The Complete Molecular Dynamics Simulation Workflow: From Foundational Concepts to Clinical Applications
This comprehensive guide details the complete molecular dynamics (MD) simulation workflow, addressing the critical needs of researchers, scientists, and drug development professionals.
The Complete Molecular Dynamics Simulation Workflow: From Foundational Concepts to Clinical Applications
Abstract
This comprehensive guide details the complete molecular dynamics (MD) simulation workflow, addressing the critical needs of researchers, scientists, and drug development professionals. It bridges foundational theory with practical application, covering the setup and execution of physically accurate simulations, advanced analysis techniques for extracting biological insights, and systematic troubleshooting for common computational challenges. Furthermore, the article provides rigorous methodologies for validating simulation results against experimental data and comparative analysis of different computational approaches, with a focus on reproducibility and best practices. By synthesizing these elements, this resource aims to empower practitioners to leverage MD simulations effectively for biomedical discovery and rational drug design.
Laying the Groundwork: Core Principles and System Setup for Robust MD Simulations
Molecular dynamics (MD) simulation is a computational technique that predicts the physical movements of atoms and molecules over time. By solving Newton's equations of motion for a system of interacting particles, MD provides insights into dynamic processes at an atomic level, serving as a virtual microscope for researchers. The power of an MD "engine" lies in its integration algorithm and the underlying force field—a mathematical model that describes the potential energy of a system as a function of its atomic coordinates. These force fields encode the physics of atomic interactions, determining the accuracy and applicability of simulations across diverse fields from drug discovery to materials science [1] [2]. This technical guide examines the core components of MD engines, focusing on the integration of Newton's equations and the formulation of modern force fields, framed within the broader context of molecular dynamics simulation workflows for scientific research.
Mathematical Foundation: Integrating Newton's Equations
At its core, a molecular dynamics simulation numerically integrates Newton's second law of motion for each atom in the system. The fundamental equation is:
F_i = m_ia_i
Where Fi is the force exerted on atom i, mi is its mass, and a_i is its acceleration. The force is the negative gradient of the potential energy function U(r^N), which depends on the positions of all N atoms:
F_i = -∇_i U(r^N)
The potential energy U(r^N) is defined by the force field. To numerically integrate these equations, MD engines typically employ the Velocity Verlet algorithm, which is time-reversible and conserves energy well for small time steps. This algorithm updates positions and velocities as follows [2]:
- r(t + Δt) = r(t) + v(t)Δt + (1/2)a(t)Δt²
- v(t + Δt/2) = v(t) + (1/2)a(t)Δt
- Compute forces F(t + Δt) and thus a(t + Δt) from the new positions
- v(t + Δt) = v(t + Δt/2) + (1/2)a(t + Δt)Δt
The choice of time step (Δt) is critical, typically ranging from 1-2 femtoseconds, to balance computational efficiency with energy conservation.
Table 1: Key Components of Newtonian Integration in MD
| Component | Mathematical Expression | Role in MD Engine | Typical Values/Examples |
|---|---|---|---|
| Time Step (Δt) | - | Determines interval between successive calculations of atomic positions | 1-2 fs |
| Integration Algorithm | Velocity Verlet equations | Updates atomic positions and velocities while conserving energy | Time-reversible, symplectic |
| Potential Energy (U) | U(r^N) | Defines total energy of the system based on atomic coordinates | Calculated from force field |
| Force (Fi ) | Fi = -∇iU(r^N) | Determines acceleration of each atom | Negative gradient of potential energy |
Force Fields: The Physics Engine of MD
Force fields are parametric mathematical functions that describe a system's potential energy. Traditional force fields decompose the potential energy into bonded terms (interactions between directly connected atoms) and non-bonded terms (interactions between all atoms) [3] [2].
Classical Force Field Formulation
The total potential energy in a classical force field is typically expressed as:
U_total = U_bonded + U_non-bonded
Where the bonded term includes:
U_bonded = U_bonds + U_angles + U_torsions
And the non-bonded term includes:
U_non-bonded = U_van der Waals + U_electrostatics
These components are calculated using specific mathematical functions:
- Bond Stretching: Harmonic potential approximating vibration between two covalently bonded atoms: U_bonds = ∑bonds kb (r - r0)_²
- Angle Bending: Harmonic potential for the angle between three connected atoms: U_angles = ∑angles kθ (θ - θ0)_²
- Torsional Dihedral: Periodic function for rotation around a central bond: U_torsions = ∑dihedrals k_φ [1 + cos(nφ - δ)]
- van der Waals: Lennard-Jones potential describing short-range repulsion and long-range dispersion: U_vdW = ∑i
- Electrostatic: Coulomb's law for interactions between partial atomic charges: U_electrostatics = ∑i
Table 2: Comparison of Major Biomolecular Force Fields
| Force Field | Chemical Coverage | Specialization | Key Features | Parameter Derivation |
|---|---|---|---|---|
| AMBER [3] | Proteins, Nucleic Acids | Structural Accuracy | RESP charges; Fewer torsional potentials | Fitted to QM electrostatic potential |
| CHARMM [3] | Proteins, Nucleic Acids | Structural Accuracy, Non-bonded Energies | - | Crystal structures, Lattice energies |
| OPLS4 [4] | Small molecules, Biologics, Materials Science | Thermodynamic Properties | Improved charged groups, sulfur moieties | State-of-the-art QM engine, experimental validation |
| GROMOS [3] | Biomolecules | Thermodynamic Properties | United-atom option for aliphatic hydrogens | Heats of vaporization, liquid densities, solvation properties |
Advanced Force Field Formulations
Beyond traditional fixed-charge force fields, several advanced formulations address specific limitations:
Polarizable Force Fields: Incorporate many-body polarization effects that vary with chemical and physical environments, addressing the limitation of fixed atomic charges. Examples include AMOEBA+, which improves electrostatic interactions by incorporating charge penetration and intermolecular charge transfer [3].
Reactive Force Fields: Enable chemical reactions within MD simulations. ReaxFF, for example, uses a bond-order formalism that allows bonds to form and break during simulations, making it particularly valuable for studying combustion, catalysis, and materials failure [1].
Machine Learning Potentials: Recent approaches use graph neural networks to replace human-curated atom typing schemes. Espaloma generates continuous atom embeddings from which force field parameters are predicted, enabling end-to-end differentiable optimization [5].
Experimental Protocols and Validation
DEFMap Protocol for Extracting Dynamics from Cryo-EM Data
The DEFMap (Dynamics Extraction From cryo-EM Map) protocol combines deep learning with MD simulations to extract protein dynamics information from cryo-EM density maps [6]:
- Dataset Preparation: Select 25 proteins with molecular weights under 200 kDa and cryo-EM resolution better than 4.5 Å
- MD Simulation Setup:
- Use GROMACS simulation package
- Prepare initial structures from PDB coordinates
- Model disordered regions (<7 residues)
- Cap non-natural termini with acetyl or formyl groups
- Run 30 nsec simulations to generate trajectories
- Feature Extraction:
- Calculate root-mean-square fluctuation (RMSF) from MD trajectories
- Extract local density subvoxels (15Å grid) centered on heavy atom positions
- Apply 5Å low-pass filter and unify grid width to 1.5Å/grid
- Data Augmentation: Rotate subvoxels by 90° in xy, xz, and yz planes
- Model Training:
- Train 3D-CNN using leave-one-out cross-validation
- Input: Processed cryo-EM density subvoxels
- Output: Logarithm of RMSF values
- Loss function: Mean squared error between predicted and MD-derived RMSF
This protocol achieved a correlation coefficient of 0.665±0.124 between predicted and actual dynamics, outperforming conventional methods using raw map intensity (0.459±0.179) or local resolution estimates (0.510±0.091) [6].
Casting Polymer Film Formation Workflow
An iterative MD workflow simulates solvent evaporation during casting of polymeric films [7]:
- System Preparation: Construct initial solvated polymer system (e.g., PVA-quercetin)
- Automated Solvent Removal:
- Perform cycles of solvent molecule removal
- Between removal stages, implement equilibration periods (NPT ensemble, 298K, 1 bar)
- Continue until target polymer concentration is achieved
- Structural Monitoring:
- Track root mean square deviation (RMSD)
- Calculate radius of gyration
- Generate density profiles
- Compute radial distribution functions (RDFs)
- Validation: Compare final configurations with experimental data for chain entanglement and solute clustering
This protocol successfully reproduced stepwise compaction events, reduction in polymer radius of gyration, elimination of solvent-rich voids, and formation of dense, homogeneous films consistent with experimental observations [7].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Software Tools for Molecular Dynamics Simulations
| Tool Name | Function | Application Context |
|---|---|---|
| OpenMM [8] | High-performance MD engine | GPU-accelerated simulations of biomolecules |
| Desmond [4] | MD engine with high throughput | Pharmaceutical research, accurate property predictions |
| LAMMPS [9] | General-purpose MD simulator | Materials science, ML-IAP-Kokkos interface for ML potentials |
| GROMACS [6] | MD simulation package | High-performance biomolecular simulations |
| MDTraj [8] | Trajectory analysis | RMSD, radius of gyration, secondary structure analysis |
| PackMol [8] | Solvent addition | Preparing solvated systems for simulation |
| PDBFixer [8] | Structure preparation | Cleaning PDB files, adding missing atoms/residues |
| Force Field Builder [4] | Parameter development | Optimizing custom torsion parameters in OPLS4 |
| Plumed [10] | Enhanced sampling | Free energy calculations, metadynamics |
| Espaloma [5] | Force field creation | Differentiable construction of molecular mechanics force fields |
MD Workflow and Force Field Parameterization
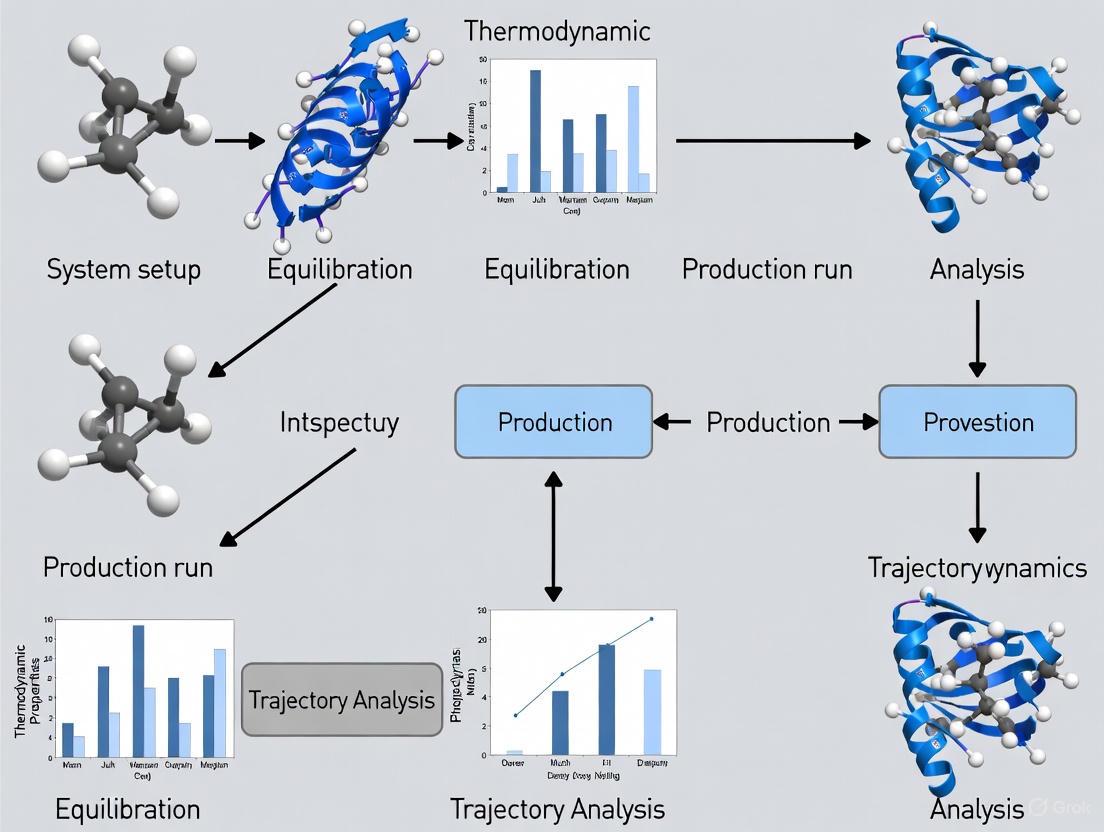
The following diagrams illustrate the core molecular dynamics workflow and the modern neural network-based approach to force field parameterization.
Core Molecular Dynamics Simulation Workflow
Neural Network Force Field Parameterization
Molecular dynamics simulations represent a powerful methodology for studying atomic-scale phenomena across scientific disciplines. The core MD engine integrates Newton's equations of motion using robust numerical algorithms, while the force field provides the essential physics describing interatomic interactions. Traditional force fields like AMBER, CHARMM, and OPLS have evolved significantly, with modern versions offering improved accuracy for challenging chemical systems. Emerging approaches, including machine learning potentials and differentiable force fields, promise to further enhance the accuracy and applicability of MD simulations. When combined with experimental data and advanced sampling techniques, MD simulations provide unprecedented insights into dynamic molecular processes, supporting research from basic science to drug discovery and materials design.
Molecular dynamics (MD) simulation is an indispensable computational tool for understanding the physical movements of atoms and molecules over time, providing atomic-level insights into biological processes and material properties [11]. In recent years, the accessibility of MD simulation tools and hardware has grown significantly, shifting the primary challenge from generating simulation data to effectively analyzing and interpreting the resulting complex trajectories [12]. This technical guide provides a comprehensive overview of the complete MD workflow, from initial structure preparation to final analysis, framed within the context of ongoing research aimed at streamlining and automating these processes for researchers, scientists, and drug development professionals. The integration of workflow optimization technologies and automated analysis platforms is revolutionizing this field, enabling more efficient and reproducible computational experiments [13] [14].
Foundational Steps in the Simulation Workflow
Initial Structure Preparation
The MD workflow begins with acquiring and preparing a initial structure, which can come from experimental sources like protein databanks or from predicted structures. The structure must be placed in a simulation box with explicit solvent molecules and ions to mimic a physiological environment. Parameters for the system are defined using a force field, which mathematically describes the potential energy of the system based on atomic interactions [15]. Tools like GROMACS include pdb2gmx for this conversion, generating the necessary topology file that defines molecular connectivity and force field parameters [15].
System Minimization and Equilibration
Before production simulation, the system must be energy-minimized to remove steric clashes and improper geometry. This is followed by a critical equilibration phase, where the system is gradually heated to the target temperature (e.g., 310 K for physiological conditions) and the pressure is stabilized around 1 bar using algorithms like the Berendsen or Parrinello-Rahman barostat [15]. This equilibration ensures the system has reached a stable thermodynamic state before data collection begins. The isothermal-isobaric (NPT) ensemble is commonly employed during this phase to maintain constant number of atoms, pressure, and temperature [15].
Production Simulation and Trajectory Generation
The production phase involves running an extended simulation to collect trajectory data for analysis. Modern implementations use integration algorithms like the leap-frog method to solve Newton's equations of motion with a typical timestep of 2 femtoseconds [15]. During this phase, atomic coordinates are saved at regular intervals (e.g., every 100 picoseconds) to create a trajectory file. Contemporary workflows may leverage cloud-native simulation platforms that offer significant computational scalability, eliminating hardware barriers for research teams [16].
Analysis Methodologies and Protocols
Core Analysis Metrics
Once simulation trajectories are generated, researchers employ a suite of analysis metrics to extract biologically or physically relevant information. The table below summarizes key properties and their significance in interpretation.
Table 1: Key Molecular Dynamics Analysis Metrics and Their Applications
| Metric | Description | Significance in Analysis |
|---|---|---|
| Root-Mean-Square Deviation (RMSD) | Measures structural deviation from a reference structure over time [17] | Quantifies system stability and conformational changes |
| Root-Mean-Square Fluctuation (RMSF) | Calculates atomic positional fluctuations around average positions [17] | Identifies flexible regions in protein structures |
| Radius of Gyration (Rg) | Measures compactness of a molecular structure [17] | Indicates folding states and structural density |
| Solvent Accessible Surface Area (SASA) | Quantifies surface area accessible to solvent molecules [15] | Probes solvent exposure and hydrophobic effects |
| Hydrogen Bonding | Tracks formation and persistence of hydrogen bonds [17] | Analyzes key molecular interactions and stability |
| Contact Frequency | Measures how often residue pairs come within a cutoff distance [12] | Maps interaction networks and allosteric pathways |
| Principal Component Analysis (PCA) | Identifies major collective motions in the system [17] | Separates large-scale conformational changes from local fluctuations |
Advanced and Specialized Analyses
Beyond these core metrics, advanced analyses provide deeper mechanistic insights. Contact frequency analysis implemented in tools like mdciao uses a modified version of the mdtraj.compute_contacts method, calculating residue-residue distances using the formula CF^(i)^~(A,B)~ = (1/N^(i)^~frames~) Σ~t~ Θ(δ - d~AB~(t)), where Θ is the contact function with cutoff distance δ, typically set at 4.5 Å as a trade-off for capturing various interaction types [12]. For drug discovery applications, MD properties combined with machine learning algorithms can predict critical properties like aqueous solubility, with studies demonstrating that Gradient Boosting algorithms can achieve R² values of 0.87 when using MD-derived features [15].
Workflow Automation and Integration Platforms
The complexity of managing interconnected simulation tasks has driven development of specialized workflow engines. Platforms like HSWAP (HPC Scientific Workflow Application Platform) encapsulate each simulation task as a component with defined attributes, managing execution through directed acyclic graphs (DAGs) that represent data dependencies between tasks [14]. This approach enables automated scheduling and execution of complex, multi-step simulation jobs while hiding underlying computational details from researchers.
Recent advances include LLM-powered assistants like MDCrow, which uses chain-of-thought reasoning over 40 expert-designed tools to automate MD workflow setup, execution, and analysis [18]. These systems can handle diverse tasks from file processing and simulation setup to literature retrieval, significantly reducing manual intervention requirements. For analysis, tools like FastMDAnalysis provide unified environments that reduce the lines of code required for standard workflows by over 90%, while ensuring reproducibility through consistent parameter management and detailed execution logging [17].
Table 2: Essential Research Reagent Solutions for Molecular Dynamics Workflows
| Tool/Category | Specific Examples | Primary Function |
|---|---|---|
| Simulation Engines | GROMACS [15] | Core MD simulation execution |
| Force Fields | GROMOS 54a7 [15] | Define potential energy functions and parameters |
| Analysis Frameworks | MDTraj, MDAnalysis, mdciao [12] [17] | Trajectory analysis and metric calculation |
| Workflow Management | HSWAP, modeFRONTIER [14] [13] | Orchestrate complex multi-step simulation processes |
| Visualization | VMD, PyMOL [12] | 3D trajectory visualization and rendering |
| Specialized Analysis | FastMDAnalysis, GetContacts [17] [12] | Automated analysis of specific properties |
Integrated Workflow Visualization
The complete MD simulation workflow, from initial structure to final analysis, can be visualized as an integrated process with multiple feedback loops for optimization. The following diagram provides a comprehensive overview of this workflow, highlighting the key stages and their interrelationships.
Diagram 1: Comprehensive MD workflow from structure to analysis (Width: 760px)
The molecular dynamics simulation workflow represents a sophisticated integration of computational techniques that extends from careful initial structure preparation through to nuanced trajectory analysis. Contemporary research in this field is increasingly focused on developing automated, reproducible workflows that leverage both specialized workflow engines like HSWAP and emerging LLM-based assistants like MDCrow [14] [18]. The ongoing development of integrated analysis platforms such as FastMDAnalysis and mdciao is simultaneously reducing technical barriers while enhancing analytical capabilities [17] [12]. For researchers in drug development and molecular sciences, mastering this complete workflow—while leveraging these emerging automation technologies—provides a powerful framework for extracting meaningful biological insights from the atomic-level dynamics simulated through computational methods.
Molecular dynamics (MD) simulations serve as a critical computational tool in structural biology, drug discovery, and materials science, enabling researchers to observe the physical movements of atoms and molecules over time. The selection of an appropriate MD software package is a foundational decision that directly impacts the accuracy, efficiency, and ultimate success of simulation-based research. Within the extensive landscape of available tools, AMBER, GROMACS, and NAMD have emerged as three of the most widely used and powerful platforms. Each possesses distinct philosophical underpinnings, algorithmic specializations, and performance characteristics tailored to different research needs. This guide provides an in-depth technical comparison of these three packages, detailing their specialized capabilities, performance benchmarks, and integration into modern computational workflows to inform researchers in selecting the optimal tool for their specific molecular investigations.
Comparative Analysis of Software Specializations
Core Characteristics and Historical Context
AMBER (Assisted Model Building with Energy Refinement): Originally developed in the late 1970s, AMBER has evolved with a strong focus on simulating biomolecular systems, particularly proteins and nucleic acids. It is renowned for its highly accurate force fields, which are often considered the gold standard for biomolecular simulation [19]. While historically optimized for CPUs, recent versions (AMBER GPU) have made significant strides in GPU acceleration [19].
GROMACS (GROningen MAchine for Chemical Simulations): Emerging in the early 1990s from the University of Groningen, GROMACS was engineered from the ground up for high performance and exceptional speed in parallel computations [19]. It is celebrated for its raw performance and scalability, making it a top choice for high-throughput studies and large systems [19] [20].
NAMD (Nanoscale Molecular Dynamics): Developed with a focus on simulating large biomolecular systems, NAMD excels at parallel scalability and is particularly effective for very large complexes (>2 million atoms) [20]. Its tight integration with the visualization package VMD offers a significant advantage for visual analysis and simulation setup [21].
Specialized Capabilities and Ideal Use Cases
Table 1: Specialized Capabilities and Recommended Use Cases for AMBER, GROMACS, and NAMD
| Software | Specialized Capabilities | Ideal Use Cases |
|---|---|---|
| AMBER | High-accuracy force fields (ff14SB, ff19SB), free energy calculations (MM/PBSA, MM/GBSA), hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) simulations, advanced parameterization for non-standard residues [19]. | Protein-ligand binding free energy calculations, detailed studies of nucleic acid conformations, enzymatic reaction mechanisms via QM/MM, force field development [19] [20]. |
| GROMACS | Exceptional simulation speed on both CPUs and GPUs, support for diverse force fields (AMBER, CHARMM, OPLS), highly efficient parallelization, extensive analysis tool suite [19] [20]. | High-throughput screening, membrane protein dynamics, large biomolecular complexes, long-timescale simulations requiring extensive sampling [19]. |
| NAMD | Extreme scalability for massive parallelization on high-performance computing clusters, superior performance with high-performance GPUs, robust collective variable (colvar) methods, seamless integration with VMD for simulation and analysis [21] [20]. | Massive systems such as viral capsids or entire chromosomes, real-time molecular dynamics simulations via VMD integration, complex free energy surfaces requiring advanced colvar sampling [21] [20]. |
Performance, Scalability, and Hardware Considerations
Performance characteristics vary significantly across the three packages, influencing hardware selection and configuration.
GROMACS is frequently recognized for its raw speed, often ranking as one of the fastest MD engines available. It is highly optimized for both CPU and GPU architectures, making it exceptionally efficient for a wide range of system sizes [19] [20]. Its performance advantage is most pronounced in standard molecular dynamics simulations.
NAMD demonstrates superior parallel scalability, especially on high-performance computing (HPC) clusters with many cores. It is engineered to efficiently leverage multiple nodes, making it a powerhouse for simulating massive systems that exceed several million atoms [20]. As one researcher notes, "NAMD demonstrates superior performance compared to GROMACS when employing high-performance GPUs" [21].
AMBER has made substantial progress in GPU acceleration with its PMEMD code. While it may not always match the raw speed of GROMACS for standard simulations, its GPU implementation is highly efficient and is the recommended platform for running AMBER simulations, particularly for free energy calculations [22] [19].
Table 2: Hardware Configuration Recommendations for Optimal Performance
| Component | AMBER | GROMACS | NAMD |
|---|---|---|---|
| Recommended CPU Strategy | Balance of clock speed and core count [22]. | Prioritize higher clock speeds over extreme core counts [22]. | High core count for massive parallelization [20]. |
| Top-Tier GPU Choices | NVIDIA RTX 6000 Ada (48 GB VRAM), NVIDIA RTX 4090 [22]. | NVIDIA RTX 4090 (High CUDA core count), NVIDIA RTX 6000 Ada [22]. | NVIDIA RTX 6000 Ada, NVIDIA RTX 4090 [22]. |
| Multi-GPU Support | Well-optimized for multi-GPU setups [22]. | Supports multi-GPU execution [22]. | Efficiently distributes computation across multiple GPUs [22]. |
Force Field Compatibility and Accuracy
Force field selection is critical for simulation accuracy, and the three packages offer different levels of flexibility and specialization.
AMBER is renowned for the accuracy and reliability of its own force fields, particularly for proteins (e.g., ff14SB, ff19SB) and nucleic acids [19]. While it primarily uses its own force fields, it is considered a benchmark for biomolecular simulation accuracy [19].
GROMACS stands out for its exceptional versatility, with native support for a wide range of force fields including AMBER, CHARMM, and OPLS [19] [20]. This allows researchers to select the most appropriate force field for their specific system without being locked into a single ecosystem.
NAMD also supports multiple force fields and is often used with CHARMM-family force fields, though it is compatible with others as well [20]. Its mature implementation of collective variable methods makes it robust for advanced sampling techniques that depend on force field accuracy [21].
Integration into a Modern MD Workflow
A typical MD simulation workflow encompasses multiple stages, from system preparation to trajectory analysis. The optimal placement of each software package within this workflow depends on its strengths.
MD Software Integration in a Simulation Workflow
System Setup and Equilibration
The initial steps of building the molecular system and achieving a stable, equilibrated state are crucial for production simulations.
AMBER utilizes the
AmberToolssuite, which includestleapfor system building and parameterization. This is particularly powerful for handling non-standard residues and small molecule ligands through the GAFF force field [19]. Theantechamberprogram facilitates the parameterization of small molecules.GROMACS provides a comprehensive set of setup utilities, with
pdb2gmxbeing a workhorse for generating topology and coordinates from an initial PDB file. Its strength lies in the straightforward setup of standard biomolecular systems with support for various force fields [19].NAMD benefits tremendously from its seamless integration with VMD. The combination allows for graphical building, parameterization, and equilibration of complex systems, which is especially valuable for large, heterogeneous systems like membrane-protein complexes [21].
