evolution Systems
In vitro directed evolution methodologies perform diversification and selection outside living organisms, offering distinct advantages for manipulating biomolecules under controlled conditions:
Pure In Vitro Systems: Methodologies like mRNA and ribosome display conduct both diversification and selection entirely in cell-free environments. mRNA display creates covalent mRNA-protein linkages using puromycin, while ribosome display maintains non-covalent protein-mRNA-ribosome complexes during selection [2]. These approaches circumvent transformation efficiency limitations, expanding screenable library sizes by several orders of magnitude compared to bacterial or phage displays [2]. Additionally, they uniquely accommodate protein sequences that prove unstable or toxic to living cells, significantly expanding the scope of evolvable biomolecules [2].
Error-Prone Artificial DNA Synthesis (epADS): This emerging approach harnesses base errors occurring during chemical oligonucleotide synthesis under specific controlled conditions as a source of random mutagenesis [27]. The process involves: (1) in silico design of overlapping oligonucleotides covering the target DNA; (2) chemical synthesis under error-prone conditions (e.g., high water content, mixed dNTP monomers); (3) assembly into double-stranded DNA via annealing or PCR; (4) cloning into suitable vectors; and (5) variant selection or screening [27]. Applied to fluorescent protein genes (EmGFP, mCherry, BFP, mBanana), epADS introduced diverse mutations including substitutions and indels randomly distributed across sequences, achieving 200-4000-fold fluorescence diversification and demonstrating particular effectiveness in optimizing regulatory genetic parts and synthetic gene circuits [27].
Library Generation Methods: Traditional in vitro diversification techniques include error-prone PCR (epPCR), which utilizes polymerases without proofreading capability to introduce random mutations, though with biases toward transitions and limited contiguous mutations [5] [27]. DNA shuffling and related methods (RACHITT, StEP, NExT) enable recombination of homologous sequences, accelerating evolution by combining beneficial mutations from different parental sequences [5] [27]. While these methods have successfully evolved numerous enzymes and pathways, they often require extensive manual intervention and multiple molecular biology steps between diversification rounds [14].
Comparative Performance Analysis
Quantitative Comparison of Evolution Platforms
The table below summarizes key performance metrics for major in vivo and in vitro directed evolution platforms, highlighting their respective capabilities and limitations:
Table 1: Performance Metrics of Directed Evolution Platforms
| Platform | Mutation Rate/Frequency | Library Size | Key Advantages | Primary Limitations |
|---|---|---|---|---|
| In Vivo Systems | ||||
| E. coli XL1-Red mutator strain | 1 mutation/2,000 bp [2] | ~10⁸-10⁹ variants [2] | Simple system; natural cellular environment; post-translational modifications [2] | Uncontrolled genome-wide mutagenesis; biased mutation spectrum [2] [5] |
| Error-prone DNA Pol I system | 8.1 × 10⁻⁴ mutations/bp (80,000× natural rate) [2] | Limited by plasmid size and replication | Preferentially mutagenizes target plasmid regions [2] | Limited to ~3kb from ColE1 origin; maximal in first 700bp [2] |
| MAGE | High efficiency (≈30% incorporation/cycle) [2] | Up to 15 billion genetic variants [2] | Multiplexed; targets specific genes/pathways; rapid cycling [2] | Complex setup; requires specialized oligonucleotide design [2] |
| Thermal-responsive system | 600× increased mutation rate [14] | Compatible with ultrahigh-throughput screening | Tunable mutagenesis; compatible with biosensor coupling [14] | Requires temperature shifts; optimization needed [14] |
| In Vitro Systems | ||||
| mRNA/ribosome display | Varies with method | 10¹²-10¹⁴ variants [2] | No transformation bottlenecks; toxic protein compatible [2] | Limited to affinity-based selection; difficult for complex functions [2] |
| Error-prone PCR | Varies with polymerase | Limited by transformation efficiency | Easy to perform; no prior knowledge needed [5] | Biased toward transitions; limited contiguous mutations [5] [27] |