Production Simulation and Advanced Sampling
This core phase of the MD workflow is where the primary data collection occurs, and the choice of software has the most significant impact on performance and capability.
For maximum throughput with standard system sizes, GROMACS is often the preferred choice due to its exceptional optimization [19] [20].
For massive systems requiring hundreds or thousands of cores, NAMD's superior parallel scalability becomes advantageous [20].
For advanced sampling methods like free energy perturbation (FEP) or QM/MM simulations, AMBER provides specialized, highly-tuned implementations that can justify potentially longer simulation times [19].
Trajectory Analysis and Visualization
The final stage involves extracting meaningful scientific insights from the simulation trajectory.
AMBER includes
CPPTRAJ, a powerful and versatile analysis program capable of calculating a vast array of structural, dynamic, and thermodynamic properties from trajectory data [19].GROMACS comes bundled with an extensive suite of analysis tools (
gmx rms,gmx rmsf,gmx gyrate,gmx hbond, etc.) that cover most standard analyses with high efficiency [19].NAMD, again through VMD, offers unparalleled visualization capabilities alongside analysis. This tight coupling enables researchers to visually identify phenomena of interest and then perform quantitative analysis on them within the same environment [21].
Table 3: Essential Research Reagents and Tools for Molecular Dynamics Simulations
| Tool/Reagent | Function | Software Association |
|---|---|---|
| Force Fields (AMBER, CHARMM, OPLS) | Defines the potential energy function governing atomic interactions; critical for simulation accuracy. | AMBER (native), GROMACS (multi-support), NAMD (multi-support) [19] [20]. |
| Solvation Models (TIP3P, TIP4P, SPC) | Represents water molecules explicitly or implicitly; significantly impacts dynamics and properties. | All packages support major explicit and implicit water models [23]. |
| Visualization Software (VMD, PyMOL) | Enables 3D visualization of molecular structures, trajectories, and analysis results. | NAMD (tight VMD integration), AMBER, GROMACS (third-party support) [21] [23]. |
| Enhanced Sampling Plugins (PLUMED) | Provides advanced sampling algorithms (metadynamics, umbrella sampling) to accelerate rare events. | Interfaces with GROMACS, AMBER, and NAMD [20]. |
Decision Framework and Future Outlook
Selection Guidelines for Specific Research Objectives
Choosing between AMBER, GROMACS, and NAMD requires a systematic assessment of research priorities, computational resources, and system characteristics.
MD Software Selection Decision Guide
The decision tree above provides a strategic starting point for software selection. Beyond these guidelines, consider these practical factors:
Licensing and Cost: GROMACS is fully open-source (GNU GPL). AMBER has a free version for academic use, but some specialized modules and commercial use require a license. NAMD is free for academic use [21] [23].
Learning Curve and Community: GROMACS is generally noted for being more beginner-friendly with extensive documentation and tutorials [19]. AMBER has a steeper learning curve but offers powerful tools for specialized research [19]. All three have active user communities, with GROMACS and AMBER having particularly large and established bases [19].
Workflow Integration: Consider existing laboratory protocols, available force field parameter sets for your molecules of interest, and compatibility with analysis scripts or pipelines already in place within your research group.
Emerging Trends and Interoperability
The field of molecular dynamics is rapidly evolving, with several trends influencing the use of traditional packages like AMBER, GROMACS, and NAMD.
Rise of OpenMM: OpenMM is gaining prominence as a highly flexible and GPU-optimized platform that is particularly strong for method development and prototyping [20]. Its Python scripting interface allows for easy experimentation with custom potentials and integration into larger computational workflows [20].
AI and Machine Learning Integration: The incorporation of machine learning potentials is becoming increasingly important for achieving quantum-mechanical accuracy at classical mechanics cost. While some packages are adapting to support these new force fields, NAMD has been noted for its existing compatibility with ML force fields [20].
Interoperability and Hybrid Workflows: A modern best practice involves leveraging the strengths of multiple packages within a single research project. A common pattern is to use AMBER tools for system parameterization, GROMACS for high-throughput production simulations, and VMD/NAMD for visualization and analysis of large complexes [19] [20]. This hybrid approach maximizes efficiency and capability across the entire simulation lifecycle.
AMBER, GROMACS, and NAMD represent three mature, powerful, and distinct platforms for molecular dynamics simulations. The optimal choice is not a matter of identifying a universally superior package, but rather of matching each software's specialized strengths to the specific demands of a research project. AMBER excels in force field accuracy and specialized biomolecular simulations, particularly for free energy calculations and QM/MM studies. GROMACS dominates in raw performance and efficiency for high-throughput simulations of standard systems. NAMD stands out for its unparalleled scalability on HPC resources and tight integration with VMD for visualizing and simulating massive molecular complexes. As the field continues to evolve with trends in GPU computing, AI, and enhanced sampling methods, the ability to strategically deploy and potentially combine these tools will remain an essential skill for computational researchers in structural biology and drug development.
Molecular dynamics (MD) simulations provide a unique computational microscope, enabling researchers to track the motion of individual atoms and molecules over time. These simulations have become indispensable in materials science, chemistry, and drug discovery, offering insights into fundamental processes that are often difficult or impossible to observe experimentally [24]. At the core of every MD simulation lies the force field—a mathematical representation of the potential energy surface that governs atomic interactions. The accuracy, reliability, and predictive power of MD simulations depend critically on the quality of these empirical potential energy functions, making them truly the "heart" of the simulation.
This technical guide examines force fields within the broader context of MD simulation workflows, focusing specifically on recent advances in reactive potentials and accelerated sampling techniques. As MD applications expand to increasingly complex systems—from drug-target interactions to advanced materials design—researchers face persistent challenges in simulating bond breaking/formation and accessing biologically relevant timescales. Innovative formulations of empirical potentials are now overcoming these limitations, opening new frontiers in computational science.
Theoretical Foundations of Empirical Potential Energy Functions
Mathematical Formalism
Force fields decompose the complex many-body potential energy of a system into simpler additive components:
[ E{\text{total}} = E{\text{bonded}} + E{\text{non-bonded}} = E{\text{bond}} + E{\text{angle}} + E{\text{torsion}} + E{\text{electrostatic}} + E{\text{van der Waals}} ]
This separation allows for computationally efficient calculations while maintaining physical fidelity. The harmonic approximation typically used for bonded terms provides excellent computational efficiency for systems near equilibrium but fails dramatically when bonds undergo significant stretching or breaking.
Advanced Potential Energy Functions
Beyond standard harmonic potentials, more sophisticated functions have been developed to better represent molecular interactions:
Morse potential: ( E(r) = De \left[1 - e^{-\alpha(r-re)}\right]^2 ) provides a more physically realistic description of bond dissociation, where ( De ) represents the dissociation energy, ( re ) the equilibrium bond distance, and ( α ) controls the potential's width [25].
Improved Manning-Rosen potential: Offers better performance for certain diatomic systems compared to traditional Morse potentials [26].
Tietz-Hua potential: Demonstrates superior accuracy in fitting experimental Rydberg-Klein-Rees (RKR) data for various diatomic molecules [26].
Table 1: Comparison of Empirical Potential Energy Functions
| Potential Type | Mathematical Form | Advantages | Limitations |
|---|---|---|---|
| Harmonic | ( E(r) = \frac{1}{2}k(r-r_e)^2 ) | Computational efficiency; Good near equilibrium | Unphysical for large displacements; No bond breaking |
| Morse | ( E(r) = De \left[1 - e^{-\alpha(r-re)}\right]^2 ) | Physically realistic dissociation; Bond breaking capability | Requires additional parameters; Slightly more computational cost |
| Improved Manning-Rosen | Complex analytical form | Better accuracy for certain diatomics | Limited testing across diverse systems |
| Tietz-Hua | Complex analytical form | Superior RKR fitting for selected molecules | Parameter availability limited |
Reactive Force Fields: Enabling Bond Breaking and Formation
The IFF-R Approach
A significant advancement in reactive simulations comes from the Reactive INTERFACE Force Field (IFF-R), which replaces harmonic bond potentials with reactive, energy-conserving Morse potentials [25]. This clean substitution maintains compatibility with existing force fields like CHARMM, PCFF, OPLS-AA, and AMBER while enabling bond dissociation through three interpretable parameters per bond type.
The key innovation of IFF-R lies in its parameterization strategy, which preserves the accuracy of non-reactive force fields for equilibrium properties while enabling bond breaking. The parameters are derived as follows:
- Equilibrium bond length (( r_e )) remains identical to the harmonic potential value
- Dissociation energy (( D_e )) obtained from experimental data or high-level quantum calculations
- Width parameter (( α )) fitted to reproduce vibrational frequencies from infrared and Raman spectroscopy
This approach maintains the exceptional accuracy of IFF for bulk and interfacial properties (typically <0.5% deviation in densities, <5% in surface energies, and <10% in mechanical properties) while enabling realistic bond dissociation [25].
Computational Efficiency
IFF-R achieves approximately 30-fold speed improvement compared to state-of-the-art bond-order potentials like ReaxFF, while maintaining high accuracy [25]. This performance advantage stems from its simpler functional form without complex bond-order calculations or numerous correction terms.
Table 2: Performance Comparison of Reactive Simulation Methods
| Method | Functional Form | Computational Speed | Bond Breaking | Bond Formation | Compatibility |
|---|---|---|---|---|---|
| Traditional Harmonic | Harmonic potentials | Fastest | No | No | Standard biomolecular force fields |
| IFF-R | Morse potentials | ~30x faster than ReaxFF | Yes | Via template methods | CHARMM, AMBER, OPLS-AA, PCFF |
| ReaxFF | Bond-order potentials | Baseline | Yes | Yes | Limited branches (combustion, aqueous) |
| AIREBO | Bond-order potentials | Moderate | Yes | Limited | Carbonaceous materials |
Diagram 1: Force Field Selection Workflow in MD Simulations
Accelerated Molecular Dynamics for Extended Timescales
Timescale Limitations in Conventional MD
Conventional MD simulations face severe timescale limitations, typically restricted to microsecond durations, while many critical biological and materials processes occur over milliseconds or longer [27]. This timescale gap has driven the development of accelerated MD methods that enhance the sampling of rare events without sacrificing atomic-level accuracy.
Shuffling Accelerated Molecular Dynamics (SAMD)
The Shuffling Accelerated Molecular Dynamics (SAMD) method addresses timescale limitations through an empirical formulation adapted from temperature-accelerated MD (TAMD) [27]. SAMD introduces the nearest neighbor off-centering absolute displacement (NNOAD) as a collective variable, defined as the deviation of an atom from the geometric center of its nearest neighbors.
The mathematical formulation of SAMD couples the NNOAD of each atom with three additional dynamic variables that follow Langevin equation dynamics, while the system itself is thermostated using Nosé-Hoover dynamics. This approach achieves acceleration factors of several orders of magnitude while maintaining kinetic consistency and accuracy in predicting microstructure evolution [27].
Parameterization and Validation
Successful application of SAMD requires careful parameter selection:
- Time step (( δt )): Comparable to or slightly smaller than conventional MD (typically 0.5-2.0 fs)
- Temperature (( T )): Set to the desired simulation temperature
- Cutoff distance (( r_D )): 2-3 times the nearest neighbor distance in the material
- Coupling constants (( κ ), ( γx ), ( γs )): Tuned to specific systems
- Effective temperature (( T_s )): 5-50 times the simulation temperature
Validation across diverse materials systems including metals, polymers, and composites has demonstrated SAMD's capability to accurately predict long-timescale microstructure evolution while maintaining excellent agreement with conventional MD where timescales overlap [27].
Validation and Accuracy Assessment
Vibrational Quantum Defect Analysis
The vibrational quantum defect (VQD) method provides a sophisticated approach for evaluating the accuracy of empirical potential functions [26]. This method quantifies deviations between calculated and experimental vibrational energy levels, offering unprecedented sensitivity in detecting potential inaccuracies.
The VQD method works by comparing the vibrational energy levels obtained from RKR experimental data with those predicted by analytical potential functions. The quantum defect is calculated as:
[ δ = v - v_{\text{RKR}} ]
where ( v ) represents the vibrational level calculated from the potential function and ( v_{\text{RKR}} ) is the experimental reference value. A perfectly accurate potential would produce a constant VQD across all energy levels, while deviations indicate limitations in the potential function [26].
Application to Diatomic Molecules
Comprehensive VQD analysis has been performed for multiple diatomic systems including ( ^7\text{Li}2 ), ( \text{Na}2 ), ( \text{K}2 ), ( \text{Cs}2 ), and CO [26]. The results demonstrate that:
- Morse potentials generally provide good accuracy but show systematic deviations at higher vibrational levels
- Improved Manning-Rosen and Tietz-Hua potentials offer superior performance for specific molecular systems
- No single potential function performs optimally across all molecular systems, highlighting the need for system-specific potential selection
Table 3: Accuracy Assessment of Potential Functions Using VQD Analysis
| Molecule | Morse Potential | Improved Manning-Rosen | Tietz-Hua Potential | Best Performer |
|---|---|---|---|---|
| Li₂ | Moderate deviations at high E | Good agreement | Excellent agreement | Tietz-Hua |
| Na₂ | Systematic deviations | Good agreement | Best overall fit | Tietz-Hua |
| K₂ | Good agreement | Moderate deviations | Excellent agreement | Tietz-Hua |
| Cs₂ | Moderate deviations | Good agreement | Best overall fit | Tietz-Hua |
| CO | Good agreement | Excellent agreement | Not tested | Improved Manning-Rosen |
Practical Implementation and Workflow Integration
Force Field Selection Protocol
Choosing the appropriate force field requires careful consideration of the research question and system properties:
- Stable systems near equilibrium: Traditional harmonic force fields (CHARMM, AMBER, OPLS-AA) provide excellent performance and computational efficiency
- Bond dissociation studies: Morse-based potentials (IFF-R) enable physically realistic bond breaking
- Complex chemical reactions: Bond-order potentials (ReaxFF) may be necessary despite computational costs
- Extended timescales: Accelerated methods (SAMD) enhance rare event sampling
Diagram 2: Integrated MD Simulation Workflow
Table 4: Essential Tools for Force Field Development and Validation
| Tool/Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| MDAnalysis | Software library | Trajectory analysis and manipulation | Post-processing of MD simulations; RMSD, RDF, H-bond analysis [28] [29] |
| MDAnalysisData | Data repository | Benchmark datasets for MD | Testing, validation, and education in biomolecular simulations [28] |
| IFF-R | Reactive force field | Bond dissociation simulations | Polymer failure, protein mechanics, composite materials [25] |
| SAMD | Accelerated MD method | Enhanced sampling of rare events | Microstructure evolution in materials [27] |
| VQD Method | Validation methodology | Accuracy assessment of potentials | Evaluating potential function performance [26] |
| RKR Data | Experimental reference | Potential energy curves | Benchmarking potential functions [26] |
Force fields and empirical potential energy functions remain the fundamental determinants of success in molecular dynamics simulations. Recent advances in reactive potentials and accelerated sampling methods have dramatically expanded the scope of addressable research questions, enabling realistic simulations of bond breaking/formation and access to biologically relevant timescales.
The emerging trend toward specialized potential functions tailored to specific scientific questions represents a paradigm shift from the one-size-fits-all approach that dominated early force field development. Coupled with rigorous validation methodologies like vibrational quantum defect analysis and integration with machine learning approaches for parameter optimization, the next generation of force fields promises unprecedented accuracy and transferability across the chemical space.
As MD simulations continue to bridge scales from electronic structure to macroscopic properties, the empirical potential energy functions at their heart will remain active areas of research and development—driving innovations in materials design, drug discovery, and fundamental science.
Within the broader workflow of molecular dynamics (MD) research, the initial steps of system preparation, solvation, and ionization are critically important. These stages transform a static molecular structure into a realistic, stable model of a chemical environment, forming the very foundation upon which all subsequent simulation data is built. The accuracy of the production simulation is contingent upon the care taken during these preparatory phases [30]. A properly constructed model ensures that the observed dynamics and thermodynamics are representative of the system under study, whether it be a protein in a biological fluid, an electrode in an electrolyte, or a novel material in solution. This guide details the core principles and detailed methodologies for building these simulation environments, providing researchers with the protocols necessary to generate robust and reliable data for their molecular investigations.
Theoretical Framework: Forces, Solvents, and Ions
The physical accuracy of an MD simulation is governed by the force field (FF), a set of mathematical functions and parameters that describe the potential energy of a system as a function of the nuclear coordinates. It encodes the physics of interatomic interactions, including bond stretching, angle bending, torsional rotations, and non-bonded van der Waals and electrostatic forces [31]. The choice of force field is a foundational decision, as different parameter sets are optimized for specific classes of molecules or properties. For instance, the GROMOS 53A5 and 53A6 parameter sets were specifically optimized to reproduce the free enthalpies of hydration and solvation, making them particularly suitable for processes like protein folding and biomolecular association [32].
Continuum solvation models offer a computationally efficient alternative to explicit solvent for certain quantum mechanical or preliminary calculations. Models like the COSMO (COnductor-like Screening Model) approximate the solvent as a polarizable continuum with a defined dielectric constant (( \epsilon )) [33]. The key parameters for such models include the dielectric constant itself, which defines the solvent's polarity, atomic radii that determine the molecular cavity's shape, and a solvent-accessible surface parameter.
For simulations requiring atomic detail, the system is immersed in a box of explicit solvent molecules. The choice of water model (e.g., SPC, TIP3P, TIP4P) and the possible addition of ions to mimic physiological or experimental salt concentrations are crucial for modeling realistic environments. The process of ionization, or adding counter-ions, is essential to neutralize the net charge of the system, which prevents unrealistic electrostatic forces from destabilizing the simulation [30].
Table 1: Common Explicit Water Models and Key Characteristics
| Water Model | Force Field Family | Number of Sites | Brief Description |
|---|---|---|---|
| SPC | GROMOS, OPLS | 3 | Simple Point Charge model; rigid geometry. |
| TIP3P | CHARMM, AMBER | 3 | Transferable Intermolecular Potential 3-Point; a standard for biomolecular simulations. |
| TIP4P | Specialized FFs | 4 | An extra dummy atom site allows for a more accurate charge distribution. |
| Flexible SPC | Specialized FFs | 3 | Includes internal degrees of freedom for O-H bond stretching and H-O-H angle bending. |
System Preparation: A Step-by-Step Methodology
The following workflow outlines the comprehensive process for preparing a molecular system for a production MD simulation. This sequence ensures that the system is structurally sound, energetically relaxed, and properly equilibrated at the desired thermodynamic state points.
Initial Structure Preparation and Topology Generation
The process begins with an initial coordinate file, often from a database like the Protein Data Bank (PDB) for biomolecules or the Materials Project for crystalline materials [24]. Raw structural data frequently requires preprocessing, as experimental structures may have missing atoms, unresolved loops, or non-standard protonation states [34]. Tools like Schrödinger's Maestro or molecular modeling suites can be used to add hydrogen atoms, assign bond orders, fill missing side chains or loops, and set correct protonation states for residues at a given pH [34].
The next critical step is generating the system's topology, which defines the chemical structure, atom types, bonded connections, and force field parameters. This can be done using tools like gmx pdb2gmx (GROMACS), tleap (AMBER), or CHARMM-GUI [30]. The topology file is the instruction manual for the force field, describing how to calculate the potential energy for the specific molecules in the simulation.
Solvation and Ionization Protocols
Once the solute's topology is defined, it is placed in a simulation box with periodic boundary conditions. The box size should be large enough to ensure the solute does not interact with its own periodic images, typically with a minimum of 1.0 to 1.2 nm between the solute and the box edge.
Solvation is then performed by filling the box with explicit solvent molecules using tools like gmx solvate [30]. The choice of water model must be consistent with the selected force field.
Following solvation, ionization is performed to neutralize the system's net charge and, if needed, achieve a specific physiological salt concentration (e.g., 150 mM NaCl). Ions are placed at energetically favorable positions, typically replacing solvent molecules. This can be accomplished using tools like gmx insert-molecules or gmx genion [30]. For accurate studies of electrolytes, the specific solvation structures and ion dynamics, such as those revealed for CaCl₂ solutions using machine learning MD, must be considered [35].
Table 2: Tools for System Setup in GROMACS
| Software/Tool | Primary Function | Application Context |
|---|---|---|
gmx pdb2gmx |
Generates topology from PDB file. | Biomolecules (proteins, DNA). |
gmx editconf |
Defines simulation box type and size. | All system types. |
gmx solvate |
Fills the box with solvent molecules. | Solvating any solute. |
gmx insert-molecules |
Inserts ions, multiple solutes, or other molecules. | Ionization, creating mixtures. |
| Automated Topology Builder | Generates parameters for small molecules. | Drug-like molecules, inhibitors (e.g., for GROMOS96 53A6). |
gmx grompp & gmx mdrun |
Pre-processes and executes the simulation. | Energy minimization and equilibration. |
Energy Minimization and System Equilibration
Even after solvation and ionization, the system may contain unfavorable atomic contacts or steric clashes. Energy minimization is a crucial energy relaxation step that employs algorithms like steepest descent or conjugate gradient to adjust atomic coordinates towards a local energy minimum [30].
After minimization, the system must be gently brought to the target temperature and pressure. This is achieved through a two-stage equilibration process:
- NVT Equilibration: The number of particles (N), volume (V), and temperature (T) are held constant. This stage, also called "isothermal," allows the particle velocities to relax to a Maxwell-Boltzmann distribution at the target temperature. Position restraints are often applied to the solute heavy atoms to prevent unfolding while the solvent equilibrates around it.
- NPT Equilibration: The number of particles (N), pressure (P), and temperature (T) are held constant. This "isothermal-isobaric" stage allows the density of the system to adjust, ensuring the simulation box has the correct size and density for the chosen temperature and pressure. The use of a robust barostat like the C-rescale in GROMACS is recommended [30].
Only after successful NPT equilibration, confirmed by stable temperature, pressure, and potential energy, should the production MD simulation begin.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Software and Resources for Simulation Environment Setup
| Tool/Resource Name | Type/Category | Primary Function in System Prep |
|---|---|---|
| GROMACS [30] | MD Simulation Engine | Performs solvation, ionization, energy minimization, equilibration, and production MD. |
| AMBER (tleap) [34] | MD Suite / Topology Builder | Generates system topologies and coordinate files for AMBER force fields. |
| CHARMM-GUI [30] | Web-Based Setup Tool | Provides a user-friendly interface to build complex systems (e.g., membrane proteins). |
| Automated Topology Builder [30] | Parameterization Service | Generates force field parameters for small organic molecules. |
| PACKMOL [35] | Packing Tool | Creates initial configurations for complex systems by packing molecules into a defined box. |
| COSMO [33] | Continuum Solvation Model | Provides efficient, approximate solvation energies for quantum chemical calculations. |
| AlphaFold2 [24] | Structure Prediction AI | Predicts 3D protein structures from amino acid sequences when experimental structures are unavailable. |
| DeePMD-kit [35] | Machine Learning Potential | Enables high-accuracy MD simulations by using neural networks to model interatomic forces. |
Advanced Topics and Emerging Methodologies
The field of system preparation is being transformed by the integration of machine learning (ML). A significant advancement is the development of Machine Learning Interatomic Potentials (MLIPs). These potentials are trained on high-fidelity quantum mechanical data and can capture complex atomic interactions with near-quantum accuracy but at a fraction of the computational cost, bridging the gap between classical and ab initio MD [24] [35]. Frameworks like the ML-IAP-Kokkos interface now allow for the seamless integration of PyTorch-based MLIPs into popular MD software like LAMMPS, enabling scalable, AI-driven simulations [9].
Furthermore, ML is revolutionizing the analysis of simulation data. Techniques like Principal Component Analysis (PCA) can sift through the high-dimensional trajectory data to extract the essential collective motions and dominant structural changes in the system [24]. This is invaluable for understanding phenomena like allosteric regulation and conformational transitions.
For studying rare events or complex free energy landscapes, advanced sampling techniques like metadynamics are essential. As demonstrated in studies of CaCl₂ aqueous electrolytes, metadynamics can be used to identify diverse metastable solvation structures and map free energy surfaces, revealing states that are difficult to access with standard MD [35]. These advanced methods, combined with robust system preparation, empower researchers to tackle increasingly complex scientific questions with greater confidence and predictive power.
From Theory to Practice: Execution, Analysis, and Biomolecular Applications
Molecular dynamics (MD) simulation is a cornerstone technique in computational chemistry, biophysics, and drug development, enabling researchers to study the structural and dynamic properties of biomolecular systems at atomic resolution. A properly executed MD workflow is essential for obtaining physically meaningful and reproducible results. This technical guide examines the three critical phases of MD simulations—energy minimization, equilibration, and production runs—within the broader context of molecular dynamics workflow research. These stages collectively ensure system stability, proper sampling of physiological conditions, and the generation of reliable trajectory data for scientific analysis. The protocols discussed herein integrate established best practices with emerging methodologies, including AI-driven automation and advanced free energy calculations, to provide researchers with a comprehensive framework for conducting robust molecular simulations [30] [36].
Foundational Principles of MD Simulation
The Newtonian equations of motion form the mathematical foundation of molecular dynamics, where the evolution of the system is determined by numerically solving $mi\frac{d^2 xi}{dt^2} = -(\nabla U)_i$ for each atom [37]. The potential energy field $U$ is described by a force field, which typically comprises bonded interactions (bonds, angles, dihedrals) and non-bonded interactions (electrostatics and van der Waals forces) [37]. The selection of an appropriate force field represents a critical decision point that must align with both the system characteristics and the research objectives, with popular choices including AMBER, CHARMM, and Martini (for coarse-grained systems) [38] [39] [37].
The integration time step must be carefully chosen based on the fastest motions in the system, typically not exceeding 1/10 of this period. For all-atom simulations, this often translates to a 2 fs time step when constraining bonds involving hydrogen atoms [37]. System preparation begins with obtaining initial coordinates, often from Protein Data Bank files, followed by separate parameterization of protein and ligand components using tools such as gmx pdb2gmx for proteins and acpype or similar tools for small molecules [30] [38].
Energy Minimization: Protocols and Best Practices
Purpose and Rationale
Energy minimization addresses structural imperfections introduced during system setup, such as atom overlaps, distorted geometries, or inappropriate side-chain conformations. This crucial initial step relieves steric clashes and strains in the initial structure that would otherwise cause instabilities or simulation failures during subsequent dynamics [30]. By finding a local minimum on the potential energy surface, minimization produces a stable starting configuration for the equilibration phase.