| DNA shuffling | Depends on homology | Limited by transformation efficiency | Recombines beneficial mutations [5] [27] | Requires high sequence homology [5] |
| epADS | 0.05%-0.17% total mutation frequency [27] | Limited by transformation efficiency | Random indels and substitutions; diversifies regulatory elements [27] | Requires oligonucleotide synthesis; optimization needed for error rate [27] |
Experimental Outcomes and Efficacy Data
Direct comparisons of in vivo and in vitro platforms across various protein engineering challenges reveal distinct performance patterns:
Table 2: Experimental Outcomes Across Evolution Platforms
| Target System | Evolution Platform | Experimental Outcome | Key Mutations/Mechanisms | Reference |
|---|---|---|---|---|
| β-lactamase | In vivo: Error-prone Pol I | 150-fold increase in aztreonam resistance [2] | Multiple substitutions in enzyme active site region [2] | [2] |
| β-lactamase | In vitro: RAISE | Improved activity with random insertions/deletions [5] | Short indels introducing structural flexibility [5] | [5] |
| Esterase | In vivo: XL1-Red | Altered substrate specificity for hindered 3-hydroxy ester [2] | Undefined but distributed across sequence [2] | [2] |
| α-Amylase | In vivo: Thermal-responsive + droplet screening | 48.3% activity improvement [14] | Selected via ultrahigh-throughput microfluidic screening [14] | [14] |
| Resveratrol pathway | In vivo: Thermal-responsive + biosensor | 1.7-fold increased production [14] | Multiple pathway mutations selected via transcription factor biosensor [14] | [14] |
| Fluorescent proteins | In vitro: epADS | 200-4000× fluorescence diversification [27] | Random substitutions and indels across sequence [27] | [27] |
Genomic and Structural Evidence
Mutation Pattern Analysis
Comparative genomic analysis of evolved strains reveals distinctive mutational signatures between in vivo and in vitro platforms. In vivo evolution often follows germline gene-defined substitution patterns, as evidenced in antibody evolution where somatic hypermutation targets not only complementarity determining regions but also framework regions and even distal protein core residues in a germline-dependent manner [54]. Analysis of IgG sequences from human bone marrow demonstrates that different immunoglobulin germline genes (IGHV1-8, IGHV3-11, IGHV5-51) exhibit unique substitution patterns extending well beyond traditional antigen-binding sites, suggesting the immune system evolves antibodies along preferred trajectories encoded within germline sequences [54].
In vitro systems typically generate more randomized mutational distributions, though with methodological biases. Error-prone PCR favors transition mutations over transversions, while DNA shuffling creates chimeric sequences with crossovers in regions of high homology [5] [27]. The epADS approach introduces more random mutation profiles including indels and substitutions broadly distributed across target sequences, as demonstrated in fluorescent protein evolution where mutations appeared throughout the genes without apparent positional bias [27].
Functional Pathway Optimization
In vivo platforms particularly excel at optimizing complex biological pathways and multi-protein systems due to their natural cellular context. The resveratrol biosynthetic pathway evolution exemplifies this advantage, where an in vivo system coupled with transcription factor biosensors successfully identified mutant combinations yielding 1.7-fold production increases [14]. Similarly, MAGE technology simultaneously optimized multiple genes in the DXP pathway, rapidly achieving fivefold lycopene yield improvements by exploring combinatorial mutations across pathway enzymes [2].
These successes highlight in vivo systems' capacity to identify mutations that optimize not only individual enzyme activities but also pathway flux, regulatory interactions, and metabolic balancing—challenges difficult to recreate in vitro. The integration of biosensors with in vivo evolution creates particularly powerful platforms for metabolic engineering, where fluorescence-activated cell sorting enables ultrahigh-throughput screening of pathway variants based on product accumulation [14].