Methodological Approaches
The GROMACS documentation recommends careful consideration of minimization parameters, including the potential use of flexible water models and avoiding bond constraints or frozen groups during this stage [30]. Position restraints and distance restraints should be evaluated carefully based on system requirements. For complex systems, it may be necessary to perform initial minimization of solute structures in vacuo before introducing solvent molecules or membrane environments [30].
Table 1: Key Parameters for Energy Minimization
| Parameter | Recommended Setting | Purpose | Considerations |
|---|---|---|---|
| Force Field | AMBER99SB-ILDN [37], CHARMM, GAFF (ligands) [38] | Defines potential energy function | Must be consistent throughout workflow |
| Algorithm | Steepest descent initially, conjugate gradient for refinement | Efficient convergence | Switch algorithms when energy decrease slows |
| Energy Tolerance | 10-100 kJ/mol/nm | Convergence criterion | Tighter tolerances for stable systems |
| Maximum Steps | 1000-50000 | Prevents infinite loops | System size dependent |
Workflow Implementation
The minimization process typically begins with the steepest descent algorithm for initial rapid energy decrease, potentially switching to conjugate gradient methods for final refinement. The protocol should include careful monitoring of the potential energy and maximum force throughout the process to ensure proper convergence. As highlighted in the High Throughput MD tutorial, energy minimization represents the first computational step after system construction, preparing the structure for subsequent dynamics [38].
Equilibration Phase: Achieving Stable Dynamics
Theoretical Framework
Equilibration prepares the minimized structure for production dynamics by gradually introducing temperature and pressure controls, allowing the system to reach the target ensemble. This phase is essential for establishing appropriate density, solvent orientation, and realistic atomic fluctuations while preventing simulation "blow-ups" that can occur from improper initialization [30]. The GROMACS documentation recommends a staged approach, potentially beginning with NVT ensemble simulation with position restraints on solvent and/or solute to approach the target temperature, followed by NPT simulation to correct density [30].
Practical Implementation
Position restraints are typically applied to heavy atoms of the solute during initial equilibration stages, gradually releasing them as the system adapts to the simulation conditions. The choice of thermostat and barostat should align with force field recommendations, with the GROMACS documentation specifically recommending the c-rescale barostat for pressure coupling [30]. For temperature coupling, modified Berendsen thermostats (such as velocity rescale) are often used during equilibration, transitioning to Nosé-Hoover during production for more rigorous canonical sampling.
Table 2: Equilibration Protocol Parameters
| Stage | Ensemble | Duration | Restraints | Thermostat/Barostat |
|---|---|---|---|---|
| NVT Initial | NVT | 50-100 ps | Heavy atoms | Velocity rescale |
| NPT Density | NPT | 100-200 ps | Backbone atoms | Velocity rescale + C-rescale |
| NPT Full | NPT | 100-500 ps | None | Nosé-Hoover + Parrinello-Rahman |
Advanced Considerations
The equilibration phase should continue until key system properties (potential energy, density, temperature, pressure) have stabilized around their target values. For challenging systems with large solvent cavities or complex molecular arrangements, multiple gentler heating stages or smaller integration time steps may be necessary to prevent instabilities [30]. Recent research on free energy calculations suggests that certain systems, such as the TYK2 dataset in binding affinity studies, may require longer equilibration times of approximately 2 ns for proper convergence [40].
Production Simulations: Data Generation for Analysis
Parameter Selection and Configuration
Production simulations represent the final data-generation phase where configurations are sampled for subsequent analysis. The simulation parameters must remain consistent with how the force field was derived, with particular attention to non-bonded interaction cutoffs, long-range electrostatics treatment (PME is standard), and temperature/pressure coupling schemes [30]. Critically, velocities should not be re-generated between equilibration and production phases to maintain proper system evolution [30]. For free energy calculations, recent research indicates that sub-nanosecond simulations can be sufficient for accurate prediction of ΔG in many systems, though this depends on the specific biological context [40].
Duration and Sampling Considerations
The required production length varies significantly with the scientific question, ranging from nanoseconds for local conformational sampling to microseconds or beyond for large-scale structural transitions. For binding free energy calculations using thermodynamic integration, recent practical guidelines indicate that most systems achieve reasonable convergence within sub-nanosecond simulations per window, though perturbations with |ΔΔG| > 2.0 kcal/mol exhibit increased errors and require careful interpretation [40]. Configuration saving frequency should balance storage constraints with the time scales of relevant motions, typically between 10-100 ps intervals.
Emerging Approaches
AI-driven approaches are transforming production MD workflows. The ML-IAP-Kokkos interface enables integration of PyTorch-based machine learning interatomic potentials with MD packages like LAMMPS, providing enhanced scalability and performance on GPU architectures [9]. Similarly, automated analysis frameworks such as FastMDAnalysis significantly reduce post-processing overhead, enabling comprehensive trajectory analysis including RMSD, RMSF, hydrogen bonding, and principal component analysis with minimal scripting [17].
Table 3: Production Run Specifications for Different System Types
| System Type | Minimum Duration | Time Step | Save Frequency | Special Considerations |
|---|---|---|---|---|
| Solvent/Ligand | 10-100 ns | 2 fs | 10-50 ps | Multiple replicates for statistics |
| Protein-Ligand | 50-500 ns | 2 fs | 50-100 ps | Enhanced sampling for binding |
| Membrane Protein | 200 ns-1 μs | 2 fs | 100-200 ps | Longer stabilization needed |
| Free Energy | 1-10 ns/window | 2 fs | 10-50 ps | Multiple λ states |
Integration with Broader Research Workflows
Automated and AI-Enhanced Protocols
Recent advances in AI and automation are creating new paradigms for MD simulation workflows. Systems like DynaMate provide modular templates for building multi-agent frameworks that automate setup, execution, and analysis of molecular simulations [41]. These frameworks leverage large language models (LLMs) in a ReAct (Reasoning and Acting) pattern, where the AI interprets queries, generates and executes code, observes outcomes, and adapts strategies accordingly [36]. Similarly, NAMD-Agent demonstrates how LLMs can automate web-based tools like CHARMM-GUI to generate simulation-ready inputs, significantly reducing setup time and minimizing manual errors [36].
Specialized Methodologies
Advanced sampling techniques represent crucial extensions to standard MD protocols. Umbrella sampling, as implemented in free energy calculations, employs harmonic restraints along a reaction coordinate to sample unfavorable states, with the weighted histogram analysis method (WHAM) used to reconstruct potential of mean force (PMF) [39]. Thermodynamic integration provides another approach for calculating free energy differences, particularly valuable in protein-ligand binding affinity prediction for drug design applications [40] [39].
Validation and Analysis Frameworks
Comprehensive analysis of production trajectories is essential for extracting scientific insights. Automated tools like FastMDAnalysis provide unified environments for end-to-end trajectory analysis, encapsulating core analyses into a single framework that ensures reproducibility through consistent parameter management and detailed execution logging [17]. These tools can perform comprehensive conformational analysis including RMSD, RMSF, radius of gyration, hydrogen bonding, solvent accessibility, secondary structure assignment, principal component analysis, and hierarchical clustering, dramatically reducing the scripting overhead for researchers [17].
Table 4: Key Software Tools for MD Simulation Workflows
| Tool | Function | Application Context |
|---|---|---|
| GROMACS [30] [38] | MD simulation engine | High-performance biomolecular simulations |
| AMBER [40] | MD simulation suite | Free energy calculations, biomolecules |
| LAMMPS [9] | MD simulation engine | Materials science, MLIP integration |
| CHARMM-GUI [30] [36] | Web-based system setup | Membrane proteins, complex assemblies |
| acpype [30] [38] | Ligand topology generation | Small molecule parameterization |
| FastMDAnalysis [17] | Trajectory analysis | Automated analysis, visualization |
| MDCrow [41] [36] | LLM-assisted workflow | Automated simulation planning |
| DynaMate [41] | Multi-agent framework | Customizable workflow automation |
| alchemlyb [40] | Free energy analysis | TI, FEP analysis |
| PyAutoFEP [36] | Automated FEP setup | Free energy perturbation |
The meticulous execution of energy minimization, equilibration, and production protocols forms the foundation of reliable molecular dynamics research. By adhering to established best practices while incorporating emerging technologies such as AI-driven automation and machine learning potentials, researchers can enhance the efficiency, reproducibility, and scientific impact of their computational studies. The continued development of integrated workflows and automated analysis frameworks promises to further democratize MD methodologies, enabling broader application across scientific domains from basic biophysics to structure-based drug design. As these tools evolve, maintaining rigorous validation of simulation protocols remains paramount for generating physically meaningful insights into molecular structure and function.
The analysis of molecular dynamics (MD) trajectories represents a critical bottleneck in computational research, often requiring researchers to chain together multiple software tools and write extensive custom scripts. This fragmented process creates significant barriers to efficiency, standardization, and reproducibility. This whitepaper examines the emerging paradigm of automated MD analysis environments, with a specific focus on FastMDAnalysis—a unified Python package that encapsulates core analyses into a single, coherent framework. We demonstrate how such tools can reduce coding requirements by over 90% while maintaining numerical accuracy, enabling researchers to focus on scientific interpretation rather than computational implementation. Within the broader context of MD workflow research, we position these developments as essential for achieving reproducible, high-throughput computational science.
Molecular dynamics simulations have become an indispensable tool across materials science, drug discovery, and biosciences, generating massive quantities of trajectory data that track the time evolution of atomic positions and velocities [24]. The critical challenge has shifted from simply running simulations to effectively analyzing the resulting complex, high-dimensional data. Traditional analysis approaches require researchers to piece together multiple specialized tools and write substantial bespoke scripting code, creating workflow fragmentation that hinders both efficiency and reproducibility [17]. This paper examines automated solutions to this challenge, with particular emphasis on integrated software environments that unify the analysis pipeline while ensuring computational robustness and methodological consistency.
FastMDAnalysis: A Unified Framework for Automated Analysis
Core Architecture and Design Principles
FastMDAnalysis addresses the fragmentation problem through a unified, automated environment for end-to-end MD trajectory analysis. Built upon the high-performance foundations of MDTraj and scikit-learn, it combines computational reliability with a simplified interface through a dual-interface design [17]. This architecture encapsulates diverse analysis algorithms into a coherent framework while maintaining the numerical accuracy of the underlying validated libraries.
A key innovation in FastMDAnalysis is its explicit focus on reproducibility, enforced through consistent parameter management, detailed execution logging, and standardized output of both publication-quality figures and machine-readable data files. This approach ensures that analyses can be precisely repeated and verified across different research environments.
Comprehensive Analysis Capabilities
The software provides a comprehensive suite of analytical techniques in a unified environment:
- Structural Metrics: Root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and radius of gyration
- Solvation Analysis: Hydrogen bonding patterns and solvent accessibility surfaces
- Structural Classification: Secondary structure assignment and conformational clustering
- Dimensionality Reduction: Principal component analysis (PCA) for identifying essential dynamics
- Advanced Sampling: Hierarchical clustering for metastable state identification [17]
Table 1: Key Analysis Modules in FastMDAnalysis
| Analysis Category | Specific Metrics | Application Context |
|---|---|---|
| Structural Stability | RMSD, RMSF, Radius of Gyration | Protein folding, conformational stability |
| Solvation & Interactions | Hydrogen Bonding, SASA | Ligand binding, solvation effects |
| Collective Dynamics | Principal Component Analysis | Functional motions, allosteric mechanisms |
| Conformational Clustering | Hierarchical Clustering | Metastable states, transition pathways |
Performance and Validation
In a validation case study analyzing a 100 ns simulation of Bovine Pancreatic Trypsin Inhibitor (BPTI), FastMDAnalysis performed a comprehensive conformational analysis with a single command, producing results in under 5 minutes [17]. The implementation demonstrated a >90% reduction in the lines of code required for standard workflows while maintaining numerical accuracy against reference implementations on benchmark systems including ubiquitin and the Trp-cage miniprotein.
Experimental Protocols and Methodologies
Workflow Automation Protocol
The following protocol outlines the standard methodology for automated trajectory analysis using unified frameworks:
- Trajectory Preparation: Input trajectory data from common MD packages (GROMACS, NAMD, AMBER) in appropriate formats
- System Setup: Define topological information and atomic selections for analysis
- Analysis Configuration: Select analytical modules and parameters through configuration file or API
- Execution: Run automated analysis pipeline with built-in optimization
- Output Generation: Collect standardized results including structural data, quantitative metrics, and visualization assets
Radial Distribution Function Calculation
The radial distribution function (RDF) provides a fundamental metric for quantifying structural features in condensed phases [24]. The RDF, g(r), is mathematically defined as:
$$g(r) = \lim{dr \to 0} \frac{p(r)}{4\pi (N{pairs}/V)r^2dr}$$
where p(r) is the average number of atom pairs found at distance between r and r+dr, V is the system volume, and Npairs is the number of unique atom pairs between selections [42].
The computationally intensive component involves histogramming interatomic distances across trajectory frames:
$$p(r) = \frac{1}{N{frame}} \sumi^{N{frame}} \sum{j \in sel1} \sum{k \in sel2; k \neq j} \sum{\kappa} d{\kappa}(r; r{ijk})$$
where $d_{\kappa}$ represents the binning function that places distances into appropriate histogram bins [42]. For high-performance applications, this calculation can be accelerated up to 92× using multi-GPU implementations [42].
Principal Component Analysis for Essential Dynamics
Principal Component Analysis identifies dominant modes of collective motion by diagonalizing the covariance matrix of atomic positional fluctuations:
- Covariance Matrix Construction: Calculate the covariance matrix of atomic coordinates after alignment
- Diagonalization: Extract eigenvectors (principal components) and eigenvalues (variances)
- Projection: Map the trajectory onto principal components to reduce dimensionality
- Cluster Analysis: Identify conformational states in the reduced space [24]
This approach enables researchers to extract large-scale conformational changes from the high-dimensional noise of atomic trajectories.
Visualization of Automated Analysis Workflows
End-to-End MD Analysis Pipeline
Automated MD Analysis Pipeline
Tool Integration Ecosystem
Tool Integration Ecosystem
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Software Solutions for Automated MD Analysis
| Tool/Resource | Function | Application Context |
|---|---|---|
| FastMDAnalysis | Unified analysis environment | End-to-end trajectory processing |
| MDTraj | High-performance trajectory operations | Core computational backend |
| scikit-learn | Machine learning algorithms | Dimensionality reduction, clustering |
| VMD | Visualization & GPU-accelerated analysis | RDF calculation, trajectory viewing |
| OMol25 Dataset | Quantum chemical training data | Neural network potential development |
| eSEN/UMA Models | Neural network potentials | Accurate force field calculations |
Advanced Applications and Emerging Directions
Integration with Machine Learning Potentials
The field is rapidly evolving with the integration of machine learning interatomic potentials (MLIPs) trained on massive quantum chemical datasets. Meta's Open Molecules 2025 (OMol25) dataset, containing over 100 million quantum chemical calculations, enables neural network potentials (NNPs) that achieve accuracy comparable to high-level density functional theory at dramatically reduced computational cost [43]. Architectures like eSEN and the Universal Model for Atoms (UMA) demonstrate essentially perfect performance on molecular energy benchmarks, representing an "AlphaFold moment" for atomistic simulation [43].
LLM-Driven Workflow Automation
Recent advances in large language models (LLMs) have enabled new paradigms for workflow automation. Systems like MDCrow use chain-of-thought reasoning over 40 expert-designed tools to handle file processing, simulation setup, output analysis, and information retrieval [18]. This approach demonstrates the potential for natural language interfaces to further reduce technical barriers in MD analysis.
Specialized Applications
Advanced analysis pipelines are enabling insights in specialized domains:
- Materials Design: MD simulations calculate stress-strain curves at the atomic scale, revealing mechanical properties including Young's modulus, yield stress, and tensile strength [24]
- Enhanced Oil Recovery: Automated analysis elucidates polymer-oil interactions at atomic scale, predicting how polymer wettability changes affect recovery efficiency [44]
- Diffusion Processes: Mean square displacement (MSD) analysis quantifies ion and molecule mobility through materials, enabling calculation of diffusion coefficients via Einstein's relation [24]
Automated analysis tools like FastMDAnalysis represent a fundamental shift in molecular dynamics workflow research, transforming trajectory analysis from a fragmented, script-heavy process into a reproducible, efficient pipeline. By encapsulating complex algorithms into unified frameworks with standardized outputs, these tools simultaneously accelerate discovery and enhance methodological rigor. As the field evolves toward tighter integration with machine learning potentials and natural language interfaces, researchers gain unprecedented capacity to extract scientific insight from the complex dynamics of molecular systems. The continued development of automated analysis frameworks will be essential for realizing the full potential of molecular dynamics simulations across chemical, biological, and materials sciences.
Molecular dynamics (MD) simulation has emerged as a powerful computational microscope, enabling researchers to observe the dynamic behavior of biological systems at an atomic level. For researchers and drug development professionals, the true value of MD lies not in running simulations, but in the subsequent analysis of the vast and complex trajectory data generated. Proper analysis transforms raw atomic coordinates into profound biological insights, revealing the intricacies of conformational change, molecular stability, and binding interactions that are critical for understanding disease mechanisms and guiding rational drug design. This guide details the core analytical techniques—RMSD, RMSF, Rg, and hydrogen bonding—that form the essential toolkit for extracting meaningful information from MD simulations within the broader context of a molecular dynamics research workflow [45] [46].
Foundational Concepts in MD Analysis
At its core, an MD simulation predicts the movement of every atom in a molecular system over time based on a physics-based model, producing a trajectory that describes the atomic-level configuration at each time step [31]. The analyses described below are performed on this trajectory data to quantify and interpret the system's behavior.
- RMSD (Root-Mean-Square Deviation) measures the average distance between the atoms of a structure (e.g., a protein) after simulation and a reference structure (usually its starting conformation). It is a standard measure of structural distance and a key indicator of structural stability over time [47] [46].
- RMSF (Root-Mean-Square Fluctuation) quantifies the deviation of a particle (like a protein residue) from its average position, rather than a reference structure. It is ideal for identifying flexible or rigid regions within a molecule [47].
- Rg (Radius of Gyration) describes the compactness of a molecular structure. It is calculated as the root-mean-square distance of all atoms in the structure from their common center of mass [45].
- Hydrogen Bonding analysis identifies and characterizes the formation of hydrogen bonds, which are crucial for maintaining secondary, tertiary, and quaternary structures of proteins and nucleic acids, as well as for molecular recognition and specificity [48] [49].
The following workflow illustrates how these analyses integrate into a typical MD simulation research pipeline:
Detailed Analytical Methodologies
Root-Mean-Square Deviation (RMSD)
Experimental Protocol: The calculation of RMSD involves a few defined steps. First, a subset of atoms for the analysis must be selected; for protein stability assessment, this is typically the protein backbone or Cα atoms [47]. The structures from the trajectory are then superposed onto the reference structure (e.g., the initial energy-minimized structure or an experimental crystal structure) to minimize the RMSD. Finally, the RMSD is calculated for every frame in the trajectory using the standard formula, which computes the square root of the average squared distance between the coordinates of the selected atoms in the simulated and reference structures [46]. The resulting time series plot reveals the convergence and stability of the simulation.
Interpretation of Results: A low and stable RMSD value indicates that the simulated structure remains close to its reference conformation, suggesting high stability. A rising RMSD curve suggests significant conformational drift, while a plateau indicates that the system has reached a stable state or a new metastable conformation. Sudden jumps in RMSD can signify large-scale conformational transitions [47] [46]. In drug discovery, the RMSD of a protein-ligand complex is often compared to that of the apo protein; a stable complex RMSD can indicate strong binding and structural integrity [45].
Root-Mean-Square Fluctuation (RMSF)
Experimental Protocol: RMSF is calculated for each residue in a protein (or nucleotide in a nucleic acid). The first step is to calculate the time-averaged position for each atom over the entire trajectory or a stable portion of it. The RMSF is then derived as the root mean square of the fluctuation of each atom from this mean position [47]. This analysis is particularly useful when performed on the Cα atoms of a protein to understand residue-level mobility.
Interpretation of Results: Peaks in an RMSF plot correspond to regions of high flexibility. In proteins, loops and terminal regions typically show higher RMSF values, while secondary structural elements like alpha-helices and beta-sheets exhibit lower fluctuations, indicating rigidity [47]. In the context of ligand binding, a reduction in RMSF for residues near the binding site can indicate a stabilization effect upon inhibitor binding [45] [50]. This can help identify key residues involved in molecular recognition.
Radius of Gyration (Rg)
Experimental Protocol: The radius of gyration is calculated for a single molecule (like a protein) at each time step of the trajectory. The calculation involves determining the center of mass of the molecule and then computing the mass-weighted square distance of each atom from this center [45]. Plotting Rg over time provides a view of the global compactness of the structure throughout the simulation.
Interpretation of Results: A stable, low Rg value suggests a compact, well-folded, and stable structure. An increasing Rg trend may indicate protein unfolding or a transition to a more extended conformation. Conversely, a decreasing Rg can signify a collapse into a more compact state. For folded proteins, Rg is expected to fluctuate around a stable value, and deviations from this can signal destabilization, potentially induced by mutations or the binding of small molecules [45].
Hydrogen Bonding Analysis
Experimental Protocol: Identifying hydrogen bonds in a trajectory typically involves applying geometric criteria to every potential donor-acceptor pair. The most common method uses a distance cutoff (e.g., less than the sum of the van der Waals radii of the atoms, often around 3.5 Å for C...O contacts) and an angle cutoff (e.g., the D-H···A angle greater than 120°), where D is the donor, H is the hydrogen, and A is the acceptor [49]. Analysis can track the total number of hydrogen bonds over time, the specific residues involved, and the lifetime or occupancy of individual bonds [46].
The following diagram illustrates the key components and geometric criteria of a hydrogen bond:
Interpretation of Results: A stable or high number of hydrogen bonds, particularly in secondary structures like beta-sheets, contributes to structural integrity [49]. In molecular recognition, the consistent presence (high occupancy) of specific hydrogen bonds between a drug candidate and its protein target is a strong indicator of binding affinity and specificity [50]. Changes in hydrogen-bonding patterns can reveal the mechanistic details of enzyme catalysis or allosteric regulation [49] [51].
The Scientist's Toolkit: Essential Research Reagents & Software
The following table catalogues key computational tools and resources essential for conducting the analyses described in this guide.
Table 1: Key Research Reagent Solutions for MD Analysis
| Tool/Resource Name | Type | Primary Function in Analysis |
|---|---|---|
| GROMACS [46] | Software Suite | High-performance MD simulation and built-in analysis tools (e.g., gmx rms, gmx hbond). |
| AMBER Force Field [50] | Force Field | Provides parameters for calculating interatomic forces and energies during simulation. |
| VMD (Visual Molecular Dynamics) [47] [46] | Visualization Software | Trajectory visualization, animation, and a platform for various analysis routines. |
| PyMOL [46] | Visualization Software | Molecular graphics for rendering structures, analyzing interactions, and creating publication-quality images. |
| Bio3D Package [47] | R Package | A specialized toolkit for analyzing biomolecular structure and trajectory data (RMSD, RMSF, PCA). |
| MDAnalysis [47] | Python Library | An object-oriented toolkit for analyzing MD trajectories in a programmable and accessible manner. |
| SwissADME [50] | Web Server | In-silico prediction of Absorption, Distribution, Metabolism, and Excretion (ADME) properties. |
Integration of Analyses: A Practical Application
The power of these tools is fully realized when they are integrated. For instance, in a study seeking DPP4 inhibitors for type 2 diabetes, researchers combined these analyses to evaluate a potential drug candidate, ZINC000003015356 [50]. The complex's stability was confirmed by a low, stable RMSD. RMSF analysis showed that the binding of the candidate drug reduced fluctuation in key regions of the enzyme compared to the unbound form, indicating stabilization. The stability was further corroborated by a steady Rg, suggesting no loss of compactness. Finally, hydrogen bond analysis identified persistent specific interactions between the inhibitor and the DPP4 active site, explaining the structural observations at an atomic level. The MM/PBSA method was then used to calculate a favorable binding free energy, quantitatively affirming the insights gained from the geometric analyses [50]. This multi-faceted approach provides a robust framework for validating and understanding the behavior of molecular systems through simulation.
Principal Component Analysis (PCA) stands as a pivotal computational technique in the analysis of Molecular Dynamics (MD) simulations, enabling researchers to decouple essential collective motions from the complex, high-dimensional noise of atomic trajectories. By reducing dimensionality, PCA facilitates the identification of large-scale, concerted movements in biomolecules that are often functionally relevant, such as domain motions and allosteric transitions. This guide details the theoretical foundation, practical implementation, and advanced applications of PCA within the standard MD workflow, providing researchers and drug development professionals with the protocols and tools necessary to leverage this powerful analysis method.
Molecular Dynamics simulations generate vast amounts of data, producing time-series of atomic coordinates with numerous degrees of freedom. The resulting trajectories depict complex dynamics that are challenging to interpret directly. Principal Component Analysis addresses this by transforming the original, correlated Cartesian coordinates into a new set of orthogonal vectors, the principal components (PCs). These PCs are ordered by the amount of variance they capture from the trajectory, with the first PC representing the single largest collective motion [52] [24].
In essence, PCA separates the signal from the noise. It reveals the dominant patterns of collective motion—such as the hinge-bending of enzyme domains or the breathing motions of binding pockets—that may be obscured in the raw trajectory data. For drug discovery, understanding these intrinsic dynamics is crucial, as they can influence ligand binding, allostery, and protein function [53] [54]. When integrated into the broader MD workflow, PCA serves as a bridge between raw simulation data and biologically meaningful insight.
Theoretical Foundation of PCA
The input to PCA in MD is typically the coordinate covariance matrix. This matrix is constructed from the Cartesian coordinates of the atoms selected for analysis (e.g., Cα atoms of a protein). Each element of the matrix represents the covariance between the displacements of a pair of atomic coordinates from their mean positions [52].
For a system with N atoms, the covariance matrix C has dimensions of [3N x 3N]. Its elements are calculated as: ( C{ij} = \langle (xi - \langle xi \rangle)(xj - \langle xj \rangle) \rangle ) where ( xi ) and ( x_j ) are atomic coordinates, and ( \langle \rangle ) denotes the average over all frames in the trajectory.