Technical Workflows and Experimental Design
In Vivo Directed Evolution Workflow
The following diagram illustrates the generalized workflow for in vivo directed evolution, integrating key steps from multiple platforms:
In Vitro Directed Evolution Workflow
The diagram below outlines the core process for in vitro directed evolution, highlighting critical differences from in vivo approaches:
Essential Research Reagents and Tools
Successful implementation of directed evolution campaigns requires specific reagents and tools optimized for each platform:
Table 3: Essential Research Reagents for Directed Evolution
| Reagent/Tool | Function | Platform Applicability | Examples/Specifications |
|---|---|---|---|
| Mutator Strains | Enhanced mutation rates for in vivo diversification | In vivo | E. coli XL1-Red (mutD, mutS, mutT deficient) [2] |
| Error-Prone Polymerases | Introduce random mutations during PCR | In vitro | Taq polymerase without proofreading; specialized mixes for transition/transversion control [5] [27] |
| Specialized Vectors | Target gene maintenance and expression | Both | ColE1-based plasmids for Pol I mutagenesis; expression vectors with inducible promoters [2] [14] |
| Biosensors | Link desired phenotype to selectable output | Primarily in vivo | Transcription factor-based fluorescent reporters for metabolic products [14] |
| Microfluidic Systems | Ultrahigh-throughput screening | Both | Droplet-based encapsulation and sorting [14] |
| Selection Markers | Enrichment of functional variants | Both | Antibiotic resistance; nutrient auxotrophy; fluorescence [2] [14] |
| DNA Synthesis Reagents | Oligonucleotide synthesis for library generation | In vitro | Controlled quality reagents for epADS; mixed nucleotide phosphoramidites [27] |
The genomic evidence comparing in vivo and in vitro evolved strains reveals complementary strengths that recommend specific applications for each platform. In vivo systems excel at optimizing complex functions requiring cellular context—including metabolic pathways, multi-protein interactions, and functional traits coupled to cellular fitness—while providing more biologically relevant post-translational modifications and folding environments [2] [14]. The emergence of integrated biosensor systems enables ultrahigh-throughput screening of complex phenotypes like metabolite production, significantly expanding in vivo evolution capabilities beyond traditional growth-based selection [14].
In vitro platforms maintain advantages for evolving biomolecules with requirements incompatible with cellular systems—including toxic proteins, components requiring non-natural substrates, or functions needing specialized reaction conditions [2]. Pure in vitro methods (mRNA/ribosome display) particularly excel at affinity maturation and optimizing molecular binding characteristics, benefiting from enormous library sizes unconstrained by transformation efficiency [2] [5].
Future developments will likely focus on orthogonal systems that restrict mutagenesis specifically to target genes without affecting host genomes, enhanced recombination technologies for efficiently exploring sequence space, and computational integration for predicting functional variants [2] [27]. As comparative genomics advances, researchers are increasingly identifying evolution patterns specific to gene families and biological contexts, enabling smarter library design that incorporates natural evolutionary trajectories [54]. These developments will further blur distinctions between in vivo and in vitro approaches, potentially enabling hybrid platforms that combine the strengths of both methodologies for more efficient biomolecule engineering.
Directed evolution, the laboratory process of mimicking natural selection to engineer biomolecules with desired traits, has become an indispensable tool in basic and applied biology. This iterative two-step process, involving genetic diversification followed by screening or selection, has traditionally been divided into two main camps: in vitro and in vivo approaches [44]. In vitro systems, such as mRNA and ribosome displays, allow for the generation of exceptionally large libraries and the evolution of proteins that might be unstable or toxic in cells [2]. Conversely, in vivo systems perform both diversification and selection within living cells, providing the distinct advantage of a natural cellular environment. This ensures proper protein folding, post-translational modifications, and functional assessment within complex multi-protein interactions, which are difficult to replicate in artificial systems [2].
The field is now undergoing a transformative shift, driven by three key technological frontiers: the integration of artificial intelligence (AI) for predictive design and optimization, the development of continuous evolution systems that enable unprecedented scalability, and the expansion into non-conventional hosts for specialized applications. This guide provides a comparative analysis of these advanced directed evolution platforms, offering experimental data, detailed protocols, and key reagent information to inform the selection of an appropriate strategy for specific research goals in drug development and protein engineering.
Comparative Performance Analysis of Directed Evolution Platforms
The following tables summarize the quantitative performance, key features, and optimal use cases for modern directed evolution platforms, providing a objective basis for comparison.
Table 1: Quantitative Performance Comparison of Directed Evolution Technologies
| Technology / System | Library Size/Diversity | Mutation Rate/Frequency | Key Performance Metrics | Experimental Evidence |
|---|---|---|---|---|
| CRISPR-based Base Editing | Limited by delivery efficiency, but highly diverse at target loci [6] | Precitable C>T and A>G conversions via cytosine (BE) and adenine (AE) base editors [39] | Successfully generated gain-of-function OsTIR1 variants (e.g., S210A) for improved AID 2.1 system [39] | Base-editing-mediated mutagenesis and functional screening in hiPSCs [39] |
| In Vivo Mutator Strains (e.g., XL1-Red) | Population of hundreds of millions to billions of cells [2] | ~1 mutation per 2,000 bp (5,000-fold increase over wild-type) [2] | Shifted optimal pH of L. gasseri ADH beta-glucuronidase from 5.0 to neutral [2] | Selection on indicator plates (Neutral Red/Crystal Violet) [2] |
| AI-Guided Evolution (DeepDE) | Iterative exploration using ~1,000 variant training libraries [11] | Utilizes triple mutants per round to explore vast sequence space [11] | 74.3-fold increase in GFP activity over 4 rounds, surpassing superfolder GFP [11] | Multiple rounds of fluorescence-based screening and model retraining [11] |
| MAGE (Multiplex Automated Genome Engineering) | Up to 15 billion genetic variants in a culture [2] | High-efficiency oligonucleotide incorporation via λ-Red β protein [2] | >5-fold increase in lycopene production in E. coli DXP pathway [2] | Repeated oligonucleotide integration and selection for pathway output [2] |
| Error-Prone Pol I System | Targeted mutagenesis within ~3 kb of plasmid origin [2] | 8.1 x 10⁻⁴ mutations/bp (80,000-fold increase) [2] | 150-fold increase in aztreonam resistance for TEM-1 β-lactamase [2] | Selection on increasing concentrations of aztreonam [2] |
Table 2: Feature Comparison of In Vivo vs. In Vitro Platform Strengths and Weaknesses
| Feature | In Vivo Platforms | In Vitro Platforms |
|---|---|---|
| Key Advantage | Functional selection in a natural cellular context; ideal for metabolic pathways and essential genes [2] | Vast library sizes (e.g., >10¹²); can evolve toxic/unstable proteins [2] [44] |
| Primary Limitation | Library size constrained by transformation/transfection efficiency [2] | Lack of cellular environment; difficult to select for complex traits like fitness [2] |
| Best Suited For | • Engineering multi-protein complexes• Optimizing biosynthetic pathways• Studying essential genes via conditional degradation [39] [2] | • Affinity maturation (e.g., antibodies)• Engineering individual enzymes for in vitro use• When protein is toxic to cells [2] [44] |
| Automation & Continuous Evolution Potential | High potential with advanced tools like MAGE and CRISPR-directed continuous evolution in chemostats [2] [6] | Inherently continuous in systems like mRNA display; easily automated with microfluidics [44] |
Experimental Protocols for Advanced Directed Evolution
Protocol: Base-Editing-Mediated Directed Evolution
This protocol, adapted from a recent study, details the use of base editors for directed protein evolution in human induced pluripotent stem cells (hiPSCs) [39].
- sgRNA Library Design: Design a custom sgRNA library tiling across all coding exons of your target gene (e.g., OsTIR1). Each sgRNA should position a targetable cytosine or adenine within the activity window of the base editor [39].
- Cell Line Preparation: Generate a stable hiPSC line expressing the target protein (e.g., an OsTIR1-based degron system) integrated into a safe harbor locus (e.g., AAVS1) [39].
- Delivery: Co-transfect the cells with plasmids encoding a cytosine base editor (BE) or an adenine base editor (ABE) and the pooled sgRNA library [39].
- Functional Selection and Screening:
- Apply Selective Pressure: For the AID system, this involves culturing cells in the presence of auxin (e.g., 5-Ph-IAA) to select for OsTIR1 variants that confer desired properties like reduced basal degradation [39].
- Enrichment: Harvest cells that survive the selective pressure over multiple passages.
- Screen Clones: Isolve single-cell clones and screen them via Western blot or functional assays to quantify improvements in the target parameters (e.g., degradation kinetics, recovery rate) [39].
- Variant Identification: Amplify the target gene from selected clones by PCR and perform Sanger sequencing to identify the underlying beneficial mutations [39].