This covariance matrix is then diagonalized to obtain its eigenvectors and eigenvalues.
- Eigenvectors represent the principal components themselves—the directions in the 3N-dimensional space along which the system fluctuates. They define a set of collective coordinates.
- Eigenvalues quantify the variance of the trajectory along each corresponding eigenvector. The eigenvalues are typically presented in descending order, meaning the first eigenvector (PC1) accounts for the largest portion of the total positional variance [52] [24].
The projection of the MD trajectory onto a principal component is computed as the scalar product between the atomic displacement vectors and the eigenvector. This converts the complex trajectory into a simple time series along a single mode of motion, facilitating analysis of conformational transitions and their frequencies.
Practical Implementation: A Step-by-Step Protocol
This section provides a detailed methodology for performing PCA on an MD trajectory using CPPTRAJ, a widely used tool within the AmberMD suite [52].
Preprocessing: Alignment and Covariance Matrix Calculation
The first critical step is to remove global translational and rotational motions, which are artifacts and do not represent internal dynamics. This is achieved by aligning every frame of the trajectory to a common reference structure, such as the first frame or the average structure.
Figure 1: The sequential workflow for performing Principal Component Analysis on an MD trajectory.
A sample CPPTRAJ script to load trajectories, perform alignment, and calculate the covariance matrix is as follows [52]:
Diagonalization and Projection
Once the covariance matrix is built, it is diagonalized to extract the eigenvectors and eigenvalues.
The resulting projection files (PC1 and PC2) contain time series data that describe the motion of the system along these dominant collective modes during the simulation.
Visualization and Analysis
The eigenvectors can be visualized as molecular motions by generating a pseudo-trajectory that oscillates along a specific PC. In CPPTRAJ [52]:
This output file (mode1.nc) can be visualized in standard molecular viewers like VMD or PyMOL to animate the dominant collective motion captured by PC1.
To understand the relative importance of each PC, the eigenvalues can be analyzed to determine the cumulative variance captured. The fraction of total variance explained by the first ( k ) PCs is the sum of their eigenvalues divided by the sum of all eigenvalues. Often, the first few PCs account for the majority of the biologically relevant motions [53].
Advanced Applications and Integration in Research
PCA extends beyond basic motion analysis into more sophisticated areas of research.
- Identifying Functional Motions: PCA has been instrumental in revealing large-scale, collective motions in proteins like calmodulin, where it helped characterize conformational changes related to calcium binding and target recognition [55].
- State Identification and Free Energy Landscapes: Projections from PCA are frequently used as reaction coordinates to construct free energy landscapes. Conformational states appear as basins in a 2D plot of PC2 vs. PC1, and the relative populations and barriers between these states can be quantified [24].
- Trajectory Compression and Visualization: The projected trajectories are vastly smaller than the original Cartesian coordinate data. This property is exploited by tools like PCAViz and pyPcazip for efficient compression, storage, and web-based visualization of long MD simulations [56] [53]. PCAViz, for instance, uses PCA to compress trajectory data into a JSON file that can be efficiently decompressed and visualized in a web browser, facilitating collaboration and sharing [53].
The Scientist's Toolkit: Essential Software for PCA
The following table details key software tools that enable PCA and related analyses for MD simulations.
| Tool Name | Language/Environment | Primary Function | Key Feature |
|---|---|---|---|
| CPPTRAJ [52] | AmberMD Suite | General MD trajectory analysis | Powerful, integrated tool for covariance matrix calculation and diagonalization. |
| pyPcazip [56] | Python | PCA-based compression and analysis | Suite of tools designed specifically for compression and analysis of MD data. |
| PCAViz [53] | Python/JavaScript | Web-based trajectory visualization | Compresses trajectories via PCA for interactive visualization in web browsers. |
Quantitative Analysis of PCA Performance
The accuracy of a PCA-based representation involves a trade-off with file size, influenced by the number of components retained and the numerical precision used.
Table 1: Impact of PCA Compression on Accuracy and File Size (LARP1 Protein Example) [53]
| Retained Variance | Rounding Precision | Avg. Non-Hydrogen Atom RMSD (Å) | Output JSON File Size (KB) |
|---|---|---|---|
| 50% | 0.001 | ~1.5 | ~200 |
| 70% | 0.001 | ~1.0 | ~260 |
| 80% | 0.001 | ~0.8 | ~300 |
| 90% | 0.001 | ~0.5 | ~380 |
| 90% | 0.01 | ~1.5 | ~150 |
Principal Component Analysis is an indispensable technique for elucidating the essential collective motions from the intricate dynamics captured in molecular simulations. Its integration into the MD workflow—from trajectory preprocessing and covariance analysis to projection and visualization—provides a powerful framework for interpreting atomic-scale motions in terms of biologically relevant, large-scale conformational changes. As MD simulations continue to grow in scale and complexity, advanced implementations and tools building on core PCA principles will remain vital for extracting clear insights from complex data, directly supporting research and drug development efforts.
Molecular dynamics (MD) simulations generate intricate trajectories that capture the temporal evolution of atomic positions, velocities, and forces within a molecular system. These trajectories contain a wealth of information about structural changes, dynamic processes, and thermodynamic properties, but their complexity presents significant challenges for interpretation. Effective visualization and analysis are therefore critical components of the MD workflow, enabling researchers to transform raw coordinate data into meaningful scientific insights. This technical guide provides a comprehensive overview of advanced techniques and tools for visualizing and analyzing MD trajectories, with particular emphasis on methodologies relevant to drug discovery and biomolecular research.
The process of trajectory analysis extends beyond simple visualization to encompass quantitative measurements of structural properties, dynamics, and interactions. As MD simulations continue to increase in temporal and spatial scale due to advances in computing hardware and algorithms, the resulting datasets have grown correspondingly large and complex. This expansion has driven the development of sophisticated analysis frameworks that can efficiently process trajectory data while providing intuitive visual representations that facilitate scientific discovery. The integration of these tools into cohesive workflows allows researchers to connect atomic-level structural details with macroscopic observables, bridging gaps between simulation and experiment.
Essential Visualization Software
Visualization programs form the foundation of MD trajectory analysis, providing researchers with intuitive graphical representations of complex three-dimensional molecular structures and their dynamics. These tools employ various rendering algorithms and user interface paradigms to help scientists comprehend spatial relationships, identify conformational changes, and communicate findings effectively.
Table 1: Primary Molecular Visualization Software
| Software | Trajectory Support | Key Features | Platform Compatibility |
|---|---|---|---|
| VMD | Native read of GROMACS trajectories | 3D graphics, built-in scripting, animation, analysis of large biomolecular systems | Windows, macOS, Linux |
| PyMOL | Requires format conversion (unless compiled with VMD plugins) | High-quality rendering, crystallography support, animations | Windows, macOS, Linux |
| Chimera | Reads GROMACS trajectories | Python-based, full-featured, extensive plugin ecosystem | Windows, macOS, Linux |
| nglview | Jupyter notebook integration | Interactive web-based visualization, ideal for inline analysis in computational workflows | Web browsers, Jupyter environments |
| MolecularNodes | Blender integration | Professional rendering, structural biology import, Geometry Nodes for atomic data | Blender (cross-platform) |
| SAMSON | Interactive visualization and simulation | AI assistant, physics-based construction, cloud computing integration | Windows, macOS, Linux |
When utilizing these visualization tools, researchers must recognize a critical limitation: visualization software determines chemical bonds for rendering purposes using internal heuristics applied to atomic coordinates, rather than consulting the topology information defined in GROMACS top or tpr files [57]. This distinction can lead to discrepancies between the visualized molecular representation and the actual bonding topology used in simulations, particularly for non-standard residues, transition states, or systems with unusual bonding patterns. Verification of critical structural features against the simulation topology is therefore recommended when bond assignment ambiguities could affect data interpretation.
Visualization tools have evolved beyond simple structure viewers into comprehensive platforms for interactive analysis. Modern systems like SAMSON incorporate AI assistants to help with molecular design tasks and provide contextual guidance [58], while virtual reality solutions such as Nanome offer immersive environments for collaborative structural analysis [59]. These advanced interfaces enable researchers to manipulate complex molecular systems more intuitively, potentially revealing structural insights that might be overlooked in traditional desktop visualization environments.
Trajectory Analysis Frameworks and Techniques
Core Analysis Approaches
Extracting scientifically meaningful information from MD trajectories requires specialized analysis techniques that can quantify structural, dynamic, and thermodynamic properties. These approaches range from simple geometric measurements to sophisticated statistical analyses that characterize ensemble behaviors and rare events.
Table 2: Trajectory Analysis Methods and Applications
| Analysis Type | Representative Tools | Key Metrics | Common Applications |
|---|---|---|---|
| Geometric Analysis | GROMACS utilities, MDAnalysis | Distance, angle, dihedral distributions; radius of gyration; solvent accessible surface area | Conformational sampling, folding/unfolding studies, compaction metrics |
| Dynamics Analysis | GROMACS, MDAnalysis, pydiffusion | Mean squared displacement; rotational correlation times; residence times | Diffusion coefficients, molecular tumbling, binding kinetics |
| Interaction Analysis | pycontact, PyInteraph | Hydrogen bonding patterns; salt bridge persistence; non-covalent interaction networks | Protein-ligand binding, allosteric communication pathways |
| Cluster Analysis | GROMACS, PENSA | Root-mean-square deviation (RMSD); structural clustering populations | Conformational classification, representative structure identification |
| Density Analysis | GROMACS spatial tool | Spatial distribution functions; density maps | Solvation structure, micelle formation, ion distribution around biomolecules |
The MDAnalysis library serves as a particularly versatile foundation for trajectory analysis, providing a Python framework that supports numerous specialized toolkits [60]. This ecosystem includes tools like LiPyphilic for lipid membrane analysis, PBxplore for protein block analysis, and taurenmd for command-line trajectory analysis. The modular nature of this ecosystem allows researchers to combine multiple analysis approaches within unified workflows, facilitating comprehensive characterization of complex molecular systems.
Extraction of Trajectory Information
Several technical approaches exist for accessing trajectory data within custom analysis routines. The GROMACS toolkit provides multiple utilities for this purpose, including gmx traj for writing data in xvg format compatible with external plotting software, and gmx dump for converting trajectory information into text formats readable by mathematical software such as MATLAB or Mathematica [57]. For researchers developing custom analysis code, the GROMACS distribution includes template C++ code (gromacs/share/template/template.cpp) that provides a starting point for trajectory processing programs [57]. These options accommodate diverse programming preferences and application requirements, from rapid prototyping with scripting languages to high-performance implementations in compiled languages.
Python has emerged as a particularly popular environment for trajectory analysis due to its rich ecosystem of scientific computing libraries. The MDAnalysis library enables researchers to load trajectory data directly into Python for analysis with packages like NumPy, SciPy, and scikit-learn [60]. This approach facilitates the implementation of sophisticated analyses including machine learning approaches for pattern recognition in conformational ensembles. For example, the acceleratedsamplingwith_autoencoder toolkit leverages deep learning to identify relevant collective variables from trajectory data [60], demonstrating how modern analysis frameworks are integrating cutting-edge computational techniques.
Essential Experimental Protocols
Micelle Clustering Protocol
The analysis of micelle formation and behavior in MD simulations requires careful preprocessing to address artifacts introduced by periodic boundary conditions. Without proper clustering, a single micelle may appear split across simulation box boundaries, leading to incorrect calculations of properties such as radius of gyration and radial distribution functions [57]. The following three-step protocol ensures correct micelle clustering:
Step 1: Initial Clustering
This command processes the first frame of the trajectory (-e 0.001 selects the first frame) and clusters molecules to ensure all lipids reside within the unit cell. The output frame (a_cluster.gro) serves as the reference for subsequent trajectory processing.
Step 2: Topology Regeneration
This step generates a new run input file (a_cluster.tpr) using the clustered structure as the reference conformation. This updated topology ensures consistent molecular definitions throughout the processing workflow.
Step 3: Trajectory Processing
The final command processes the entire trajectory using the newly created topology, applying the "nojump" correction to prevent molecules from appearing to jump across periodic boundaries. The resulting trajectory (a_cluster.xtc) is suitable for subsequent analysis of micelle properties [57].
This protocol is essential for obtaining physically meaningful results from micelle simulations, as it ensures continuous tracking of molecular associations throughout the trajectory. Similar approaches may be adapted for other self-assembling systems where maintaining cluster integrity across periodic boundaries is critical for accurate analysis.
Workflow for Comprehensive Trajectory Analysis
A systematic approach to trajectory analysis ensures consistent and reproducible characterization of MD simulation data. The following workflow diagram illustrates the key stages in this process, from initial trajectory processing to final visualization and interpretation:
This workflow encompasses both the computational processing of trajectory data and the scientific interpretation of results. Quality control steps verify the physical reasonableness of simulations before proceeding to detailed analysis, preventing misinterpretation of artifacts resulting from simulation issues. The multidimensional analysis phase incorporates structural, dynamic, energetic, and system-specific approaches to develop a comprehensive understanding of the molecular system under investigation.
Visualization and Plotting Methodologies
Data Plotting Software
Quantitative analysis of MD trajectories typically generates numerical data that requires visualization through specialized plotting software. The GROMACS analysis toolkit produces output in xvg format, which can be processed by various graphing applications depending on researcher preference and required capabilities.
Table 3: Software for Visualization of Analysis Data
| Software | Platform | Key Features | XVG File Compatibility |
|---|---|---|---|
| Grace | Unix-like systems | WYSIWYG 2D plotting, Motif/Lesstif UI | Native support (including embedded formatting codes) |
| gnuplot | Cross-platform | Command-driven, interactive, extensive output formats | Requires set datafile commentschars "#@&" to ignore formatting |
| Matplotlib | Python | Programmatic generation, publication-quality output, extensive customization | Compatible via NumPy loadtxt with comment character specification |
| R | Cross-platform | Statistical computing environment, advanced graphing capabilities | Requires preprocessing to handle Grace formatting |
| Microsoft Excel | Windows, macOS | Widely available, familiar interface | Requires file extension change to .csv and data cleaning |
For researchers using Python, Matplotlib provides a powerful and flexible environment for creating publication-quality figures. The following code example demonstrates basic trajectory data plotting:
This approach can be extended to create multi-panel figures, overlayed datasets, and specialized visualizations that integrate directly with analysis scripts, facilitating reproducible research workflows.
Advanced and Interactive Visualization
Modern visualization approaches extend beyond static images to include interactive and immersive experiences that enhance researcher understanding of complex molecular behaviors. Tools like MDSrv stream and visualize MD trajectories interactively within web browsers, enabling collaborative analysis across geographically distributed research teams [60]. Virtual reality platforms such as Nanome provide spatially intuitive environments for examining molecular structures and dynamics, with users reporting significant improvements in their ability to identify key structural features and communicate findings [59].
The MolecularNodes plugin for Blender represents another advanced visualization approach, leveraging professional rendering capabilities to create publication-quality images and animations [60]. This tool facilitates the creation of visually compelling representations that effectively communicate complex structural concepts, while its integration with Blender's Geometry Nodes system enables sophisticated procedural visualization workflows. These advanced visualization technologies are particularly valuable in drug discovery contexts, where understanding subtle conformational changes and interaction patterns can guide optimization of therapeutic compounds.
The Scientist's Toolkit: Essential Research Reagents
Successful analysis of MD trajectories requires both software tools and conceptual frameworks for interpreting complex data. The following table catalogues essential "research reagents" – computational tools and methodologies – that form the foundation of effective trajectory analysis workflows.
Table 4: Essential Research Reagents for MD Trajectory Analysis
| Tool/Technique | Category | Primary Function | Example Applications |
|---|---|---|---|
| MDAnalysis Library | Analysis Framework | Python toolkit for trajectory loading and processing | Foundation for custom analysis scripts; integration with scientific Python ecosystem |
| GROMACS Analysis Utilities | Analysis Suite | Built-in trajectory analysis algorithms | Calculation of RMSD, RDF, SASA; clustering; hydrogen bond analysis |
| PyContact | Interaction Analysis | Quantification of non-covalent interactions | Identification of persistent protein-ligand contacts; allosteric pathway analysis |
| LiPyphilic | Specialized Analysis | Lipid membrane system characterization | Lipid diffusion; phase behavior; domain formation in membranes |
| RotamerConvolveMD | Spectroscopy Integration | DEER spectroscopy prediction from trajectories | Validation of simulations against experimental distance measurements |
| MDTraj | Lightweight Analysis | Fast trajectory operations with minimal dependencies | Rapid prototyping of analysis methods; large-scale trajectory processing |
| chemiscope | Visualization | Interactive structure/property explorer | Correlation of structural features with calculated properties in materials |
| Free Energy Tools | Energetic Analysis | Binding free energy calculations | Drug discovery prioritization; protein-ligand affinity prediction |
These computational reagents enable researchers to connect atomic-level simulation data with experimentally observable phenomena, creating bridges between simulation and experimental domains. Specialized tools like RotamerConvolveMD facilitate direct comparison with spectroscopic data [60], while free energy calculators provide predictions of binding affinities relevant to drug discovery programs. The integration of these diverse tools into cohesive workflows allows for comprehensive characterization of molecular systems across multiple temporal and spatial scales.
The field of MD trajectory visualization and analysis has evolved dramatically from basic structure viewers to integrated platforms that combine advanced visualization with quantitative analysis capabilities. This evolution has been driven by both the increasing scale and complexity of MD simulations and the growing sophistication of scientific questions being addressed through computational approaches. Modern analysis workflows seamlessly integrate multiple specialized tools, leveraging the strengths of each to develop comprehensive understandings of molecular systems.
Looking forward, several trends are likely to shape the future of trajectory analysis. Machine learning approaches are increasingly being applied to identify relevant features and patterns in high-dimensional trajectory data, potentially revealing subtle correlations that might escape conventional analysis methods. Interactive and immersive visualization technologies continue to mature, offering new paradigms for exploring complex structural dynamics. Furthermore, the growing emphasis on reproducibility and transparency in computational science is driving the development of workflow management systems that capture and preserve analysis procedures. By staying abreast of these developments while maintaining a firm foundation in the established techniques and tools described in this guide, researchers can effectively harness the full scientific potential of molecular dynamics simulations.
Molecular dynamics (MD) simulations have evolved into powerful computational tools for studying biomolecular systems at atomic resolution. This whitepaper provides an in-depth technical examination of MD methodologies applied to three critical areas: protein folding mechanisms, drug-binding prediction, and allosteric regulation analysis. By integrating advanced approaches such as graph theory and machine learning with traditional MD frameworks, researchers can now capture complex biological phenomena with unprecedented detail. This guide presents current case studies, detailed experimental protocols, and quantitative analyses aimed at equipping researchers and drug development professionals with practical frameworks for implementing these techniques within a comprehensive MD simulation workflow.
Molecular dynamics simulations provide a computational microscope for observing atomic-level motions in biomolecular systems. Modern MD workflows have matured beyond simple trajectory calculation to encompass sophisticated analysis frameworks that bridge simulation data with experimental validation. The core strength of MD lies in its ability to generate atomic-resolution structural models for states that are only indirectly accessible by experiment, providing a molecular framework for interpreting experimental data and fleshing out complex biological processes [61]. As simulations extend into microsecond timescales and beyond, researchers can now directly observe phenomena like protein folding pathways and drug-binding events that were previously inferable only through indirect means.
The integration of MD with other computational approaches has created powerful hybrid methodologies. Graph theory approaches, for instance, provide a robust framework for analysing the structural and functional properties of biomolecules by representing atoms or groups of atoms as nodes and their dynamic interactions as edges [62]. Similarly, machine learning potentials are increasing simulation accuracy and speed. These advanced workflows are transforming MD from a specialized computational technique into an essential component of structural biology and drug discovery pipelines.
Protein Folding Studies
Case Studies and Quantitative Results
Protein folding remains one of the most challenging and insightful applications of MD simulations. Several small model systems have been successfully studied using long-timescale MD, providing atomic-level insights into folding mechanisms.
Table 1: Protein Folding Case Studies and MD Results
| System | Simulation Protocol | Refolding Criteria | Key Findings | Reference |
|---|---|---|---|---|
| HP-35 Villin Headpiece | 20 simulations of 1.0 μs at 300K | Global Cα RMSD | Sub-1.0 Å RMSD structures reached in 7 trajectories; identified critical stability region | [61] |
| Trp-cage | Multiple 20 ns at 325K | Force field energy | Successful refolding prediction with correct intramolecular contacts | [61] |
| Chymotrypsin Inhibitor 2 (CI2) | 1 simulation of 200 ns at 348K (Tm) | Global Cα RMSD, contacts, topology | Demonstrated microscopic reversibility; same contacts gained/lost in folding/unfolding | [61] |
| HP-35 Variant | 410 μs aggregate at 300K | Individual helix RMSD, hydrophobic core contacts | Refolding observed from multiple thermally denatured starting conformations | [61] |
| FBP28 WW Domain | Multiple unfolding simulations | Φ-value comparison | Transition state structure identification; first turn native-like but lacking correct H-bonds | [61] |
Experimental Protocols and Methodologies
Residual Structure Analysis in Denatured States
Objective: Characterize residual structure in chemically denatured proteins and compare with experimental data. Methodology:
- System Preparation: Generate extended protein structures using molecular modeling tools
- Solvation: Employ explicit solvent models (e.g., TIP3P water) with ion concentrations matching experimental conditions
- Equilibration: Perform energy minimization followed by gradual heating to target temperature (typically 300K)
- Production Simulation: Run multiple independent trajectories (5-20) for hundreds of nanoseconds to microseconds
- Analysis: Calculate radius of gyration (Rg) as function of simulation time and compare with FRET and SAXS experimental data
Key Considerations: Dye molecules in FRET experiments may perturb denatured states; simulations help interpret experimental discrepancies by providing pure protein structural evolution without experimental artifacts [61].
Refolding from Extended States
Objective: Observe complete refolding pathways from fully extended conformations. Methodology:
- Initial Structure: Generate fully extended conformation using internal coordinate manipulation
- Force Field Selection: Choose appropriate force field (e.g., AMBER, CHARMM) with explicit water representation
- Simulation Parameters: Run at physiological temperature (300K) with periodic boundary conditions
- Multiple Trajectories: Initiate 10-20 independent simulations from same extended state with different random velocity distributions
- Assessment Metrics: Monitor Cα RMSD, secondary structure content, native contact formation, and specific hydrophobic core interactions
Validation: Compare final refolded structures with experimental NMR or X-ray data using pairwise contact maps and NOE validation [61].
Research Reagent Solutions
Table 2: Essential Research Tools for Protein Folding MD Studies
| Tool/Software | Type | Function | Application Example |
|---|---|---|---|
| MDTraj | Software Library | Trajectory analysis | RMSD calculations, secondary structure assignment, order parameter extraction [63] |
| AMBER | Force Field/Software | Molecular mechanics parameters | Providing energy functions for protein folding simulations [61] |
| CHARMM | Force Field/Software | Molecular mechanics parameters | Alternative force field for folding studies [61] |
| Graph Theory Analysis | Analytical Framework | Network analysis of dynamic correlations | Identifying key residues in folding pathways [62] |
| Cross-Correlation Analysis | Analytical Method | Identifying correlated motions | Revealing patterns of coordinated motion during folding [62] |
Drug Binding Prediction
Ensemble Docking Approaches
Traditional rigid receptor docking approaches often fail to account for protein flexibility, limiting their predictive accuracy for drug binding. MD-enhanced ensemble docking addresses this limitation by incorporating target flexibility through multiple receptor conformations.
Methodology:
- MD Simulation: Perform one-tenth nanosecond MD simulations of the drug target
- Conformational Clustering: Group sampled structures into representative clusters
- Ensemble Docking: Screen compounds against all cluster representatives
- Binding Affinity Prediction: Calculate average binding affinity across ensembles
This approach captures binding-induced conformational changes and produces sufficient flexibility information about the structure and configuration of drug binding [64]. Compared to rigid receptor docking, ensemble docking properly accounts for protein topology and conformational changes while remaining computationally feasible through parallelization at either conformation or compound-level dimensions.
Signaling Pathway and Workflow Visualization
Graph Theory Approaches for Biomolecular Analysis
Network Models of Allosteric Regulation
Graph theory provides a mathematical framework for representing biomolecular structures as networks of nodes (atoms or residues) and edges (interactions), enabling quantitative analysis of allosteric mechanisms and signal propagation.
Node Representation: Amino acids represented by Cα atoms; nucleotides by P atoms (purines N1, pyrimidines N9) [62] Edge Definition: Dynamic correlations between nodes calculated through cross-correlation or generalized correlation methods
Correlation Analysis Methodologies
Cross-Correlation Analysis
Cross-correlation analysis identifies coordinated motions by computing Pearson's correlations between atomic fluctuations:
[CC{ij} = \frac{\langle \Delta \vec{ri}(t) \cdot \Delta \vec{rj}(t) \rangle}{\sqrt{\langle \Delta \vec{ri}(t)^2 \rangle \langle \Delta \vec{r_j}(t)^2 \rangle}}]
Where ( \Delta \vec{ri} ) and ( \Delta \vec{rj} ) are fluctuation vectors of atoms i and j, respectively. Positive CCij values indicate correlated motion, while negative values describe anticorrelated motions [62].
Generalized Correlation Method
For capturing non-linear correlations, the generalized correlation method assesses mutual information between residues:
[MI(xi, xj) = \iint p(xi, xj) \ln \left[ \frac{p(xi, xj)}{p(xi) \cdot p(xj)} \right] dxi dxj]
Normalized generalized correlation coefficients are then defined as:
[GC{ij}(xi, xj) = \left[ 1 - \exp \left( -\frac{2MI(xi, x_j)}{d} \right) \right]^{1/2}]
Where d=3 represents the dimensionality of xi and xj [62]. This approach identifies any form of dependence in atomic motions, irrespective of directional relationships.
Graph Theory Workflow Visualization
Case Studies in Allosteric Mechanisms
Graph theory approaches combined with MD have revealed allosteric mechanisms in several biologically significant systems:
- tRNA Synthase: Early implementation identifying dynamic allosteric networks in aminoacylation [62]
- Transcription Initiation Complexes: Graphs defining signal transduction during transcription initiation [62]
- Nucleosome Core Particle: Revealing how allosteric signals propagate through histone proteins to influence DNA accessibility [62]
- CRISPR-Cas9 System: Identifying critical residues and conformational changes governing catalysis and selectivity [62]
Integrated MD Workflow for Comprehensive Analysis
Unified MD Simulation Framework
Advanced Integration with Machine Learning
Machine learning frameworks are increasingly integrated with MD simulations to enhance sampling efficiency and accuracy. Network models trained on protein families provide better physical priors during learning until reaching steady state [64]. These approaches implement accurate, fast, and flexible soft potentials within ML frameworks, though challenges remain in serial implementation speedup and end-to-end optimization through gradients.
Molecular dynamics simulations have transformed from computational novelties into essential tools for understanding biomolecular function. The integration of graph theory analysis, ensemble docking approaches, and machine learning frameworks with traditional MD workflows has created a powerful multidisciplinary approach for studying protein folding, drug binding, and allosteric regulation. As simulation timescales continue to expand and methodologies refine, MD will play an increasingly central role in bridging theoretical biophysics with practical drug design and biomolecular engineering. The case studies and methodologies presented here provide researchers with a foundation for implementing these advanced techniques within their own molecular simulation workflows.
Navigating Computational Challenges: A Guide to Common Errors and Performance Optimization
Energy minimization (EM) represents a critical, foundational step in the molecular dynamics (MD) simulation workflow. Its primary role is to relieve adverse atomic contacts, resolve steric clashes, and relax strained interactions inherent in experimentally derived or model-built initial structures. Without proper minimization, the large, unphysical forces present in an unrelaxed structure can cause instantaneous simulation failure or introduce significant artifacts during subsequent equilibration phases. Within the broader context of molecular dynamics simulation workflow research, robust energy minimization is not merely a technical prelude but a determinant of thermodynamic relevance, ensuring that the system resides in a reasonable starting configuration before exploring its conformational landscape. This guide deciphers two common yet critical EM errors—'Forces Not Converged' and 'Stepsize Too Small'—providing researchers with the diagnostic and corrective methodologies essential for reliable simulation science.
Understanding the Errors: A Diagnostic Guide
The errors "Energy minimization has stopped, but the forces have not converged..." and the algorithm stopping because "the algorithm tried to make a new step whose size was too small" are intimately related. Both indicate that the minimizer cannot find a direction to move atoms that lowers the potential energy while simultaneously reducing the maximum force (Fmax) below the user-specified tolerance (emtol).
The "Stepsize too small" condition is often a symptom of the minimizer being trapped in a region of the potential energy surface where any infinitesimal movement increases the energy, a classic sign of being in, or very near, a local minimum. However, the precision of this local minimum is insufficient, meaning the forces remain unacceptably high for the desired production simulation. This frequently stems from a handful of atoms experiencing extreme forces, as evidenced in a case where the maximum force was 1.5580516e+04 kJ mol⁻¹ nm⁻¹ on a single atom, preventing convergence despite 2062 steps of steepest descent [65].
Table: Interpreting Common Energy Minimization Errors
| Error Message | Primary Meaning | Common Underlying Causes |
|---|---|---|
| Forces Not Converged | The maximum force (Fmax) on any atom remains above the set tolerance (emtol) after the minimizer exhausts its options. |
Severe steric clashes, incorrect topology, missing atoms, problematic constraints, an emtol value that is unrealistically tight. |
| Stepsize Too Small | The minimizer cannot find a displacement for any atom that results in a lower potential energy. | The system is in a local minimum (but forces are still high), a few atoms have extreme localized forces, constraints are preventing necessary movement. |
The GROMACS documentation clarifies that when these errors occur, the minimization is considered "converged to within the available machine precision, given your starting configuration and EM parameters" [66] [65]. This signifies that the algorithm has done all it can within its numerical limits, and the onus is on the researcher to alter the system's configuration or the minimization parameters.
Systematic Troubleshooting and Solutions
A systematic approach to resolving EM failures moves from simple parameter adjustments to more fundamental checks of the molecular system itself.
Adjusting Minimization Parameters and Algorithms
The initial response to convergence failure should be to reevaluate the minimization protocol. The choice of algorithm is crucial. The steepest descent algorithm is robust for poorly structured starting configurations and is recommended for the initial stages of minimization, often for the first 50-100 steps [67]. Its stability far from a minimum makes it ideal for relieving the largest steric clashes. For final convergence to a tighter tolerance, more efficient algorithms like conjugate gradients or L-BFGS are preferred [67] [68]. Note that conjugate gradients cannot be used with constraints in GROMACS [68].
Key parameters to review include:
emtol: The force tolerance. For preliminary relaxation before dynamics, a value of 100-1000 kJ mol⁻¹ nm⁻¹ is often sufficient, rather than the default 10.0 [67]. The enormousFmaxvalues in the examples (1.9e+05 [66] and 1.6e+04 [65]) indicate that achieving anemtolof 10 or 500 was impossible, and a larger value should be used.nsteps: The maximum number of steps. Ensure this is high enough (e.g., 5000-100000) to allow for slow convergence.constraints: As the error message suggests, "You might need to increase your constraint accuracy, or turn off constraints altogether" [66]. For initial minimization, settingconstraints = nonecan be highly beneficial. If constraints are necessary, usinglincs_iterandlincs_ordercan increase the accuracy of the LINCS algorithm.
Investigating and Correcting System Topology
If parameter adjustment fails, the problem likely lies in the molecular system itself.
- Severe Steric Clashes and Missing Atoms: Initial structures, especially those from homology modeling or with manually inserted ligands, can have atoms placed far too close together, creating gigantic repulsive forces. Visually inspecting the structure in a program like VMD or PyMol can identify these atomic overlaps. Furthermore, the
pdb2gmxutility will issue warnings like "WARNING: atom X is missing in residue XXX," which must be addressed before minimization can succeed [69]. Missing atoms must be modeled back in using specialized software. - Ligand and Residue Topologies: A common failure point is an incorrect topology for a non-standard residue or ligand. GROMACS will fail if a residue is not found in the force field's residue topology database (
rtpfile) [69]. Ensuring that all molecules have correct, consistent topologies is paramount. This may involve manually generating topology files using tools likex2topor external programs. - Position Restraints and Includes: The order of
#includestatements in the topology file matters. An#includedirective for a position restraint file must appear immediately after the#includefor the corresponding molecule's topology [69]. Incorrect ordering can lead to errors like "Atom index in position_restraints out of bounds."
Table: Summary of Core Minimization Algorithms in GROMACS
| Algorithm | Best Use Case | Advantages | Limitations |
|---|---|---|---|
| Steepest Descent | Initial stages of minimization; highly distorted structures [67]. | Robust, stable when far from a minimum. | Slow convergence near a minimum. |
| Conjugate Gradient | Final convergence to a tight force tolerance [67] [68]. | More efficient than steepest descent near a minimum. | Cannot be used with constraints in GROMACS [68]. |
| L-BFGS | Final convergence for large systems [68]. | Faster convergence than conjugate gradients; low memory requirements. | Not yet parallelized in GROMACS. |
A Protocol for Structured System Relaxation
For complex systems, particularly those derived from crystallographic data with potential disorder, a multi-stage relaxation protocol is more effective than a single minimization step. This approach progressively relaxes the system, preventing large, artifactual movements [67].
A recommended protocol is:
- Fix heavy atoms, minimize hydrogens and solvent: Apply position restraints to all protein heavy atoms and minimize. This allows added hydrogens, water, and ions to relax around a static framework.
- Restrain the backbone, minimize side chains: Apply position restraints to the protein backbone atoms (Cα, C, N) and minimize. This lets side chains adjust to relieve clashes.
- Progressively release restraints and minimize: Gradually reduce the force constant of the position restraints on the backbone in a series of minimizations until the system can be minimized with no restraints.
At each stage, the steepest descent algorithm can be used initially, switching to a more efficient algorithm like L-BFGS as the system approaches the minimum for that stage [67].
The Scientist's Toolkit: Essential Research Reagents
Successful energy minimization and MD workflows rely on a suite of software tools and resources.
Table: Key Software Solutions for Molecular Modeling and Simulation
| Tool / Resource | Category | Primary Function in Workflow |
|---|---|---|
| GROMACS [68] [70] | MD Simulation Engine | High-performance package for running energy minimization, equilibration, and production MD simulations. |
| MOE (Molecular Operating Environment) [71] | Comprehensive Modeling | An all-in-one platform for structure-based drug design, molecular docking, and QSAR modeling; useful for pre-processing structures. |
| Schrödinger Suite [71] | Advanced Modeling | Platform integrating quantum mechanics and machine learning for molecular modeling; includes tools for free energy calculations. |
| Cresset Flare [71] | Protein-Ligand Modeling | Offers advanced capabilities for protein-ligand modeling, including Free Energy Perturbation (FEP) and molecular dynamics. |
| VMD / PyMOL | Visualization | Critical for visually diagnosing structural problems, analyzing minimized structures, and visualizing trajectories. |
| pdb2gmx / x2top | Topology Generation | GROMACS utilities for generating molecular topologies from coordinate files. |
The following diagram summarizes the integrated decision-making process for diagnosing and resolving energy minimization failures, encapsulating the strategies detailed in this guide.
Diagnosing Energy Minimization Failures
In conclusion, "Forces Not Converged" and "Stepsize Too Small" errors are not terminal failures but diagnostic signals. They call for a systematic approach that balances numerical parameters with physical realism. By understanding the core algorithms, meticulously preparing the molecular system, and employing a structured relaxation protocol, researchers can reliably overcome these hurdles. Mastering energy minimization is a cornerstone of robust molecular dynamics workflow, ensuring that subsequent simulation stages are founded upon a stable, physically meaningful atomic configuration, thereby unlocking the full predictive power of computational biochemistry and drug discovery.
Within the broader research context of molecular dynamics (MD) simulation workflows, system instability represents a critical bottleneck that compromises data integrity and research progress. LINCS and SETTLE warnings, alongside 'range checking' errors, serve as primary indicators of underlying simulation pathologies that must be systematically diagnosed and resolved. These algorithms—LINCS (Linear Constraint Solver) and SETTLE—are fundamental to maintaining numerical stability by preserving bond lengths and geometries during integration, with SETTLE specifically optimized for rigid water molecules [72]. When these constraint solvers generate warnings, they signal that the simulation is deviating from physically realistic states, potentially leading to catastrophic failure or biologically irrelevant trajectories. For researchers in drug development and protein engineering, where MD simulations provide critical insights into conformational dynamics and binding mechanisms [73] [74], addressing these instabilities is not merely technical troubleshooting but a fundamental prerequisite for generating reliable, publication-quality results.
Theoretical Foundation of Constraint Algorithms
Mathematical Principles of LINCS and SETTLE
Constraint algorithms in MD simulations enforce holonomic constraints, typically fixed bond lengths, that would otherwise require impractically small integration time steps to capture accurately. The mathematical formulation begins with a system of (N) particles subject to (K) constraint equations:
[ \sigmak(\mathbf{r}1 \ldots \mathbf{r}_N) = 0; \;\; k=1 \ldots K ]
For example, a bond constraint between atoms (i) and (j) takes the form ((\mathbf{r}i - \mathbf{r}j)^2 - b^2 = 0) [72]. The forces required to maintain these constraints are derived from Lagrange multipliers:
[ \mathbf{G}i = -\sum{k=1}^K \lambdak \frac{\partial \sigmak}{\partial \mathbf{r}_i} ]
where (\mathbf{G}i) represents the constraint force on particle (i), and (\lambdak) are the Lagrange multipliers solved iteratively by SHAKE or matrix inversion by LINCS [72].
The LINCS algorithm operates through a two-stage process: first, it removes the projection of new bonds onto old bond directions, and second, it applies a correction for bond lengthening due to rotational effects [72]. This is mathematically expressed as:
[ \mathbf{r}{n+1}=(\mathbf{I}-\mathbf{T}n \mathbf{B}n) \mathbf{r}{n+1}^{unc} + {\mathbf{T}}_n \mathbf{d} ]
where (\mathbf{B}) is the gradient matrix of constraint equations, (\mathbf{T} = {{\mathbf{M}}^{-1}}\mathbf{B}^T ({\mathbf{B}}{{\mathbf{M}}^{-1}}\mathbf{B}^T)^{-1}), and (\mathbf{d}) is the vector of constraint distances [72].
The SETTLE algorithm provides an analytical solution for constraining rigid water molecules, specifically avoiding the calculation of the center of mass to reduce rounding errors [72]. This makes it particularly valuable for large systems where water molecules may constitute over 80% of the simulated atoms.
Algorithm Selection and System Compatibility
The choice between constraint algorithms depends critically on system composition and research objectives. LINCS is generally preferred for its speed and stability but is incompatible with coupled angle constraints, as the resulting high connectivity leads to large eigenvalues in the constraint coupling matrix that violate the convergence criterion for the matrix inversion [72]. SETTLE remains the exclusive option for rigid water models, while SHAKE offers broader compatibility for systems with complex coupled constraints at the cost of iterative solving and potential convergence issues.
Table 1: Constraint Algorithm Properties and Applications
| Algorithm | Mathematical Approach | Optimal Use Cases | Key Limitations |
|---|---|---|---|
| LINCS | Matrix inversion with expansion [72] | Standard biomolecular systems, bond constraints only [72] | Cannot handle coupled angle constraints [72] |
| SETTLE | Analytical solution [72] | Rigid water molecules [72] | Limited to specific triatomic geometries |
| SHAKE | Iterative Lagrange multiplier solution [72] | Systems with coupled constraints | Slower convergence, tolerance-dependent |
Systematic Diagnosis of LINCS/SETTLE Warnings
Error Classification and Root Cause Analysis
LINCS warnings manifest when the algorithm cannot adequately maintain bond lengths according to specified constraints, typically due to highly unusual forces or atomic displacements [75]. These warnings frequently correlate with 'range checking' errors, which occur when atomic positions exceed reasonable physical boundaries. Through analysis of troubleshooting forums and technical documentation, we have identified a systematic classification of these errors and their underlying causes.
Table 2: LINCS Warning Classification and Root Causes
| Warning Type | Common Error Messages | Primary Root Causes | Associated Simulation Artifacts |
|---|---|---|---|
| Minor Drift | "LINCS warning (1-5% drift)" | Slightly too large timestep, mild steric clashes [75] | Acceptable for short simulations, but accumulates over time |
| Major Drift | "LINCS warning (>5% drift)" | Significant force imbalances, improper equilibration [76] | Visible bond distortion, energy explosions |
| Catastrophic Failure | "There were X LINCS warnings," followed by crash [76] | Atomic overlaps, gross parameter mismatch [75] [76] | Simulation termination, 'range checking' errors |
The following diagnostic workflow provides a systematic approach for identifying the specific pathology underlying constraint warnings:
Diagnostic Workflow for LINCS/SETTLE Warnings
Quantitative Diagnostic Metrics
Effective diagnosis requires monitoring specific quantitative metrics during simulation initialization and early dynamics:
- Constraint Deviation: Monitor the maximum relative constraint deviation reported by LINCS, with values exceeding 0.0001 indicating potential instability [72].
- Energy Components: Track potential energy, angle energy, and dihedral energy for sudden jumps suggesting parameter mismatch.
- Pressure Anomalies: Abnormally high or oscillating pressure may indicate LJ parameter issues or incorrect cutoffs [75].
- Bond Length Distribution: Compare actual bond lengths with force field reference values, particularly for newly parameterized molecules.
Methodologies for System Stabilization
Pre-simulation Protocols: Equilibration and Parameter Validation
Proper system equilibration represents the most effective strategy for preventing constraint warnings. A structured equilibration protocol, validated through benchmarking studies [77], significantly reduces instability:
Energy Minimization: Begin with steepest descent or conjugate gradient minimization until the maximum force falls below 1000 kJ/mol/nm [75]. This resolves atomic overlaps and irregular bond lengths that would otherwise trigger LINCS warnings during dynamics.
Staged Temperature Coupling: Gradually heat the system to the target temperature while maintaining positional restraints on solute atoms (force constant of 1000 kJ/mol/nm²). Implement in stages: 100K increments with 100ps simulation at each stage [75].
Pressure Equilibration: After temperature stabilization, conduct NPT equilibration with maintained restraints, ensuring proper density convergence (fluctuations within ±5% of experimental values) [75].
Force field validation is equally critical. Inaccurate parameters—particularly for bonded terms—generate unrealistic forces that constraint algorithms cannot compensate [75]. Cross-reference bond lengths and angles with quantum mechanical calculations or crystallographic data for novel molecules, and ensure compatibility between force field versions and topology files.
Simulation Parameter Optimization
Strategic adjustment of simulation parameters can resolve persistent constraint warnings without compromising physical accuracy:
Timestep Optimization: For all-atom systems, reduce timestep to 1fs when LINCS warnings persist, particularly with stiff bonds [75]. For united-atom systems, 2fs typically represents the safe maximum.
LINCS Parameter Enhancement: Increase the LINCS expansion order (lincs-order) from the default 4 to 6 for improved accuracy, particularly for molecules with constrained angles [72]. Simultaneously, increase the number of iteration steps (lincs-iter) from 1 to 2-4 for complex systems.
Cutoff Scheme Adjustment: Verify non-bonded interaction cutoffs match force field specifications, typically 1.0-1.2nm for modern biomolecular force fields. Implement potential modifiers for Lennard-Jones interactions to prevent energy drift [78].
Table 3: Critical Simulation Parameters for Stability
| Parameter | Default Value | Optimized Value | Impact on Stability |
|---|---|---|---|
| dt (timestep) | 2.0 fs | 1.0-2.0 fs | Smaller values prevent large displacements [75] |
| lincs-order | 4 | 6-8 | Improves constraint accuracy for connected bonds [72] |
| lincs-iter | 1 | 2-4 | Enables convergence for complex topologies [72] |
| rcoulomb | 1.0 nm | Force field specific | Prevents missing interactions [78] |
| rvdw | 1.0 nm | Force field specific | Prevents missing interactions [78] |
| constraints | all-bonds | h-bonds only | Reduces constraint network complexity [75] |
Case Studies in Error Resolution
Pharmaceutical Development: Protein-Ligand Complex Stability
In CADD applications, LINCS warnings frequently emerge during protein-ligand simulation, particularly with novel chemical entities. A representative case from the literature involved persistent LINCS warnings during simulation of a kinase-inhibitor complex [73]. Diagnosis revealed incorrect bond parameters for a sulfonamide moiety in the ligand topology. Resolution required:
- Ligand Parameter Validation: Re-parameterization using GAFF with HF/6-31G* optimization
- Staged Restraint Release: Gradual removal of positional restraints on binding site residues
- Solvent Equilibration: Extended water relaxation (500ps) with restrained protein-ligand coordinates
This protocol eliminated constraint warnings while preserving binding pocket geometry, enabling accurate calculation of binding free energies [73].
Enhanced Sampling: Managing Instabilities in Metadynamics
Advanced sampling techniques like metadynamics introduce additional energy terms that can disrupt constraint satisfaction. A study employing Gaussian-accelerated MD for protein folding observed escalating LINCS warnings as boost potential increased [73]. The resolution integrated:
- Adaptive LINCS Iteration: Dynamic increase of lincs-iter based on constraint deviation monitoring
- Collective Variable Design: Selection of CVs that minimally perturb local bond geometry
- Boost Potential Tuning: Careful calibration of acceleration parameters to prevent excessive force generation
This approach maintained sampling efficiency while eliminating constraint violations, demonstrating the importance of algorithm interoperability in advanced simulation workflows [73].
Table 4: Research Reagent Solutions for Stable MD Simulations
| Tool/Category | Specific Examples | Function in Addressing Instabilities |
|---|---|---|
| Force Fields | CHARMM36, AMBER, GROMOS [78] | Provide validated parameters minimizing energy strain |
| Topology Generators | CGenFF, ACPYPE, MATCH | Create chemically accurate initial topologies |
| Validation Tools | VMD, MDAnalysis, GROMAT | Identify structural anomalies pre-simulation |
| Specialized Hardware | Anton 3, GPU clusters [73] | Enable longer equilibration and smaller timesteps |
| Constraint Algorithms | P-LINCS, SHAKE, SETTLE [72] | Maintain bond geometry with system-appropriate methods |
LINCS and SETTLE warnings serve as valuable diagnostic signals within the molecular dynamics workflow, highlighting deviations from physical realism that demand researcher intervention. Through systematic diagnosis and targeted intervention—spanning careful equilibration, parameter validation, and algorithm selection—researchers can transform unstable simulations into robust computational experiments. As MD applications expand toward more complex systems in drug discovery [73] and materials science [24], these stabilization protocols become increasingly essential for producing reliable, reproducible results. Future developments in machine-learned force fields [24] and specialized hardware [73] promise to alleviate some inherent instabilities, but the fundamental principles of careful system preparation and parameter validation will remain cornerstone practices in computational molecular science.
Within the broader thesis of molecular dynamics simulation workflow research, the generation and management of simulation artifacts represent a critical challenge that can compromise data integrity and scientific conclusions. Artifacts—unphysical behaviors arising from computational approximations rather than true physics—are endemic in molecular dynamics (MD) simulations, particularly at interfacial regions and in complex biomolecular systems. This technical guide examines three prevalent categories of artifacts: unphysical forces from truncated potentials, bad contacts from structural distortions, and water settlement issues at interfaces. Proper management of these artifacts is essential for producing reliable, reproducible simulations that accurately reflect underlying biological and physical processes, rather than numerical aberrations. The systematic approach to identifying, mitigating, and validating against these artifacts forms a crucial component of robust simulation workflow design [79].
Artifact Type 1: Unphysical Forces from Truncated Non-Bonded Potentials
Underlying Mechanism and System Impact
A primary source of unphysical forces in MD simulations stems from the truncation of non-bonded potentials. For computational efficiency, non-bonded interactions (both van der Waals and electrostatic) are typically calculated explicitly within a cutoff radius, while interactions beyond this distance are approximated or omitted. This common practice can induce significant artifacts, particularly at interfaces between different dielectric media (e.g., lipid-water interfaces, protein-solvent boundaries) [80] [81].
The fundamental issue arises from the discontinuous change in force at the cutoff distance. When particle pairs cross this boundary, they experience an abrupt change in interaction energy, violating energy conservation. Furthermore, truncation distorts the true long-range nature of electrostatic forces, which theoretically extend to infinity. At interfaces, this disruption of delicate force balances can dramatically alter system behavior. Research demonstrates that these artifacts are not merely quantitative errors but can induce qualitative changes in system behavior—for instance, altering the character of a wetting transition from continuous to first-order by creating artificial metastable states [81]. Common correction methods like force-switching functions or potential shifting do not fully resolve these issues.
Experimental Protocols for Identification and Mitigation
Identification Protocol:
- Energy Drift Analysis: Monitor total energy in an NVE (microcanonical) ensemble simulation. Significant energy drift suggests force discontinuities from truncation.
- Interface Analysis: Compare density profiles of different components (e.g., water, lipids) at interfaces using different cutoffs. Artifactual density accumulation or depletion at the cutoff distance indicates truncation issues.
- Radial Distribution Function (RDF) Examination: Calculate RDFs for key atom pairs. Unphysical peaks or discontinuities near the cutoff distance signify truncation problems.
Mitigation Protocol:
- Implement Long-Range Electrostatics: For electrostatic interactions, replace simple truncation with mesh-based methods like Particle Mesh Ewald (PME) that properly account for long-range forces.
- Increase Cutoff Distance: Where computational resources allow, increase non-bonded cutoffs (e.g., to 1.2 nm or greater) to minimize truncation effects, while ensuring consistency with pairlist update frequency [80].
- Validate with Known Systems: Test simulation parameters on systems with known properties (e.g., bulk water density, interfacial tension) before applying to novel systems.
- Avoid TR/CFA Schemes: The Twin-Range constant force application scheme exacerbates these artifacts; prefer Single-Range treatment of nonbonded interactions [80].
Table 1: Numerical Parameters for Managing Truncation Artifacts
| Parameter | Problematic Value | Recommended Value | Rationale |
|---|---|---|---|
| Nonbonded Cutoff | < 0.9 nm | 1.0 - 1.2 nm | Balances accuracy with computational cost |
| Pairlist Update | Every 20-40 fs | Every 10-20 fs | Prevents missed interactions from particle diffusion |
| Electrostatics Method | Reaction Field (RF) | PME or PPPM | Properly handles long-range forces |
| TR Scheme Usage | Rs < Rl, nm > 1 | Rs = Rl, nm = 1 | Avoids force splitting artifacts |
Artifact Type 2: Water Settlement and Density Artifacts at Interfaces
Multiple Time-Step Integration Artifacts
Water settlement anomalies and unphysical density accumulations at interfaces represent particularly insidious artifacts that can dramatically alter simulation outcomes, especially in systems mimicking biological membranes or multi-phase environments. These artifacts are frequently induced by the use of multiple time-step (MTS) integration methods, such as the Twin-Range (TR) scheme, which aim to improve computational efficiency by updating computationally expensive non-bonded forces less frequently than bonded interactions [80].
In TR schemes, non-bonded forces within the long-range cutoff (Rl) are split into short-range (within Rs) and mid-range (between Rs and Rl) contributions. The short-range forces update every time step (inner time step), while mid-range forces update less frequently (outer time step). This force splitting, when combined with certain thermostats, generates resonance effects that manifest as artificial density increases at interfaces between polar and apolar media. Research demonstrates these artifacts are particularly pronounced in systems like water-chloroform layers, water-cyclohexane interfaces, and droplet systems—simplified models for biological membranes [80].
Thermostat and Integration Scheme Dependencies
The severity of interfacial density artifacts strongly depends on thermostat selection and integration methodology:
- Weak Coupling (WC) and Nosé-Hoover (chain) Thermostats: Exhibit significant density artifacts when used with TR schemes due to resonance effects from force splitting.
- Stochastic Dynamics (SD) Thermostats: Effectively suppress resonance artifacts, allowing for longer pairlist-update periods (up to 40 fs) without significant density distortions.
- RESPA Integration: Merely shifts artifact occurrence to larger outer time steps rather than eliminating them.
- Single-Range (SR) Treatment: Completely resolves density issues by eliminating force splitting, maintaining accurate density profiles even with extended pairlist-update periods [80].
Experimental Protocol for Detection and Resolution
Detection Protocol:
- Density Profile Analysis: Calculate mass density profiles normal to interfaces. Look for unnatural peaks or accumulations precisely at the interface that persist across simulation replicates.
- Thermostat Comparison: Run identical systems with different thermostats (WC, NH, SD) and compare density profiles. Consistent profiles indicate physical behavior, while discrepancies suggest artifacts.
- Time-Step Dependence: Test with different outer time-step values. Artifacts that diminish with smaller outer time steps indicate MTS resonance issues.