Protocol: AI-Guided Iterative Protein Evolution with DeepDE
This protocol outlines the DeepDE algorithm for directed evolution, which uses deep learning to guide the exploration of protein sequence space [11].
- Initial Library Construction: Create a diverse mutant library of the target protein (e.g., GFP) using random mutagenesis or saturation mutagenesis [11].
- First-Round Screening: Screen approximately 1,000 variants from the library for the desired activity (e.g., fluorescence intensity at 488 nm). This dataset (sequence + activity) forms the initial training set [11].
- Model Training: Train a deep learning model (DeepDE) on the collected sequence-activity data to learn the complex mapping between sequence and function [11].
- In Silico Prediction and Selection: Use the trained model to predict the activity of a vast number of virtual triple mutants. Select a new set of ~1,000 top-predicted triple mutants for experimental synthesis and testing [11].
- Iterative Evolution: The data from the newly tested variants is added to the training set, and the process (steps 3-5) is repeated for multiple rounds. With each iteration, the model becomes more accurate at predicting highly active sequences, leading to rapid performance improvements [11].
AI-Guided Directed Evolution Workflow
The Scientist's Toolkit: Essential Research Reagents
Successful execution of modern directed evolution experiments relies on a suite of specialized reagents and tools. The following table details key solutions for various stages of the workflow.
Table 3: Key Research Reagent Solutions for Directed Evolution
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| Cytosine Base Editor (BE) | Mediates precise C•G to T•A conversions in DNA without causing double-strand breaks [39]. | Creating diverse mutant libraries for directed protein evolution in hiPSCs [39]. |
| Adenine Base Editor (ABE) | Mediates precise A•T to G•C conversions in DNA without causing double-strand breaks [39]. | Creating diverse mutant libraries for directed protein evolution in hiPSCs [39]. |
| CRISPR/Cas9 System | RNA-guided nuclease that induces double-strand breaks (DSBs) for precise genome editing [6]. | Enabling targeted gene knock-in of degron tags or donor DNA templates via HDR [39]. |
| dTAG Ligands (e.g., dTAG13) | Bifunctional molecule that binds FKBP12F36V-degron and CRBN E3 ligase, inducing target degradation [39]. | A chemical tool for rapid protein degradation in the dTAG system; used in comparative degron studies [39]. |
| Auxin Analogues (e.g., 5-Ph-IAA) | Plant hormone derivative that induces interaction between OsTIR1/AFB2 and AID-tagged proteins, leading to degradation [39]. | The inducing ligand for the auxin-inducible degron (AID) system; used for selective pressure in evolution experiments [39]. |
| HaloPROTAC3 | Bifunctional ligand that binds HaloTag7-fusion proteins and VHL E3 ligase, inducing degradation [39]. | A chemical tool for rapid protein degradation in the HaloPROTAC system; used in comparative degron studies [39]. |
| DeepDE Algorithm | A deep learning model that uses data from ~1,000 variants to predict highly active protein sequences for the next round of evolution [11]. | Guiding iterative protein engineering to achieve dramatic improvements in activity, as demonstrated with GFP [11]. |
Integrated Workflows and Visualizing the Experimental Pipeline
A typical advanced directed evolution campaign, particularly one leveraging in vivo platforms in non-conventional hosts, integrates multiple modern technologies. The workflow begins with the selection of an appropriate host organism, which could range from traditional E. coli to hiPSCs, depending on the protein's requirements. The genetic diversity is then introduced using a method like CRISPR-base editing, which allows for targeted and efficient mutagenesis without double-strand breaks. The resulting cellular library is then subjected to a functional selection, such as survival under pressure from a ligand that induces protein degradation, to enrich for improved variants. Selected clones are screened with medium-to-high throughput, and the best hits are sequenced. The sequence and functional data from these hits can optionally be used to train an AI model, which predicts the next, more refined library to test, creating a powerful, iterative cycle of improvement.