Resolution Protocol:
- Thermostat Selection: Implement stochastic dynamics (Langevin) thermostats with appropriate friction coefficients (e.g., γ = 10 ps-1) for interface-containing systems [80].
- Single-Range Scheme: Adopt SR treatment of non-bonded interactions (Rs = Rl) with pairlist updates every 10-20 fs.
- Consistent Pairlist Updates: Ensure pairlist update frequency matches mid-range force evaluation period in TR schemes (np = nm).
- Validation Systems: Test simulation parameters on simple interface systems (e.g., water-chloroform layers) before applying to complex biological membranes.
Diagram 1: Workflow for identifying and mitigating water settlement artifacts. The decision tree guides users through systematic diagnosis and resolution steps.
Artifact Type 3: Bad Contacts and Structural Distortions
Bad contacts refer to unphysical atomic overlaps or distorted molecular geometries that violate steric constraints or proper chemical bonding. These artifacts arise from multiple sources, including insufficient energy minimization before dynamics, improper constraint algorithms, faulty temperature/pressure coupling, or inadequate sampling of complex energy landscapes. In drug discovery applications, such artifacts can severely compromise binding pose predictions, protein-ligand interaction energies, and free energy calculations [82].
Global optimization challenges present a related source of structural artifacts. As molecular systems become more complex, the potential energy surface (PES) develops numerous local minima, with the number scaling exponentially with system size (Nmin(N) = exp(ξN), where ξ is a system-dependent constant) [83]. Without proper sampling techniques, simulations can become trapped in non-physical conformational states that represent force field artifacts rather than true biological conformations.
Protocols for Identification and Correction
Identification Protocol:
- Steric Clash Detection: Use tools like VMD's "bad contacts" utility or CHARMM's "contact" command to identify atom pairs unrealistically close (e.g., < 80% of sum of van der Waals radii).
- Energy Component Monitoring: Track individual energy terms (bond, angle, dihedral, van der Waals, electrostatic) for unusually high values indicating structural strain.
- Constraint Algorithm Verification: For systems with bond constraints (SHAKE, LINCS), monitor constraint satisfaction and algorithm convergence.
- Conformational Sampling Assessment: Employ collective variable analysis or RMSD clustering to ensure adequate sampling of conformational space rather than single-basin trapping.
Correction Protocol:
- Enhanced Sampling Methods: Implement advanced sampling techniques like parallel tempering (REMD), metadynamics, or accelerated MD to escape local minima and explore global configuration space [83].
- Multi-Stage Equilibration: Employ progressive equilibration with careful temperature and pressure coupling, starting with strong position restraints that are gradually released.
- Constraint Algorithm Optimization: Ensure proper tolerance settings for constraint algorithms (e.g., 10−4 for SHAKE) and verify hydrogen mass repartitioning when using large time steps.
- Hybrid Global Optimization: Combine stochastic methods (genetic algorithms, simulated annealing) with local refinement for complex systems with rugged energy landscapes [83].
Table 2: Research Reagent Solutions for Artifact Management
| Tool/Category | Specific Examples | Function/Purpose | Application Context |
|---|---|---|---|
| Simulation Software | GROMACS [70] [15], GROMOS [80] | MD engine with force field implementations | Core simulation execution |
| Force Fields | GROMOS 53A6 [80], GROMOS 54a7 [15] | Molecular mechanical parameter sets | Defining interaction potentials |
| Thermostats | Stochastic Dynamics (Langevin) [80], Nosé-Hoover | Temperature control algorithms | Regulating system temperature |
| Global Optimization | Basin Hopping [83], Genetic Algorithms | Locating global minima on PES | Initial structure determination |
| Analysis Tools | VMD, PyMOL, MDAnalysis | Trajectory analysis and visualization | Artifact detection and validation |
Integrated Workflow for Comprehensive Artifact Management
Systematic Metadata Practices for Reproducibility
Robust artifact management requires systematic metadata practices throughout the simulation workflow. As highlighted in recent studies, comprehensive metadata collection is essential for replicability (same results with same setup) and reproducibility (same results with different setup) [79]. The generic knowledge production workflow encompasses three sub-workflows: (1) abstract simulation experiment with data and metadata generation, (2) metadata post-processing into meaningful formats, and (3) usage of enriched data for analysis and sharing.
Critical metadata for artifact tracking includes:
- Software Environment: Exact software versions, compilation flags, and algorithmic settings (e.g., integrator type, thermostat parameters).
- Hardware Configuration: System specifications that may affect numerical precision or performance.
- Force Field Details: Complete force field parameters, modification sources, and water models.
- Simulation Parameters: All numerical settings (cutoffs, time steps, constraints) documented with their rationales.
- Analysis Protocols: Specific methods and tools used for artifact detection and validation.
Implementation tools like Archivist (Python-based metadata processing) can help structure this heterogeneous metadata into unified formats, supporting sustainable numerical workflows and facilitating artifact diagnosis across research groups [79].
Unified Experimental Protocol for Artifact Prevention
Pre-Simulation Preparation:
- System Setup: Build initial coordinates using global optimization methods where needed to ensure proper initial geometry [83].
- Parameter Selection: Choose appropriate cutoffs (≥1.0 nm), PME for electrostatics, Single-Range nonbonded treatment, and stochastic thermostats for interfaces.
- Metadata Template: Prepare structured metadata template to document all simulation parameters and environmental factors.
Equilibration Protocol:
- Energy Minimization: Perform sufficient minimization steps (until maximum force < 1000 kJ/mol/nm) to remove bad contacts.
- Staged Equilibration: Implement stepwise equilibration with gradually released position restraints (backbone → sidechains).
- Stabilization Monitoring: Verify stability of temperature, pressure, density, and potential energy before production phase.
Production and Validation:
- Artifact Screening: Regularly check for interface density anomalies, energy conservation, and steric clashes throughout production run.
- Control Simulations: Compare key properties with different integration schemes or thermostats to identify potential artifacts.
- Metadata Export: Finalize metadata recording with comprehensive runtime information and validation results.
Diagram 2: Integrated workflow for comprehensive artifact prevention. The protocol emphasizes systematic parameter selection, staged equilibration, and regular artifact screening throughout the simulation lifecycle.
Effective management of simulation artifacts is not merely a technical exercise but a fundamental requirement for producing reliable molecular dynamics research. This guide has outlined specific strategies for addressing three critical artifact categories: unphysical forces from truncated potentials, water settlement issues from multiple time-step integrations, and bad contacts from structural distortions. The common theme across all mitigation approaches is the need for systematic parameter selection, appropriate algorithm choices, and comprehensive validation protocols. By implementing these practices within a rigorous metadata framework, researchers can significantly enhance the reproducibility and physical accuracy of their simulations, advancing the broader thesis of robust molecular dynamics workflow research. Future directions will likely involve increased integration of machine learning approaches for artifact detection and the development of more automated validation pipelines, further embedding artifact management as an essential component of the simulation lifecycle.
Within the broader thesis of molecular dynamics (MD) simulation workflow research, the pursuit of both computational efficiency and physical accuracy presents a central challenge. MD serves as a critical tool for investigating chemical reactions, material properties, and biological interactions at the atomic level across diverse fields, from drug development to materials science [9]. The core of this challenge lies in the inherent trade-offs between the speed of a simulation and the fidelity of its results. Key parameters under the researcher's control—the integration timestep, the application of constraints, and the schemes for handling non-bonded interactions—directly and interactively influence this balance. An optimized MD workflow must therefore navigate these interdependent factors. This technical guide examines the latest advances in integration schemes, constraint algorithms, and cutoff methods, providing a structured framework for researchers to enhance their simulation protocols without compromising scientific rigor, ultimately contributing to more reliable and scalable computational workflows.
Core Concepts and Definitions
The performance and accuracy of an MD simulation are governed by its numerical foundation. The following core concepts are essential for understanding the optimization strategies discussed in this guide.
- Hamiltonian System: The classical mechanical system being simulated, described by its Hamiltonian, ( H(\boldsymbol{p}, \boldsymbol{q}) ), which defines the total energy as a function of particle momenta (( \boldsymbol{p} )) and positions (( \boldsymbol{q} )) [84]. For many systems, this takes the form ( H(\boldsymbol{p}, \boldsymbol{q}) = \sum{i=1}^{F} \frac{pi^2}{2m_i} + V(\boldsymbol{q}) ), where ( V(\boldsymbol{q}) ) is the potential energy.
- Symplectic Integrator: A numerical integration algorithm that preserves the geometric structure of the Hamiltonian flow. This property guarantees the long-term stability of the simulation and near-conservation of energy, as it ensures the existence of a "shadow Hamiltonian" that the numerical solution closely follows [84].
- Time-Reversibility: A property of an integrator where the equations of motion are symmetric with respect to time. Time-reversible methods further improve energy conservation and accuracy [84].
- Neighbor List (Pair List): A list of atom pairs that are within a specified cut-off distance, used to efficiently compute non-bonded forces without checking every possible pair at every step. Modern schemes use a buffered Verlet list to update this list infrequently [85].
- Neural Network Potential (NNP): A machine-learning model trained on quantum mechanical data that provides a potential energy surface. NNPs offer near-quantum accuracy but at a higher computational cost per evaluation than classical force fields [86] [87].
Optimizing the Integration Timestep
The choice of integration timestep is perhaps the most direct lever for controlling simulation performance. A larger timestep allows for probing longer timescales with the same computational resources but risks numerical instability and energy drift if it exceeds the period of the system's fastest motions.
Traditional Limits and Structure-Preserving Integrators
Conventional MD simulations are typically limited to timesteps of 1-2 femtoseconds (fs) to accurately resolve the highest frequency vibrations, such as bond stretching involving hydrogen atoms. Exceeding this limit can lead to catastrophic simulation failure. However, recent research focuses on structure-preserving maps that enable significantly larger timesteps while maintaining stability.
A promising approach involves learning a symplectic and time-reversible map directly from data. This is equivalent to learning the mechanical action of the system. One implementation parametrizes a generating function, ( S^3(\bar{\boldsymbol{p}}, \bar{\boldsymbol{q}}) ), where ( \bar{\boldsymbol{p}} ) and ( \bar{\boldsymbol{q}} ) are the mid-step momenta and positions. The symplectic transformation is then defined by: [ \Delta \boldsymbol{p} = -\frac{\partial S^{3}}{\partial \bar{\boldsymbol{q}}}, \quad \Delta \boldsymbol{q} = \frac{\partial S^{3}}{\partial \bar{\boldsymbol{p}}} ] This method eliminates pathological behaviors like energy drift and loss of equipartition that plague non-structure-preserving machine learning predictors, allowing for stable simulations with timesteps orders of magnitude larger than conventional limits [84].
Multi-Time-Step (MTS) and Machine Learning Integration
The Multi-Time-Step (MTS) approach, exemplified by the Reference System Propagator Algorithm (RESPA), strategically uses different timesteps for different components of the force. Fast-varying forces (e.g., bonded interactions) are integrated with a small inner timestep, while slow-varying forces (e.g., long-range electrostatics) are integrated with a larger outer timestep [87].
This principle has been successfully adapted for use with computationally expensive Neural Network Potentials (NNPs). A dual-level NNP MTS strategy employs a fast, distilled model to propagate the dynamics with a small timestep (e.g., 1 fs), while a large, accurate foundation model is called less frequently (e.g., every 3-6 fs) to apply a correction. This hybrid RESPA-like scheme, such as the BAOAB-RESPA integrator, can yield significant speedups—4-fold in homogeneous systems and 2.7-fold in large solvated proteins—while preserving both static and dynamical properties [87].
Table 1: Comparison of Timestep Optimization Strategies
| Strategy | Key Principle | Reported Performance Gain | Key Considerations |
|---|---|---|---|
| Structure-Preserving ML Integrators [84] | Learns a symplectic map equivalent to the system's action. | Enables timesteps ~100x larger than stability limit. | Eliminates energy drift; requires training data. |
| MTS with NNPs (RESPA-like) [87] | Uses a fast, distilled NNP for inner steps and a large NNP for outer corrections. | 2.7x to 4x speedup over 1 fs integration. | Preserves dynamics; requires a pre-trained fast model. |
| Conventional Symplectic Integrators | Uses fixed, small timesteps with algorithms like leap-frog. | N/A (Baseline) | Stable for small (1-2 fs) steps; fails with larger steps. |
Managing Constraints and System Rigidity
Constraints are used to freeze the fastest degrees of freedom (e.g., bonds involving hydrogen atoms), thereby allowing a larger integration timestep. The choice of constraint algorithm and its parameters directly impacts performance and stability.
Optimizer Performance in Geometry Optimization
While often associated with energy minimization, the performance of geometry optimizers is highly relevant for constrained dynamics and pre-equilibration. The choice of optimizer significantly affects the success rate, speed, and quality of the resulting structures when using Neural Network Potentials (NNPs).
Table 2: Optimizer Performance with Various Neural Network Potentials (Success Rate / Avg. Steps to Converge)
| Optimizer | OrbMol | OMol25 eSEN | AIMNet2 | Egret-1 | GFN2-xTB |
|---|---|---|---|---|---|
| ASE/L-BFGS | 22/108.8 | 23/99.9 | 25/1.2 | 23/112.2 | 24/120.0 |
| ASE/FIRE | 20/109.4 | 20/105.0 | 25/1.5 | 20/112.6 | 15/159.3 |
| Sella (internal) | 20/23.3 | 25/14.88 | 25/1.2 | 22/16.0 | 25/13.8 |
| geomeTRIC (tric) | 1/11 | 20/114.1 | 14/49.7 | 1/13 | 25/103.5 |
Key Insights from Benchmark Data:
- Sella with internal coordinates demonstrates a high success rate and a low average number of steps, making it a robust choice for finding minima [88].
- L-BFGS is a reliable and consistent performer across multiple NNP types, though it may require more steps than Sella (internal) [88].
- The performance of geomeTRIC is highly dependent on the coordinate system and the specific NNP, showing great variability [88].
- The number of imaginary frequencies in optimized structures (a measure of how close the structure is to a true minimum) also varies with the optimizer, with Sella (internal) and L-BFGS generally producing better results [88].
Cutoff Schemes and Neighbor Searching
Efficiently calculating non-bonded interactions, which scale quadratically with atom count, is paramount for performance. This is managed through cutoff schemes and neighbor lists.
The Verlet Cutoff Scheme and List Buffering
Modern MD packages like GROMACS employ a buffered Verlet cutoff scheme. The non-bonded pair list is created with a cut-off radius, ( r\ell ), that is larger than the interaction cut-off, ( rc ), by a buffer size, ( rb ): ( r\ell = rc + rb ) [85].
This buffer accounts for the displacement of atoms between pair list updates. The pair list is updated infrequently (every 10-20 steps), and the buffer ensures that all atoms within the interaction cut-off at each step are still included in the list. The required buffer size can be determined automatically based on a user-defined tolerance for energy drift, which is linked to atomic displacements and the temperature of the system [85].
Dynamic List Pruning and Cluster-Based Searching
To further enhance performance, dynamic pair list pruning is used. Although the list is built with a large buffer, a fast kernel periodically prunes out cluster-pairs that are outside the interaction cut-off range for the entire lifetime of the list. This reduces the number of pairs that need to be evaluated in the main interaction kernel. On GPUs, this pruning can often be overlapped with the CPU integration step, making it virtually free [85].
Furthermore, the neighbor search algorithm uses clusters of particles (e.g., 4 or 8 atoms). The list is constructed based on cluster-pairs rather than atom-pairs, which is significantly faster. The non-bonded force calculation kernel then processes multiple particle-pair interactions at once, efficiently mapping to SIMD or SIMT units on modern CPUs and GPUs [85].
Integrated Experimental Protocols
Protocol: Implementing a Multi-Time-Step Scheme with Foundation NNPs
This protocol details the steps for implementing the RESPA-like MTS scheme to accelerate simulations using a foundation neural network model [87].
Model Preparation:
- Select a Reference NNP: Choose a large, accurate foundation model (e.g., FeNNix-Bio1(M)) as the target potential whose dynamics you wish to recover.
- Obtain a Fast, Distilled Model: This model can be either:
- System-Specific: Generate a reference dataset by running a short MD simulation ( <1 ns) with the reference model on your system of interest. Train a smaller, architecturally simplified model (e.g., with a shorter receptive field) on the energies and forces from the reference model.
- Generic: Use a pre-trained generic fast model that has been distilled on a diverse dataset (e.g., from the SPICE2 dataset) for rapid deployment.
Integration Setup (BAOAB-RESPA):
- Implement the BAOAB-RESPA numerical integrator as described in Algorithm 1.
- Define the outer timestep (( \Delta t )), which dictates the frequency of correction from the reference model. This can range from 2 fs to 6 fs depending on the system.
- Define the number of inner steps (( n{\text{slow}} )), which determines how many times the fast model is called per outer step. The inner timestep is ( \Delta t / n{\text{slow}} ) (e.g., 1 fs).
Simulation Execution:
- For each inner step, propagate the dynamics using forces from the fast, distilled model (
FENNIX_small). - At each outer step, compute the force difference ( \mathbf{F}{\text{large}}(x) - \mathbf{F}{\text{small}}(x) ) and apply this correction to the dynamics.
- The simulation trajectory will statistically approximate the dynamics of the large, reference NNP.
- For each inner step, propagate the dynamics using forces from the fast, distilled model (
Validation and Analysis:
- Monitor key thermodynamic (potential energy, temperature) and dynamic (diffusion coefficient) observables.
- Compare these properties against a baseline simulation run with a 1 fs timestep using only the reference model to ensure accuracy is preserved.
Protocol: Optimizing Neighbor List Parameters in GROMACS
This protocol outlines the process for optimizing the pair list update frequency and buffer size in GROMACS to maximize performance while controlling energy drift [85].
Baseline Simulation:
- Run a short simulation (e.g., 100 ps) of your system with the default
rlistandnstlistvalues. - Use the
gmx energytool to calculate the total energy drift over the course of the simulation.
- Run a short simulation (e.g., 100 ps) of your system with the default
Automatic Buffer Tuning:
- In your GROMACS
.mdpfile, setverlet-buffer-tolerance = 0.005(the default, in kJ/mol/ps). This defines the acceptable energy drift per particle per picosecond. - Set
nstlistto a value between 40 and 100 (e.g., 80). GROMACS will automatically calculate the required buffer size for this update frequency and tolerance. - Run the simulation again and verify the energy drift is within acceptable limits.
- In your GROMACS
Manual Fine-Tuning (Optional):
- If performance is still critical, manually test increasing
nstlist(e.g., to 100 or 120) while slightly increasingrlist(e.g., by 0.05 nm). - For each configuration, run a short simulation and monitor:
- Performance: The nanoseconds simulated per day.
- Energy Conservation: The total energy drift.
- Select the parameter set that offers the best performance while maintaining negligible energy drift for your application.
- If performance is still critical, manually test increasing
The Scientist's Toolkit
Table 3: Essential Software and Algorithmic Tools for MD Workflow Optimization
| Tool Name / Category | Type | Primary Function in Optimization | Key Feature |
|---|---|---|---|
| GROMACS [85] | MD Simulation Engine | Implements efficient Verlet cutoff scheme, dynamic list pruning, and automatic buffer tuning. | Highly optimized for CPU and GPU architectures; extensive benchmarking. |
| Tinker-HP with Deep-HP [87] | MD Simulation Engine & Interface | Enables multi-time-step integrations with NNPs via the BAOAB-RESPA scheme. | Scalable, GPU-accelerated platform for ML-driven MD. |
| LAMMPS with ML-IAP-Kokkos [9] | MD Simulation Engine & Interface | Provides a seamless interface for running PyTorch-based NNPs with GPU acceleration. | Uses Cython to bridge Python and C++, enabling end-to-end GPU acceleration. |
| Sella [88] | Geometry Optimizer | Optimizes molecular structures to minima; useful for pre-simulation structure preparation. | Uses internal coordinates and is effective for transition states and minima. |
| geomeTRIC [88] | Geometry Optimizer | Optimizes molecular structures using translation-rotation internal coordinates (TRIC). | Robust for a wide range of molecular systems. |
| Bayesian Optimization (BO) [89] | Optimization Algorithm | Refines coarse-grained force field parameters (e.g., bonded terms in Martini3) against target data. | Efficiently explores parameter space with few evaluations; ideal for costly CGMD simulations. |
Optimizing molecular dynamics workflows requires a holistic and nuanced understanding of the interplay between timestep, constraints, and cutoff schemes. The advent of machine learning potentials has not only raised the ceiling for accuracy but also introduced new paradigms for performance gains, such as multi-time-step integration with distilled models. Simultaneously, advances in traditional algorithms—from symplectic machine-learned integrators to highly efficient, cluster-based neighbor searching with dynamic pruning—continue to push the boundaries of what is computationally feasible. By systematically applying the strategies and protocols outlined in this guide, researchers can make informed decisions to tailor their simulation parameters. This enables the achievement of an optimal balance, allowing for faster, larger-scale simulations without sacrificing the physical accuracy that is paramount in scientific and drug discovery applications.
In molecular dynamics (MD) simulations, a "blow-up"—a catastrophic failure where the simulation becomes unstable and atoms fly apart—is a common yet critical challenge. These failures often originate in the initial system setup, specifically from inaccuracies in the molecular topology and force field parameters. Within the broader workflow of molecular dynamics research, the steps of system topology refinement and parameter correction are fundamental to achieving physically meaningful and stable results. This guide provides an in-depth technical overview of methodologies for diagnosing and correcting problematic parameters, ensuring robust simulations for applications in drug development and biomolecular research.
Core Principles: Topology and Force Fields
The stability of an MD simulation is governed by the interplay between system topology and the force field.
- System Topology: A topology defines the chemical structure of a molecular system. It includes all atomic coordinates, bonds, angles, dihedrals, and non-bonded interactions. An incomplete or incorrect topology—such as missing atoms, improper bond lengths, or overlapping van der Waals radii—introduces unrealistically high energy states that the integrator cannot resolve, leading to instability [10].
- Force Field Parameters: The force field provides the mathematical functions and parameters that describe the potential energy of the system. Key components include bond-stretching and angle-bending potentials, torsional potentials, and van der Waals and electrostatic interactions [90]. Assigning incorrect atom types or parameters that do not match the chemical context is a primary cause of simulation blow-up.
The GōMartini Framework
Coarse-grained (CG) models, such as GōMartini, offer a powerful approach by simplifying the system to enhance computational efficiency while maintaining accuracy. The GōMartini model combines a structure-based Gō model with the physics-based Martini force field [90].
The potential energy in a Gō model is constructed from the known native structure of the protein:
U_Gō = Σ_(i<j)^NC V(σ_ij, ε)
where NC is the set of native contacts, and V(σ_ij, ε) is a Lennard-Jones (LJ) 12-6 potential with its energy scale and distance parameters based on the native structure [90]. This combination allows for the study of large conformational changes while mitigating the "stickiness" and protein-protein interaction inaccuracies found in earlier Martini iterations [90].
Table 1: Key Components of the GōMartini 3 Model
| Component | Description | Role in Preventing Instability |
|---|---|---|
| Virtual Sites | Beads placed at the center of mass of multiple atoms rather than on a single atom. | Smoothens the potential energy landscape, allows for larger integration time steps. |
| Native Contacts | Defined based on a known folded structure, using a Lennard-Jones potential. | Maintains structural integrity without requiring overly rigid harmonic restraints. |
| Martini 3 Backbone | Uses a P2 bead type for most backbones, placed at the center of mass of N, Cα, Cβ, and O atoms. | Improves chemical specificity and reduces spurious interactions that cause aggregation. |
| Environmental Bias | An optional correction applied to interactions with water beads. | Corrects for inaccuracies such as underestimated dimensions of disordered proteins. |
Quantitative Parameter Tables for System Stability
Correct parameterization is non-negotiable for stable simulations. The tables below summarize critical parameters and their common resolutions.
Table 2: Critical Bonded Parameters and Common Corrections
| Parameter Type | Typical Stable Range | Common 'Blow-Up' Values | Correction Methodology |
|---|---|---|---|
| Bond Length | 0.10 - 0.15 nm (C-C bond) | < 0.08 nm or > 0.2 nm | Use Shake/Lincs algorithms; check pre-processing tool outputs. |
| Angle Force Constant | 300-600 kJ/(mol·rad²) | < 50 kJ/(mol·rad²) | Derive from quantum mechanics (QM) calculations or reference force fields. |
| Dihedral Force Constant | 1-50 kJ/mol | Excessively high (>100 kJ/mol) | Parametrize dihedrals via QM potential energy surface scans. |
| Time Step (Δt) | 1-2 fs (AA), 20-30 fs (CG) | > 2 fs for all-atom with flexible bonds | Reduce Δt; use virtual sites in CG models for stability with larger Δt [90]. |
Table 3: Critical Non-Bonded Parameters and Common Corrections
| Parameter | Function | Blow-Up Source & Fix |
|---|---|---|
| Van der Waals Radius (σ) | Defines atomic size and repulsive wall. | Source: Overlapping atoms (σ too small).Fix: Use SHAKE; check and regenerate topology. |
| Van der Waals Well Depth (ε) | Defines attraction strength. | Source: Excessively deep wells (ε too high) cause "sticking."Fix: Apply standard combination rules. |
| Charge (q) | Defines electrostatic interaction. | Source: Overly high partial charges on adjacent atoms.Fix: Ensure charge neutrality; use RESP fits. |
| 1-4 Interactions | Scales non-bonded interactions for atoms separated by three bonds. | Source: Incorrect 1-4 scaling factors create huge forces.Fix: Explicitly define in topology according to force field. |
Experimental Protocols for Parameterization
Protocol: Parametrizing a Novel Ligand
A robust protocol for parametrizing a molecule absent from standard force field libraries is essential.
- Geometry Optimization: Perform a quantum mechanics (QM) calculation (e.g., HF/6-31G*) to optimize the ligand's geometry and obtain its electrostatic potential.
- Charge Derivation: Calculate electrostatic potential-derived charges (e.g., using the RESP method) to determine atomic partial charges.
- Bonded Parameters: Assign bond, angle, and dihedral parameters by analogy to existing parameters in the chosen force field. For missing dihedrals, conduct a QM potential energy surface scan by rotating the dihedral and fit the resulting profile to a periodic function.
- Van der Waals Parameters: Assign from the standard force field based on atom type.
- Validation in Solvent: Place the parametrized ligand in a water box and run a short MD simulation. Monitor potential energy, temperature, and pressure for stability. Validate against experimental data (e.g., density, hydration free energy) if available.
Protocol: Correcting a Topology with Atomic Overlaps
Atomic overlaps, indicated by enormous forces and pressure, require a step-wise relaxation.
- Energy Minimization: Perform steepest descent energy minimization, monitoring the potential energy and maximum force.
- Solvation and Ionization: Solvate the system in an appropriate water model and add ions to neutralize the system's total charge.
- Solvent Relaxation: Run a short MD simulation with heavy solute atoms restrained (force constant of 1000 kJ/(mol·nm²)). This allows the solvent to relax around the solute.
- Full System Equilibration: Gradually release the restraints in subsequent equilibration steps, first with a weaker restraint (e.g., 400 kJ/(mol·nm²)), and then with no restraints.
Workflow and Signaling Pathways for Topology Refinement
The following diagram illustrates a systematic workflow for diagnosing and correcting simulation instabilities, integrating the protocols and tools discussed.
Simulation Debugging Workflow
The Scientist's Toolkit: Research Reagent Solutions
A successful MD simulation relies on a suite of software tools and force fields.
Table 4: Essential Tools for Topology Building and Validation
| Tool/Resource | Function | Application Context |
|---|---|---|
| GAUSSIAN | Quantum chemistry software package. | Deriving accurate bonded parameters and partial charges for novel molecules. |
| ACPYPE | AnteChamber PYthon Parser interfacE. | Automated generation of topologies for small molecules for AMBER and GROMACS. |
| CHARMM-GUI | Web-based platform for simulation setup. | Building complex systems (membranes, proteins with ligands) with correct topologies. |
GROMACS gmx check |
Command-line tool within GROMACS. | Checking topology file for inconsistencies and violations before simulation. |
| Martini Force Field | Coarse-grained force field. | Simulating large systems and long timescales; used in GōMartini model [90]. |
| VMD | Visual Molecular Dynamics. | Visualizing initial structures and trajectories to detect overlaps and artifacts. |
| PyMOL | Molecular visualization system. | Preparing and inspecting molecular structures prior to topology building. |
AMBER tleap |
Module in the AMBER suite. | Building systems and generating topologies for simulations with AMBER force fields. |
GROMACS pdb2gmx |
Command-line tool within GROMACS. | Generating a topology for a protein from a PDB file. |
| SAMSON | Integrative molecular design platform. | Utilizing discrete color palettes like Okabe-Ito for accessible, clear visualization of complex molecules [91]. |
Ensuring Scientific Rigor: Validation Against Experiment and Comparative Methodologies
Molecular dynamics (MD) simulations provide a "virtual molecular microscope," enabling researchers to probe the dynamical properties of atomistic systems with unparalleled detail [92]. However, the predictive capability of MD is constrained by two fundamental challenges: the sampling problem, where lengthy simulations may be required to describe certain dynamical properties, and the accuracy problem, where insufficient mathematical descriptions of physical and chemical forces can yield biologically meaningless results [92].
The empirical nature of force fields, combined with variations in implementation across simulation packages, introduces potential discrepancies between simulation outcomes and physical reality. As noted in a benchmark study, even when different MD packages reproduced experimental observables equally well for proteins like engrailed homeodomain and RNase H at room temperature, "there were subtle differences in the underlying conformational distributions and the extent of conformational sampling obtained" [92]. This ambiguity necessitates rigorous validation against experimental data to ensure simulations generate meaningful, trustworthy results. Without such validation, MD simulations risk producing quantitatively appealing but physically incorrect trajectories, potentially leading to flawed scientific conclusions.
Key Experimental Observables for Validation
A diverse array of experimental techniques provides critical data for benchmarking MD simulations, each offering unique insights into structural and dynamic properties.
Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR spectroscopy offers multiple independent observables for validating MD simulations, including nuclear Overhauser effects (NOEs), scalar couplings (J-couplings), and residual dipolar couplings (RDCs) [93] [94]. These parameters report on local atomic environments, distances between nuclei, and bond orientations, providing exquisite sensitivity to conformational dynamics at atomic resolution. NMR is particularly valuable for characterizing disordered states and conformational heterogeneity that may be obscured in static structural models [94].
Small- and Wide-Angle X-Ray Scattering (SAXS/WAXS)
Solution X-ray scattering techniques provide low-resolution structural information about the global shape and dimensions of biomolecules [94] [95]. SAXS is especially powerful for detecting large-scale conformational changes and quantifying populations of extended versus compact states in heterogeneous ensembles [94]. The forward calculation of SAXS profiles from simulation trajectories must properly account for solvent contributions and ensemble averaging to enable meaningful comparisons [94].
Cryo-Electron Microscopy (Cryo-EM)
Cryo-EM has emerged as a powerful structural biology technique that can capture multiple conformational states, particularly for large macromolecular complexes [94] [95]. While traditionally used for high-resolution structure determination, cry-EM density maps can inform flexible fitting MD simulations to refine structures against experimental maps [94]. However, the quantitative relationship between conformational dynamics and cryo-EM density remains challenging to establish precisely.
Chemical Probing and Single-Molecule FRET
Chemical probing methods provide information on solvent accessibility and hydrogen bonding of specific nucleotide or amino acid residues [94] [95]. Single-molecule Förster resonance energy transfer (smFRET) measures distances and distance distributions between specific labeling sites, offering direct insight into conformational heterogeneity and dynamics on biologically relevant timescales [94].
Table 1: Key Experimental Techniques for Validating MD Simulations
| Technique | Spatial Resolution | Temporal Resolution | Key Observables | Strengths |
|---|---|---|---|---|
| NMR Spectroscopy | Atomic | Picoseconds to milliseconds | NOEs, J-couplings, RDCs, chemical shifts | Atomic-level detail, sensitive to dynamics and disorder |
| SAXS/WAXS | Molecular (~1 nm) | Ensemble average | Scattering profile, radius of gyration | Sensitive to global shape and compaction |
| Cryo-EM | Near-atomic to sub-nanometer | Static snapshots | 3D density maps | Captures multiple states, suitable for large complexes |
| smFRET | 3-10 nm | Milliseconds to seconds | Distance distributions | Probes heterogeneity and dynamics in solution |
| Chemical Probing | Nucleotide/Amino acid | Ensemble average | Solvent accessibility, base pairing | Maps secondary structure and flexibility |
Implementing Robust Validation Protocols
Forward Models and Back-Calculation
A critical aspect of validation involves using forward models to calculate experimental observables directly from simulation trajectories [94]. These mathematical frameworks enable quantitative comparison between simulation and experiment by accounting for how atomic coordinates map to experimental signals. For example, SAXS forward models must incorporate hydration layers and ensemble averaging, while NMR chemical shift predictors often employ empirical parameterizations trained against structural databases [92] [94]. The accuracy of these forward models directly impacts validation reliability, and systematic errors in forward models can lead to incorrect assessment of force field accuracy [94].
Ensemble Validation Strategies
Experimental observables typically represent ensemble averages over both time and molecular populations [92] [94]. Validating MD simulations therefore requires approaches that account for this averaging:
- Maximum Entropy Reweighting: Adjusts weights of structures in simulated ensembles to match experimental data while minimizing bias [94]
- Bayesian/Maximum Parsimony Methods: Select subsets of structures from simulations that collectively reproduce experimental observables [94]
- Ensemble Refinement Simulations: Incorporate experimental data as restraints during simulation to guide sampling toward experimentally consistent regions [94]
Each approach balances the trade-off between faithfully reproducing experiments and maintaining physical realism of the force field.
Statistical Considerations and Error Analysis
Proper validation requires careful attention to statistical uncertainties in both simulations and experiments [93] [94]. Agreement between simulation and experiment that is better than experimental uncertainty may indicate overfitting, particularly when reweighting strategies are employed [94]. Additionally, the finite sampling in MD simulations introduces statistical errors in back-calculated observables, necessitating convergence tests to ensure robust comparisons [92] [96].
Workflow for Methodological Rigor
Implementing a systematic workflow ensures comprehensive validation of MD simulations against experimental data. The process begins with establishing simulation reliability and proceeds through iterative comparison and refinement.
Diagram 1: MD Simulation Validation Workflow. This flowchart outlines the iterative process of validating molecular dynamics simulations against experimental data, emphasizing convergence assessment and refinement cycles.
The Scientist's Toolkit: Essential Research Reagents
Successful validation requires appropriate selection of computational tools and experimental interfaces. The table below summarizes key resources for implementing robust validation protocols.
Table 2: Essential Research Reagents for Simulation Validation
| Tool Category | Specific Examples | Function in Validation | Considerations |
|---|---|---|---|
| Simulation Packages | AMBER, GROMACS, NAMD, ilmm [92] | Generate MD trajectories using empirical force fields | Different packages may produce varying results even with same force field [92] |
| Force Fields | AMBER ff99SB-ILDN, CHARMM36, Levitt et al. [92] | Define potential energy functions and parameters | Performance system-dependent; requires benchmarking [92] |
| Water Models | TIP4P-EW, TIP3P, SPC/E [92] | Represent solvation environment | Significantly impacts protein dynamics and conformational sampling [92] |
| Forward Model Software | SHIFTX2, PALES, CRYSOL, FOXS [93] | Calculate experimental observables from structures | Accuracy limits validation reliability [94] |
| Enhanced Sampling Methods | Replica Exchange, Metadynamics, Umbrella Sampling [94] | Improve sampling of conformational space | Necessary for slow processes beyond reach of conventional MD [94] |
Best Practices for Reliable and Reproducible Simulations
Convergence and Sampling Assessment
Without proper convergence analysis, simulation results remain compromised [96]. Best practices include:
- Multiple independent replicates: Conducting at least three independent simulations starting from different initial conditions enables statistical assessment of convergence [96]
- Time-course analysis: Monitoring observables as a function of simulation time identifies potential drifts and establishes stability of measured properties [96]
- Enhanced sampling for rare events: For slow processes exceeding conventional MD timescales, employing advanced sampling methods with proper convergence checks is essential [96]
Reproducibility and Documentation
Maximizing the value of MD simulations to the research community requires comprehensive documentation:
- Parameter transparency: Detailed reporting of simulation parameters, including force field versions, water models, integration algorithms, and nonbonded interaction treatments [92] [96]
- Input file availability: Depositing simulation input files and final coordinate files in public repositories enables reproduction and extension of simulations [96]
- Custom code sharing: Making specialized analysis scripts and tools publicly accessible supports community standards and methodological advancement [96]
The integration of MD simulations with experimental data represents a powerful paradigm for advancing molecular science. As force fields continue to evolve and computational resources expand, the validation imperative remains constant: simulations must be grounded in experimental reality to yield meaningful biological insights. By adopting rigorous validation frameworks, employing multiple complementary experimental observables, and adhering to reproducibility standards, the MD community can maximize the predictive power and scientific impact of this transformative technology. The future of molecular simulation lies not merely in faster computations or larger systems, but in more faithful representations of biomolecular reality—a goal achievable only through relentless, rigorous validation against experimental data.
{Abstract:} Molecular dynamics (MD) simulations are indispensable in biomedical research, where the accuracy of results is fundamentally tied to the choice of the empirical force field. This technical analysis evaluates the performance of three major biomolecular force fields—AMBER, CHARMM, and GROMOS—by synthesizing findings from recent benchmarking studies. We provide a structured comparison of their accuracy in reproducing experimental structures and thermodynamic properties for diverse systems, including folded proteins, intrinsically disordered peptides (IDPs), non-natural peptidomimetics, and small organic molecules. The results indicate that force field performance is highly system-dependent; no single force field universally outperforms the others. CHARMM36m and recent AMBER refinements (e.g., ff99SBws, ff03w-sc) show a more balanced treatment of folded and disordered proteins, while CHARMM extensions demonstrate superior accuracy for non-natural β-peptides. GROMOS, though less accurate in some peptide systems, remains a robust choice for specific setups. This guide outlines detailed benchmarking methodologies and provides actionable recommendations to help researchers select and validate the most appropriate force field for their specific molecular simulation workflow.
Molecular dynamics (MD) simulation is a cornerstone technique in computational chemistry, biophysics, and drug discovery, enabling the atomistic-level investigation of biomolecular structure, dynamics, and interactions [97] [98]. The empirical force field—a set of mathematical functions and parameters describing potential energy as a function of atomic coordinates—is the foundation upon which the physical accuracy of any simulation rests [99]. The development and refinement of force fields have been active areas of research for decades, resulting in several major families, including AMBER, CHARMM, and GROMOS.
A persistent challenge in the field is that force fields parameterized to excel for one class of molecules (e.g., folded globular proteins) may perform poorly for others (e.g., intrinsically disordered proteins or non-natural peptides) [100] [97]. This variability makes the informed selection of a force field a critical first step in the MD workflow. An erroneous choice can lead to conformational artifacts, incorrect stability predictions, or misleading mechanistic insights, ultimately derailing subsequent experimental efforts [100].
This review provides a comparative analysis of the AMBER, CHARMM, and GROMOS force fields, framed within the context of establishing a reliable molecular dynamics simulation workflow. We focus on benchmarking data from recent literature to evaluate their performance across a spectrum of biologically and pharmacologically relevant systems. By consolidating quantitative results, experimental protocols, and practical recommendations, this guide aims to equip researchers with the necessary knowledge to make evidence-based decisions in their simulation projects.
Force Field Families: Origins and Philosophies
The AMBER, CHARMM, and GROMOS force fields share a common functional form for the potential energy, which typically includes terms for bond stretching, angle bending, torsional rotations, and non-bonded van der Waals and electrostatic interactions [99]. However, they differ in their parameterization philosophies and target data, leading to differences in performance.
- AMBER: The AMBER (Assisted Model Building with Energy Refinement) force field were originally developed for proteins and nucleic acids. Their parameterization heavily relies on fitting to high-level quantum mechanical data for small molecule analogs and reproducing experimental data for liquid-state properties and crystallographic structures [101] [99]. Recent versions have focused on achieving a balance between modeling folded proteins and intrinsically disordered regions (IDRs) [97].
- CHARMM: The CHARMM (Chemistry at HARvard Macromolecular Mechanics) force fields are parameterized using a combination of quantum mechanical calculations and experimental data, with a strong emphasis on reproducing condensed-phase properties and interaction energies [101] [99]. The CHARMM36m extension includes improved treatment of IDRs and membrane proteins [97].
- GROMOS: The GROMOS (GROningen MOlecular Simulation) force fields are united-atom force fields parameterized primarily to reproduce thermodynamic properties of pure liquids and solvation free energies [101] [102] [103]. The GROMOS-96 54A7 and 54A8 parameter sets are the most recent and include explicit support for β-peptides [100]. It is important to note that these force fields were parametrized with a specific twin-range cut-off scheme, and their use with a single-range cut-off can lead to deviations from intended physical properties [103].
Comparative Performance Benchmarks
Folded Proteins and Intrinsically Disordered Peptides (IDPs)
A primary challenge for modern force fields is to simultaneously stabilize the folded state of structured proteins while accurately capturing the expanded, dynamic ensembles of IDPs. Recent refinements to the AMBER and CHARMM force fields have targeted this balance by adjusting protein-water interactions and backbone torsional potentials [97].
Table 1: Performance of Force Fields for Folded and Disordered Protein Systems
| Force Field | Folded Protein Stability | IDP Chain Dimensions | Key Characteristics and Refinements |
|---|---|---|---|
| AMBER ff03ws | Instability observed in Ubiquitin and Villin HP35 over µs-timescales [97] | Accurate for many IDPs, but overestimated dimensions for RS peptide [97] | Combined with TIP4P2005 water; 10% upscaled protein-water interactions [97] |
| AMBER ff99SBws | Maintained stability of Ubiquitin and Villin HP35 [97] | Accurate for many IDPs [97] | Similar to ff03ws but with different backbone torsion adjustments [97] |
| AMBER ff03w-sc | Improved stability over ff03ws [97] | Accurate against SAXS/NMR data [97] | Selective upscaling of protein-water interactions [97] |
| AMBER ff99SBws-STQ′ | Maintained stability [97] | Accurate against SAXS/NMR data [97] | Targeted torsional refinements for glutamine [97] |
| CHARMM36m | Stable for folded proteins [97] | Improved description of IDPs [97] | Modified TIP3P water with extra LJ parameters on hydrogen atoms [97] |
As the data shows, refined versions like ff03w-sc and ff99SBws-STQ′ successfully combine accurate IDP ensembles with stable folded domains. CHARMM36m also achieves a balanced performance through its modified water model.
Non-Natural Peptidomimetics: β-Peptides
β-peptides, homologues of natural peptides with an extra carbon in the amino acid backbone, are a rigorous test for force fields due to their diverse non-native secondary structures. A 2023 study provided a direct comparison of AMBER, CHARMM, and GROMOS for seven different β-peptide sequences [100].
Table 2: Performance of Force Fields for β-Peptide Simulations [100]
| Force Field | Monomeric Structures | Oligomer Formation/Stability | Notes and Specific Variants |
|---|---|---|---|
| CHARMM | Accurately reproduced experimental structures in all monomeric simulations [100] | Correctly described all oligomeric examples [100] | Used a version with backbone torsions derived from ab initio matching [100] |
| AMBER | Reproduced experimental structures for 4 out of 7 peptides [100] | Held together pre-formed associates, but failed at spontaneous oligomer formation [100] | Best for peptides containing cyclic β-amino acids [100] |
| GROMOS | Reproduced experimental structures for 4 out of 7 peptides [100] | Performance not detailed; had the lowest performance overall [100] | Supported β-peptides "out of the box" (54A7/54A8) [100] |
The study concluded that the tested CHARMM extension performed best overall, highlighting the importance of specific parameterization against high-level quantum-chemical data for non-natural building blocks [100].
Solvation Free Energies and Liquid Properties
The ability to accurately model solvation is critical for simulating biomolecules in physiological environments. A large-scale benchmark evaluated nine force fields against a matrix of experimental cross-solvation free energies [102].
Table 3: Accuracy for Cross-Solvation Free Energies (RMSE in kJ mol⁻¹) [102]
| Force Field Family | Specific Force Field | RMSE |
|---|---|---|
| GROMOS | GROMOS-2016H66 | 2.9 |
| OPLS | OPLS-AA | 2.9 |
| OPLS | OPLS-LBCC | 3.3 - 3.6 |
| AMBER | AMBER-GAFF2 | 3.3 - 3.6 |
| AMBER | AMBER-GAFF | 3.3 - 3.6 |
| OpenFF | OpenFF | 3.3 - 3.6 |
| GROMOS | GROMOS-54A7 | 4.0 - 4.8 |
| CHARMM | CHARMM-CGenFF | 4.0 - 4.8 |
| GROMOS | GROMOS-ATB | 4.0 - 4.8 |
For liquid thermodynamic properties, a study on diisopropyl ether (DIPE) found that CHARMM36 and COMPASS provided accurate density and viscosity values, while GAFF and OPLS-AA overestimated them [104]. An older benchmark for vapor-liquid coexistence also highlighted that CHARMM22 performed nearly as well as the top-performing TraPPE force field for liquid densities of organic molecules [101].
Experimental Benchmarking Protocols
To ensure the validity of simulation results, benchmarking a chosen force field against available experimental data for the system of interest is highly recommended. The following workflow outlines a general protocol for such validation.
Diagram Title: Force Field Benchmarking Workflow
Select Experimental Observables
The first step is to identify relevant, quantitative experimental data for comparison. Key data sources include:
- NMR Spectroscopy: Chemical shifts, scalar couplings (J-couplings), residual dipolar couplings (RDCs), and spin relaxation rates provide information on local conformation and dynamics [97] [98].
- Small-Angle X-ray Scattering (SAXS): The scattering profile yields the radius of gyration (Rg) and the pair distribution function P(r), which report on the global dimensions and shape of a molecule in solution [97].
- X-ray Crystallography: Room-temperature crystal structures can be used to assess a force field's ability to maintain a folded protein's native state [98].
Force Field Selection and Simulation Setup
- Select one or more candidate force fields based on the system type (e.g.,
ff99SBws-STQ′orCHARMM36mfor mixed folded/IDP systems, specific CHARMM extensions for β-peptides) [100] [97]. - Choose a water model consistent with the force field's parameterization (e.g., TIP4P2005 for
ff99SBws, modified TIP3P forCHARMM36m) [97]. - Prepare the initial structure, solvate it in a sufficiently large water box, and add ions to neutralize the system and achieve a physiologically relevant salt concentration (e.g., 50-150 mM NaCl) [100] [105].
MD Simulation Production
- Energy Minimization: Remove steric clashes using a steepest descent algorithm [100].
- Equilibration: Gradually relax the system through short simulations with position restraints on solute heavy atoms, first in the NVT ensemble (constant particle number, volume, and temperature) and then in the NPT ensemble (constant particle number, pressure, and temperature) [100].
- Production Run: Perform multiple independent, long-timescale MD simulations (hundreds of ns to µs) without restraints. The use of replicates is crucial for assessing convergence and robustness [100] [97].
Compute Simulation-Derived Observables and Compare
- Process simulation trajectories to compute the equivalent experimental observables.
- For NMR, calculate chemical shifts and scalar couplings from simulated structures using empirical programs like SHIFTX2 or PALES [98].
- For SAXS, compute theoretical scattering profiles from simulation snapshots using tools like CRYSOL [97].
- Perform a quantitative comparison using metrics like root-mean-square deviation (RMSD) between calculated and experimental data, correlation coefficients, and direct statistical comparison of distributions (e.g., Rg) [102] [97].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Software and Computational Tools for Force Field Benchmarking
| Tool Name | Function/Purpose | Relevance to Benchmarking |
|---|---|---|
| GROMACS [100] [103] | High-performance MD simulation engine. | Used as a common platform for impartial force field comparison; supports AMBER, CHARMM, and GROMOS. |
| MCCCS Towhee [101] | Monte Carlo simulation program. | Used for computing fluid phase equilibria and liquid densities to validate force fields. |
| PyMOL [100] | Molecular visualization and modeling system. | Used for building initial molecular models and visualizing simulation trajectories. |
| gmxbatch [100] | Custom Python package for GROMACS. | Automates run preparation and trajectory analysis, facilitating high-throughput benchmarking. |
| VOTCA [103] | Toolkit for coarse-graining applications. | Provides tools for systematic coarse-graining and force field validation against atomistic simulations or experimental data. |
| CRYSOL [97] | Computes SAXS profiles from atomic models. | For validating simulated structural ensembles against experimental SAXS data. |
| SHIFTX2 / PALES [98] | Predicts NMR chemical shifts and RDCs. | For validating simulated conformational ensembles against NMR observables. |
This analysis demonstrates that the performance of AMBER, CHARMM, and GROMOS is highly system-dependent. Based on the synthesized benchmarking data, we propose the following recommendations for researchers:
- For Folded Proteins with Intrinsically Disordered Regions (IDRs): Use refined force fields that balance protein-protein and protein-solvent interactions, such as AMBER ff99SBws-STQ′, AMBER ff03w-sc, or CHARMM36m, paired with their recommended four-site or modified water models [97].
- For Non-Natural Peptides (e.g., β-peptides): The CHARMM family, with specific extensions parameterized against ab initio quantum-chemical data, currently demonstrates superior performance for both monomeric and oligomeric systems [100].
- For Solvation and Liquid Properties: For organic small molecules, OPLS-AA and GROMOS-2016H66 show the best accuracy for solvation free energies [102], while CHARMM36 performs well for liquid densities and viscosities of ethers [104].
- General Workflow Advice: Always initiate a new project by running short benchmark simulations of your system and, if possible, comparable control systems with known experimental data. Use the workflow and tools outlined in this guide to quantitatively compare results with experimental observables like NMR parameters or SAXS profiles before committing to large-scale production runs [97] [98].
The ongoing development of force fields is a dynamic field. The emergence of extensive quantum-chemical datasets like OMol25 and neural network potentials (NNPs) promises a new generation of highly accurate, transferable models [43]. Until then, a careful, evidence-based selection from among traditional force fields, guided by the principles outlined in this review, remains essential for generating reliable and impactful molecular dynamics simulations.
The pursuit of scientific reproducibility represents a cornerstone of rigorous computational research, yet it poses a significant challenge in the field of molecular dynamics (MD) simulations. As noted in a survey highlighted by Nature, over 70% of researchers have struggled to reproduce another scientist's experiments, and 52% have been unable to reproduce their own work [106]. In MD simulations, this reproducibility crisis is compounded by the complex ecosystem of software platforms, each with distinct algorithms, force field implementations, and numerical precision handling.
Cross-platform validation—the process of verifying that MD simulations produce consistent physical results across different software packages—serves as a critical methodology for addressing these challenges. When AMBER, GROMACS, and NAMD yield equivalent results for identical systems under matching conditions, researchers gain confidence in the robustness of their findings. This technical guide establishes a comprehensive framework for cross-platform validation, positioning it within the broader context of ensuring reliability in molecular dynamics workflow research for drug development and basic science applications.
The Reproducibility Challenge in Molecular Dynamics
Defining Reproducibility in Computational Science
Reproducibility in MD simulations operates on two distinct levels: computational reproducibility and scientific reproducibility. Computational reproducibility refers to the ability to run an identical analysis pipeline and obtain byte-for-byte identical output (excluding incidental metadata such as timestamps). Scientific reproducibility, while more flexible in its implementation, requires that independent analyses—potentially using different tools or algorithms—support the same scientific conclusions [106].
Several factors complicate reproducibility in MD workflows:
- Software Complexity: MD software packages encompass thousands of parameters and configuration options
- Numerical Precision: Differences in floating-point implementation across hardware and software platforms
- Force Field Implementation: Variations in how force field terms are calculated and approximated
- Random Number Generation: Inconsistent initialization of stochastic processes
The Ecosystem of MD Software
The MD software landscape includes hundreds of specialized applications developed globally in academic research groups, resulting in heterogeneous programming languages, licensing models, and dependency requirements [106]. AMBER, GROMACS, and NAMD represent three widely adopted MD engines with distinct architectural approaches:
- AMBER: Specializes in biomolecular simulations with particular strengths in force field development
- GROMACS: Emphasizes high performance and efficiency through extensive code optimization
- NAMD: Designed for parallel scalability on large-scale computing systems
Despite their shared purpose, these packages employ different numerical algorithms, integration methods, and parallelization strategies that can lead to variations in simulation outcomes.