Integrated Directed Evolution Workflow
Conclusion
The choice between in vivo and in vitro directed evolution is not a matter of superiority, but of strategic alignment with project goals. In vivo platforms offer unparalleled physiological relevance for evolving proteins destined for therapeutic use in mammalian systems or for optimizing complex cellular phenotypes. In contrast, in vitro methods provide unmatched control and library diversity for rigorous mechanistic studies and evolving properties like binding affinity. The future of directed evolution lies in the intelligent integration of both approaches, leveraging the strengths of each in a complementary workflow. Emerging technologies—such as CRISPR-based diversification, continuous evolution in multi-host systems, and machine learning-guided library design—are blurring the lines between these platforms, promising to accelerate the discovery of next-generation enzymes, therapeutics, and biosynthetic pathways. For researchers, mastering this comparative landscape is essential for efficiently navigating the protein fitness landscape and delivering innovative solutions in biomedicine and industrial biotechnology.
References
- 1. A Technical Guide to Directed Evolution for Enhancing ... [https://www.ranomics.com/a-technical-guide-to-directed-evolution-for-enhancing-protein-stability-and-function]
- 2. Technologies of directed protein evolution in vivo - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11115086/]
- 3. A chimeric viral platform for directed evolution in ... [https://www.nature.com/articles/s41467-025-59438-2]
- 4. -based discovery of ligands for Directed restimulation... evolution in vivo [https://www.nature.com/articles/s41551-025-01470-0?error=cookies_not_supported&code=ea10c651-38dc-4cc8-a3e1-658593ab3c0a]
- 5. A primer to directed evolution: current methodologies and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10074555/]
- 6. Advanced research and exploration of CRISPR technology ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975025001193]
- 7. Frontiers | Immune counter- evolution : immortalized B cell clones can... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1648717/full]
- 8. A novel directed evolution approach for co-evolution of β- ... [https://www.sciencedirect.com/science/article/abs/pii/S0168165625000422]
- 9. Directed evolution of hydrocarbon-producing enzymes [https://biotechnologyforbiofuels.biomedcentral.com/articles/10.1186/s13068-025-02689-4]
- 10. Directed evolution [https://en.wikipedia.org/wiki/Directed_evolution]
- 11. An iterative deep learning-guided algorithm for directed ... [https://www.sciencedirect.com/science/article/pii/S2589004225015858]
- 12. Directed evolution of engineered virus-like particles with ... [https://www.nature.com/articles/s41587-024-02467-x]
- 13. In vitro continuous protein evolution empowered by ... [https://www.sciencedirect.com/science/article/pii/S2405471223001151]
- 14. Ultrahigh-throughput screening-assisted in vivo directed evolution for enzyme engineering | Biotechnology for Biofuels and Bioproducts | Full Text [https://biotechnologyforbiofuels.biomedcentral.com/articles/10.1186/s13068-024-02457-w]
- 15. An in vivo target mutagenesis system for multiple hosts [https://www.sciencedirect.com/science/article/abs/pii/S0167779925001325]
- 16. Navigating directed evolution efficiently: optimizing ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2024.1439259/full]
- 17. In vivo hypermutation and continuous evolution | Nature Reviews Methods Primers [https://www.nature.com/articles/s43586-022-00119-5]
- 18. Growth-coupled continuous directed evolution by MutaT7 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12016552/]
- 19. Advancing protein evolution with inverse folding models ... [https://www.sciencedirect.com/science/article/abs/pii/S0092867425006804]
- 20. A chimeric viral platform for directed evolution in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12059018/]
- 21. A Rapid and Scalable Platform for In Planta Directed ... [http://gaolab.genetics.ac.cn/en/new/202510/t20251003_780459.html]
- 22. Directed evolution of engineered virus-like particles with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12085157/]
- 23. In vivo hypermutation and continuous evolution - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10108624/]
- 24. Directed Evolution in Mammalian Cells - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC8063495/]
- 25. VEGAS as a Platform for Facile Directed Evolution in ... [https://www.sciencedirect.com/science/article/pii/S0092867419306221]
- 26. VEGAS as a Platform for Facile Directed Evolution in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6660416/]
- 27. In vitro generation of genetic diversity for directed evolution ... [https://www.nature.com/articles/s42003-024-06340-0]
- 28. Rediscovering Inosine-EpPCR to Create Starting Libraries for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12293787/]
- 29. Reducing mutational bias in random protein libraries [https://www.sciencedirect.com/science/article/abs/pii/S0003269704009364]
- 30. CRISPR-StAR enables high-resolution genetic screening ... [https://www.nature.com/articles/s41587-024-02512-9]
- 31. Applications of multiplexed CRISPR–Cas for genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322214/]
- 32. Comparing Gene Editing Platforms: CRISPR vs. Traditional ... [https://blog.crownbio.com/comparing-gene-editing-platforms-crispr-vs-traditional-methods]
- 33. CMN Weekly (26 September 2025) [https://crisprmedicinenews.com/news/cmn-weekly-26-september-2025-your-weekly-crispr-medicine-news/]
- 34. CRISPR Clinical Trials to Follow [https://www.synthego.com/blog/crispr-clinical-trials/]
- 35. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 36. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 37. Biocatalysis in Transforming Biofuel Technologies [https://pubmed.ncbi.nlm.nih.gov/40613142/]
- 38. Enzyme Engineering Shaping The Future Of Sustainable ... [https://www.longdom.org/open-access/-1103227.html]
- 39. Systematic comparison and base-editing-mediated ... [https://www.nature.com/articles/s41467-025-61848-1]
- 40. Therapeutic Proteins Market Report 2025 [https://www.researchandmarkets.com/reports/5733857/therapeutic-proteins-market-report?srsltid=AfmBOootZjyHwK6eENun7-N6QlvG4WIU0YZYrOulliLQjxKN1slnhYi7]
- 41. Protein Therapeutics Market Share & Opportunities 2025- ... [https://www.coherentmarketinsights.com/industry-reports/protein-therapeutics-market]
- 42. Why Biopharma Companies are Prioritizing Affinity ... [https://precisionantibody.com/why-biopharma-companies-are-prioritizing-affinity-maturation-2025/]
- 43. Automated in vivo enzyme engineering accelerates ... [https://www.nature.com/articles/s41467-024-46574-4]
- 44. Directed Evolution: Past, Present and Future - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4344831/]
- 45. Nickase fidelity drives EvolvR-mediated diversification in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12009436/]
- 46. Strategies of microbial cheater control [https://www.sciencedirect.com/science/article/abs/pii/S0966842X03003366]
- 47. In Vitro vs In Vivo: Complete Comparison + Selection Guide [https://www.assaygenie.com/in-vitro-vs-in-vivo-complete-comparison-selection-guide-research-methods/?srsltid=AfmBOorlBERVNBvOd9lOzE4ViOdPyUgaEfSox_kvT5IiqglSq67lmK49]
- 48. Complex in vitro model: A transformative model in drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10828975/]
- 49. Characterizing and engineering post-translational ... [https://www.nature.com/articles/s41467-025-60526-6]
- 50. Crosstalk between protein post-translational modifications and ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-023-01380-1]
- 51. Advanced In Vitro Models: Opportunities and Challenges ... [https://www.lek.com/insights/hea/us/ei/advanced-vitro-models-opportunities-and-challenges-drug-development]
- 52. Integrating deep learning for post-translational ... [https://www.sciencedirect.com/science/article/pii/S0021925825023701]
- 53. How evolution builds up complexity?: In vitro ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8938154/]
- 54. In Vitro Evolution of Antibodies Inspired by In Vivo Evolution [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2018.01391/full]
- 55. In vivo continuous evolution via phenotypic sorting to ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925003440]
- 56. Directed evolution of aminoacyl-tRNA synthetases through ... [https://www.nature.com/articles/s41467-025-60120-w]
- 57. AI-powered high-throughput digital colony picker platform ... [https://www.nature.com/articles/s41467-025-63929-7]
- 58. Computational scoring and experimental evaluation of ... [https://www.nature.com/articles/s41587-024-02214-2]
- 59. Evaluation of machine learning-assisted directed evolution ... [https://www.sciencedirect.com/science/article/abs/pii/S2405471225002200]
- 60. climbing fitness peaks one amino acid at a time [https://pmc.ncbi.nlm.nih.gov/articles/PMC2703427/]
- 61. Rapid Mutation & Continuous Directed Evolution in Vivo [https://www.broadinstitute.org/videos/rapid-mutation-continuous-directed-evolution-vivo-continuous-directed-evolution]