Foundational Principles for Cross-Platform Validation
Computational Reproducibility Framework
Achieving reliable cross-platform validation requires strict adherence to standardized computational environments. The SBGrid Consortium addresses this through a three-pillar approach encompassing Software, Data, and Infrastructure [106]. Their platform provides:
- A curated collection of >620 scientific applications with version-controlled execution
- Conflict-free software deployment through Capsules execution environment
- Open data publication via the SBGrid Data Bank
- Scalable computational infrastructure through SBCloud
This integrated approach reduces computational friction and ensures consistent software environments—a prerequisite for meaningful cross-platform comparison.
FAIR Data Principles
The FAIR (Findable, Accessible, Interoperable, and Reusable) principles provide a framework for managing MD simulation data to enhance reproducibility [107]. Implementing FAIR principles involves:
- Rich Metadata: Comprehensive description of simulation parameters, initial conditions, and software versions
- Standardized Formats: Use of community-accepted file formats for trajectories and topologies
- Persistent Identifiers: Digital object identifiers (DOIs) for simulation datasets
- Clear Licensing: Explicit usage rights for shared simulation data
The MDverse project has demonstrated the challenges of "dark matter" in MD simulations—data that is technically accessible but neither indexed, curated, nor easily searchable [107]. Their analysis of approximately 250,000 files from generalist repositories revealed significant metadata inconsistencies that complicate reuse and validation.
Methodological Framework for Cross-Platform Validation
System Preparation Protocols
Consistent system preparation is essential for meaningful cross-platform comparison. The following workflow outlines a standardized approach:
Standardized System Preparation Workflow
Initial Structure Selection and Preparation
The initial structure serves as the foundation for all subsequent validation work. Recommended practices include:
- Source from Reputable Databases: Obtain structures from the Protein Data Bank (PDB), Materials Project, or other community-curated repositories [24]
- Structure Completion: Address missing atoms, residues, or loops using tools like PDBFixer or Modeller [8]
- Protonation State Assignment: Consistently assign protonation states for ionizable residues using tools like PROPKA at physiological pH (7.4)
- Validation: Verify structural quality using tools like MolProbity or VADAR before proceeding
System Assembly and Minimization
Consistent system setup across platforms requires standardized protocols for:
- Solvation: Use identical water models (TIP3P, TIP4P, etc.) and box dimensions with buffer distances of at least 1.0 nm from the solute
- Neutralization: Add ions to achieve physiological concentration (150 mM NaCl) and neutralize system charge
- Energy Minimization: Apply equivalent minimization criteria (maximum force < 1000 kJ/mol/nm) using the same algorithm (steepest descent followed by conjugate gradient)
Simulation Parameter Alignment
Achieving comparable simulations across AMBER, GROMACS, and NAMD requires meticulous parameter mapping. Key considerations include:
Table 1: Critical Simulation Parameters for Cross-Platform Validation
| Parameter Category | AMBER Implementation | GROMACS Implementation | NAMD Implementation | Validation Standard |
|---|---|---|---|---|
| Time Integration | leap-frog (default) | leap-frog (default) | Verlet (default) | All: 2-fs time step |
| Temperature Coupling | Langevin thermostat | Berendsen/Nose-Hoover | Langevin thermostat | Identical coupling constant |
| Pressure Coupling | Monte Carlo barostat | Parrinello-Rahman | Langevin piston | Identical compressibility |
| Non-bonded Cutoffs | PME with 1.0nm cutoff | PME with 1.0nm cutoff | PME with 1.0nm cutoff | Identical switching functions |
| Constraint Algorithm | SHAKE | LINCS | SHAKE | Equivalent tolerance |
Validation Metrics and Analysis Protocols
A comprehensive validation strategy incorporates multiple quantitative metrics to assess consistency across platforms:
Structural Metrics
- Root Mean Square Deviation (RMSD): Measure structural drift from starting conformation using backbone atoms
- Root Mean Square Fluctuation (RMSF): Quantify per-residue flexibility across the trajectory
- Radius of Gyration: Assess global compactness of protein structures
Dynamic Properties
- Diffusion Coefficients: Calculate from mean square displacement (MSD) using Einstein relation [24]
- Principal Component Analysis (PCA): Identify essential collective motions from covariance matrices of atomic positions [24]
- Hydrogen Bonding Analysis: Quantify persistent hydrogen bonds using consistent geometric criteria (distance < 3.5Å, angle > 120°)
Energetic Consistency
- Potential Energy Time Series: Monitor stability and convergence of total potential energy
- Enthalpy Contributions: Decompose energy terms (bonded, non-bonded, electrostatic) for direct comparison
Tools like FastMDAnalysis provide automated, standardized analysis pipelines that can reduce code requirements by >90% while ensuring consistent metric implementation [17].
Experimental Protocols for Cross-Platform Validation
Benchmark System Selection
Validation should encompass systems of varying complexity to assess platform performance across different scenarios:
Table 2: Recommended Benchmark Systems for Cross-Platform Validation
| System Type | Example | Simulation Length | Key Validation Metrics | Special Considerations |
|---|---|---|---|---|
| Small Globular Protein | BPTI, Ubiquitin | 100-500 ns | RMSD, RMSF, Rg, Hbonds | Standard folding stability |
| Membrane Protein | GPCR, Ion Channel | 200-1000 ns | RMSD, Lipid contacts | Membrane composition consistency |
| Nucleic Acids | DNA dodecamer | 100-200 ns | Helical parameters, Backbone torsions | Ion atmosphere effects |
| Protein-Ligand Complex | Enzyme-inhibitor | 50-200 ns | Ligand RMSD, Interaction fingerprints | Identical initial ligand placement |
Execution Protocol
A standardized execution protocol ensures comparable simulation production:
Equilibration Phase
- 100 ps of solvent equilibration with protein heavy atoms restrained
- 1 ns of full system equilibration with gradually released restraints
- Monitor temperature, pressure, and energy stability before proceeding
Production Phase
- Execute three independent replicates for each system-platform combination
- Use different random seeds for each replicate to assess stochastic variance
- Save trajectories at consistent intervals (10-100 ps) for analysis
Validation Checks
- Verify energy conservation in microcanonical (NVE) ensembles
- Confirm correct temperature and pressure maintenance
- Monitor drift in potential energy and density
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Tools for Cross-Platform Validation Workflows
| Tool Name | Category | Function in Validation | Implementation Considerations |
|---|---|---|---|
| MDCrow [8] | LLM Agent | Automated workflow setup and analysis | 40+ expert-designed tools for MD tasks |
| FastMDAnalysis [17] | Analysis Package | Unified trajectory analysis framework | Built on MDTraj and scikit-learn |
| SBGrid Capsules [106] | Execution Environment | Conflict-free, version-controlled software | Ensures identical software versions |
| MDverse [107] | Data Search | Discovery of existing simulation data | Indexes ~250,000 MD files from repositories |
| CHARMM-GUI [36] | System Preparation | Web-based input generation for multiple MD engines | Standardized system building |
| ML-IAP-Kokkos [9] | Potential Interface | Integration of ML potentials with LAMMPS | Enables AI-driven simulation validation |
Analysis and Interpretation Framework
Statistical Comparison Methodology
Quantitative comparison across platforms requires rigorous statistical approaches:
- Bland-Altman Analysis: Assess agreement between paired measurements from different platforms
- Correlation Coefficients: Calculate Pearson (linear) and Spearman (monotonic) correlations
- Statistical Significance Testing: Apply paired t-tests or ANOVA for replicated measurements
- Effect Size Calculation: Determine practical significance of observed differences
Acceptable Variance Thresholds
Establishing acceptable variance levels is essential for practical validation:
- Structural Metrics: Backbone RMSD differences < 0.1 nm generally indicate good agreement
- Dynamic Properties: RMSF correlations > 0.8 suggest consistent flexibility profiles
- Energetic Properties: Potential energy differences < 1% of total energy indicate numerical consistency
- Convergence Behavior: Similar timescales for observable convergence (within 20%)
Workflow Integration and Automation
Modern validation approaches increasingly leverage automation and artificial intelligence:
Automated Validation Workflow
Tools like DynaMate demonstrate how modular AI agents can automate MD workflow setup, execution, and analysis through customizable Python functions [41]. Similarly, MDCrow implements a ReAct (Reasoning and Acting) agent that interleaves reasoning steps with tool usage to complete complex MD tasks [8]. These approaches reduce manual intervention while increasing standardization.
Documentation and Reporting Standards
Essential Metadata Documentation
Comprehensive reporting requires capturing critical experimental metadata:
- Software Versions: Exact version numbers of AMBER, GROMACS, and NAMD
- Force Field Details: Specific force field version and modification
- Hardware Configuration: CPU/GPU models, memory, and precision settings
- Parameter Files: Complete configuration/input files for all packages
- Analysis Parameters: Specific settings for all validation metrics
Reproducibility Package Structure
A complete reproducibility package should include:
- README with experimental overview and execution instructions
- Input Files for all software platforms
- Analysis Scripts with version information for all tools
- Raw Data for key validation metrics
- Processed Results including statistical comparisons
- Visualization Code for all figures and charts
Cross-platform validation represents an essential methodology for establishing reliability in molecular dynamics research. By implementing the standardized protocols, metrics, and analysis frameworks outlined in this guide, researchers can systematically quantify and document the reproducibility of their simulations across AMBER, GROMACS, and NAMD. This rigorous approach not only strengthens confidence in individual research findings but also contributes to the broader effort to enhance reproducibility across computational structural biology.
The increasing availability of automated validation tools and standardized datasets promises to make comprehensive cross-platform testing more accessible to the research community. As MD simulations continue to play an expanding role in drug discovery and materials design, robust validation practices will ensure these computational methods deliver reliable, actionable insights for scientific advancement.
The advent of artificial intelligence (AI) models like AlphaFold has revolutionized structural biology by enabling highly accurate protein structure prediction from amino acid sequences alone [108] [109]. However, these AI-based methods predominantly provide static structural snapshots, whereas protein function often emerges from dynamic transitions between conformational states and their probability distributions [110]. Molecular dynamics (MD) simulations serve as a powerful validation tool for AI predictions by providing atomic-level insights into structural stability, conformational dynamics, and thermodynamic properties that static models cannot capture. This technical guide explores integrated methodologies leveraging MD simulations to validate, refine, and improve AI-based structure predictions, creating a synergistic framework that enhances the reliability and biological relevance of computational structural biology.
AI-Based Structural Prediction: Capabilities and Limitations
Current State of AI Protein Structure Prediction
AlphaFold2 represents a landmark achievement in computational biology, employing a novel neural network architecture that incorporates physical and biological knowledge about protein structure while leveraging multi-sequence alignments [108]. The system utilizes two main components: the Evoformer, which processes input sequences through attention mechanisms to generate paired representations, and the structure module, which refines these into accurate 3D atomic coordinates [108]. This approach achieves atomic accuracy competitive with experimental methods in most cases, with a median backbone accuracy of 0.96 Å RMSD95 as demonstrated in CASP14 [108].
Subsequent developments have further expanded these capabilities. AlphaFold3 extends predictions to include DNA, RNA, and ligands, while RoseTTAFold All-Atom employs a three-track network to handle assemblies containing proteins, nucleic acids, small molecules, and metals [109]. The recently introduced BioEmu system represents a significant advancement for simulating protein equilibrium ensembles using generative AI, achieving a 4-5 order of magnitude speedup for equilibrium distributions in folding and native-state transitions compared to traditional MD [110].
Critical Limitations Necessitating MD Validation
Despite these advances, fundamental limitations persist in AI-based structure prediction:
- Static Representations: AI models primarily generate single, static conformations, while proteins exist as dynamic ensembles of states [110]
- Limited Functional Insights: Static structures cannot fully reveal mechanisms involving conformational changes, such as enzyme catalysis, ligand binding, and signal transduction [110]
- Rare Event Sampling: Biologically crucial but low-probability states (e.g., intermediate folding states, cryptic pockets) may be missed [110]
- Energy Landscape Blindness: Traditional AI models lack inherent understanding of thermodynamic stability and free energy landscapes [110]
Table 1: Key Limitations of AI Structure Prediction and MD Validation Approaches
| Limitation | Impact on Prediction | MD Validation Approach |
|---|---|---|
| Static conformational sampling | Misses functional dynamics | Time-dependent trajectory analysis |
| Unknown thermodynamic stability | Limited utility for mutation studies | Free energy calculations and entropy analysis |
| Hidden allosteric sites | Overlooks potential drug targets | Cryptic pocket detection through sampling |
| Unverified folding pathways | Uncertain mechanism understanding | Pathway analysis using order parameters |
| Solvent interaction modeling | Limited implicit solvation | Explicit solvent simulations |
Integrated MD Validation Framework
Theoretical Foundation for MD Validation
Molecular dynamics simulations provide the physical foundation for validating AI predictions by numerically solving Newton's equations of motion for all atoms in a system, typically using empirical force fields. This approach captures the time-dependent behavior of macromolecules, including fluctuations and conformational changes that underlie biological function [111]. The integration of MD simulations with AI predictions creates a powerful feedback loop where AI provides initial structural hypotheses and MD validates their physical plausibility and refines dynamic properties.
The BioEmu system bridges these domains by combining AlphaFold2's Evoformer module for sequence representation with a generative diffusion model to sample protein equilibrium ensembles [110]. This hybrid approach achieves remarkable thermodynamic accuracy (within 1 kcal/mol) while maintaining computational efficiency, demonstrating the power of integrating AI architectural advances with physics-based sampling methodologies [110].
Experimental Protocol for MD Validation of AI Predictions
System Preparation and Equilibration
- Structure Initialization: Begin with AI-predicted structures in PDB format
- Solvation: Embed the protein in an appropriate explicit solvent box (e.g., TIP3P water) with minimum 10-12 Å padding
- Neutralization: Add ions to neutralize system charge, followed by physiological ion concentration (e.g., 150mM NaCl)
- Energy Minimization: Perform steepest descent minimization (5,000 steps) followed by conjugate gradient (5,000 steps) to remove steric clashes
- Equilibration: Conduct gradual heating from 0K to target temperature (e.g., 300K) over 100ps in NVT ensemble, followed by density equilibration (100ps NPT)
Production Simulation and Analysis
- Production MD: Run extended simulations (typically 100ns-1μs depending on system size and dynamics) using a stable integrator (e.g., leap-frog) with 2-fs time step
- Trajectory Analysis: Employ automated analysis pipelines (e.g., FastMDAnalysis) to compute:
- Root-mean-square deviation (RMSD) to assess structural stability
- Root-mean-square fluctuation (RMSF) to identify flexible regions
- Radius of gyration to monitor compactness
- Hydrogen bonding patterns and persistence
- Solvent accessible surface area
- Secondary structure evolution
- Principal component analysis to identify essential dynamics
- Advanced Analyses: Perform Markov State Model construction to identify metastable states, free energy landscape calculation, and cryptic pocket detection
Table 2: Key Validation Metrics and Their Interpretation
| Validation Metric | Calculation Method | Interpretation Guidelines |
|---|---|---|
| Backbone RMSD | Cα atom deviation after alignment | <2-3Å indicates structural integrity; >4-5Å suggests significant deviation |
| RMSF per residue | Standard deviation of atomic positions | Identifies flexible regions and potential functional domains |
| Radius of Gyration | Mass-weighted root-mean-square distance from center | Measures compactness; deviations may indicate unfolding |
| Hydrogen Bond Count | Geometric criteria (distance/angle) | Stability indicator; consistent patterns suggest stable folding |
| Secondary Structure Persistence | DSSP or STRIDE assignment | Maintenance of predicted elements validates fold stability |
| Free Energy Landscape | PCA-based or dihedral angle sampling | Identifies stable states and barriers between conformations |
Workflow Implementation and Automation
Integrated Validation Pipeline
The following diagram illustrates the comprehensive workflow for validating and refining AI-based structure predictions using molecular dynamics simulations:
Multi-Agent Automation Systems
Recent advances in AI-agent frameworks have enabled automation of the complex workflow involved in MD validation. Systems like DynaMate leverage large language models (LLMs) to create modular, multi-agent frameworks capable of coordinating the setup, execution, and analysis of molecular simulations [41]. These systems use a scheduler agent that determines which specialized agent has the appropriate tools for each step in the validation pipeline, significantly reducing manual intervention and increasing reproducibility.
The DynaMate implementation demonstrates how customized Python functions can be encapsulated as tools for LLM agents, allowing researchers to automate repetitive tasks while maintaining flexibility for specialized analyses [41]. This approach is particularly valuable for high-throughput validation of AI predictions across multiple protein targets.
Case Studies and Applications
Domain Motion and Conformational Changes
BioEmu has demonstrated remarkable capability in sampling large-scale conformational transitions, achieving 55-90% success rates in covering reference experimental structures for domain motions [110]. In benchmark studies focusing on domain motions—which enable essential functions like enzyme catalysis and ligand binding—BioEmu effectively sampled open-closed transitions with RMSD values within 3Å of experimental references [110]. This performance significantly surpassed baseline methods like AFCluster and DiG, particularly for out-of-distribution proteins with limited homologous structures.
MD validation of these AI-predicted conformational changes involves running extended simulations starting from different predicted states and monitoring transitions using collective variables such as inter-domain distances or dihedral angles. Markov State Models constructed from these simulations can quantify transition probabilities and identify kinetic bottlenecks, providing insights into functional mechanisms.
Cryptic Pocket Prediction for Drug Discovery
Cryptic pockets—potential drug binding sites that are not apparent in static structures—represent a particularly promising application of combined AI-MD approaches. BioEmu has demonstrated capability in predicting open states of cryptic pockets in proteins like the sialic acid-binding factor and Fascin, with sampling success rates of 55-80% [110]. These predictions enable structure-based drug design targeting previously inaccessible sites.
For Fascin, an actin-bundling protein implicated in tumor cell migration and metastasis, the open state revealed by AI-MD simulations exposes new binding sites for inhibitor design [110]. MD validation confirms the stability of these pockets and enables virtual screening to identify compounds that disrupt Fascin's bundling function, demonstrating the therapeutic potential of this approach.
Thermodynamic Accuracy and Stability Prediction
A crucial advancement in AI-MD integration is the achievement of thermodynamic accuracy. BioEmu incorporates Property Prediction Fine-Tuning (PPFT), which fine-tunes the model on hundreds of thousands of experimental stability measurements [110]. This approach enables the model to optimize ensemble distributions by minimizing discrepancies between predicted and experimental values, ensuring generated structures are both diverse and thermodynamically constrained.
The PPFT algorithm enables the use of unstructured experimental data through joint optimization of property prediction and diffusion loss, minimizing KL divergence between generated sample properties and experimental labels [110]. This ensures thermodynamic consistency in the distribution and improves generalization to unseen sequences, addressing a key limitation of previous protein generation models.
Research Reagent Solutions
Table 3: Essential Tools and Software for AI-MD Validation Workflows
| Tool Name | Type | Primary Function | Application in Validation |
|---|---|---|---|
| AlphaFold2 [108] [109] | AI Structure Prediction | Predicts 3D protein structures from sequence | Provides initial structural models for MD validation |
| BioEmu [110] | Generative AI Dynamics | Samples protein equilibrium ensembles | Accelerates conformational sampling beyond traditional MD |
| FastMDAnalysis [17] | Analysis Package | Unified, automated MD trajectory analysis | Streamlines calculation of RMSD, RMSF, Rg, and other metrics |
| DynaMate [41] | Multi-Agent Framework | Automates simulation setup and analysis | Coordinates complex validation workflows through AI agents |
| RoseTTAFold All-Atom [109] | Biomolecular Modeling | Predicts structures of protein complexes with ligands/nucleic acids | Expands validation to multi-component biological assemblies |
| OpenFold [109] | Trainable Implementation | Fully trainable AlphaFold2 implementation | Enables model customization for specific protein families |
Future Directions and Challenges
While the integration of MD and AI has yielded significant advances, several challenges remain. Generalization to larger complexes (≥500 residues), multi-chain systems, and membrane proteins requires further optimization [110]. The scarcity of high-quality thermodynamic data for training and validation continues to limit model accuracy, though community efforts to expand datasets are underway.
Future developments will likely focus on integrating multimodal experimental data, such as cryo-EM maps and single-molecule fluorescence, to further constrain and validate models [110]. Additionally, more efficient sampling algorithms and specialized hardware will extend simulation timescales to directly observe rare biological events currently inaccessible to both pure MD and AI approaches.
The emerging paradigm of AI-driven physical simulation, exemplified by BioEmu's 4-5 order of magnitude speedup, suggests a future where MD validation becomes nearly instantaneous, enabling real-time refinement of AI predictions and truly interactive structural biology [110]. This acceleration will be particularly transformative for drug discovery, where rapid assessment of binding site dynamics and drug-target interactions can significantly shorten development timelines.
The integration of molecular dynamics simulations with AI-based structure prediction creates a powerful synergistic framework for computational structural biology. MD provides the physical validation necessary to assess the dynamic stability and functional relevance of AI predictions, while AI offers rapid structural hypotheses and accelerated sampling of conformational space. As both fields continue to advance, their integration will become increasingly seamless, enabling researchers to move beyond static structures toward dynamic, mechanistic understanding of protein function. The automated workflows and validation protocols outlined in this guide provide a roadmap for researchers seeking to leverage these complementary technologies for robust, biologically relevant structural predictions.
Molecular dynamics (MD) simulation is a cornerstone technique in computational biology, chemistry, and materials science, providing atomic-level insights into the behavior of biomolecules, chemical reactions, and material properties. The reliability of these insights, however, hinges on a critical and often overlooked assumption: that the simulation has achieved sufficient convergence and sampling to yield statistically meaningful results. An unconverged simulation trajectory can lead to erroneous interpretations and unreliable predictions, potentially invalidating the scientific conclusions drawn from the computational experiment [112].
This technical guide examines the challenges of assessing convergence and sampling in molecular dynamics simulations, providing a comprehensive framework of methodologies and best practices to ensure statistical reliability. Within the broader context of molecular dynamics simulation workflow research, establishing robust convergence criteria is not merely a supplementary step but a fundamental prerequisite for producing trustworthy, reproducible scientific data. We explore quantitative metrics, statistical frameworks, and experimental protocols that researchers can employ to validate their simulations, with particular attention to the distinct requirements for different molecular systems and properties of interest.
The Challenge of Convergence in Molecular Dynamics
The fundamental challenge in MD simulations is the sampling problem: biomolecular systems possess extraordinarily complex conformational landscapes with numerous local minima separated by energy barriers. The limited timescales accessible to even state-of-the-art simulations mean that complete exploration of this landscape is often computationally prohibitive [55]. Consequently, simulations may become trapped in metastable states, providing a skewed representation of the true equilibrium distribution.
A crucial conceptual distinction exists between partial equilibrium, where some properties have stabilized to consistent average values, and true thermodynamic equilibrium, which requires thorough exploration of the entire accessible conformational space, including low-probability regions [112]. Properties with the most biological interest, such as average distances between domains, often depend predominantly on high-probability regions of conformational space and may converge in multi-microsecond trajectories. In contrast, properties like transition rates between conformational states or the absolute free energy depend explicitly on low-probability regions and may require substantially longer simulation times to converge [112].
A common but flawed practice in the field is relying solely on visual inspection of the root mean square deviation (RMSD) to determine convergence. A controlled survey revealed significant variability in how different researchers identify the convergence point from identical RMSD plots, demonstrating that this subjective approach is unreliable and heavily influenced by presentation factors like graph scale and color [113]. Furthermore, RMSD has been shown to be an unsuitable convergence descriptor for systems featuring surfaces and interfaces [114].
Quantitative Metrics for Assessing Convergence
A robust assessment of convergence requires monitoring multiple complementary metrics that probe different aspects of the system's behavior and sampling. The table below summarizes key metrics and their interpretation.
Table 1: Key Metrics for Assessing Convergence in MD Simulations
| Metric | Description | What it Measures | Interpretation of Convergence |
|---|---|---|---|
| Potential Energy | Total potential energy of the system [112] | Energetic stability of the simulation | Fluctuations around a stable average value with no drift |
| Root Mean Square Deviation (RMSD) | Average displacement of atomic positions relative to a reference structure [112] [113] | Structural stability of the biomolecule | Plateau in the time-series plot (use with caution [113]) |
| Radius of Gyration (Rg) | Measure of the compactness of a protein structure [55] | Global shape and volume | Stable average value across independent replicates |
| Linear Partial Density | Density profiles of system components along a dimension [114] | Structure and equilibration of interfaces and layered systems | Stable correlation function between density profiles from different time blocks |
| Property Autocorrelation Function | Correlation of a property with itself at different time offsets [112] | Dynamical relaxation and memory | Decay to zero indicates loss of memory and sufficient sampling of transitions |
For intrinsically disordered proteins (IDRs), which explore a vast conformational space, convergence assessment requires specialized approaches. Standard metrics like RMSD are often insufficient. Instead, researchers should monitor the stability of experimentally verifiable ensemble properties, such as NMR chemical shifts, scalar couplings, residual dipolar couplings (RDCs), and SAXS/SANS profiles [115]. Convergence is indicated when these averaged observables remain stable with additional sampling and are consistent across multiple independent simulation replicates.
Statistical Frameworks for Reliability
Given the stochastic nature of MD, a single simulation trajectory is merely one sample from a distribution of possible trajectories. Statistical rigor requires multiple independent replicates to quantify uncertainty and ensure reliability.
The Replication Principle
A foundational practice is running at least three independent simulations starting from different initial conditions [96]. For example, a study on calmodulin employed 35 independent simulations (20 wild-type and 15 mutant) initiated from "different but equally plausible initial conditions" [55]. This approach allows researchers to distinguish true structural effects of a mutation from random variations due to incomplete sampling. Statistical tests (e.g., t-tests for normally distributed properties like radius of gyration) can then be applied for quantitative comparison [55].
Uncertainty Quantification
All reported observables from MD simulations should be accompanied by uncertainty estimates, such as confidence intervals or standard errors. This is critically important when comparing states or systems. For instance, investigations into purported box-size effects on solvation free energies showed that apparent trends disappeared when the uncertainty of the calculations (from multiple repeats, N=20) was properly accounted for [116]. Anecdotal evidence from single or few realizations can lead to unfounded claims.
Enhanced Sampling and Convergence
For complex biomolecular processes with high energy barriers (e.g., protein-ligand binding, conformational changes), standard MD sampling is often insufficient. Enhanced sampling methods, such as umbrella sampling, metadynamics, and replica exchange, are employed to accelerate the exploration of phase space.
When using these methods, convergence must be demonstrated for the calculated free energies or potentials of mean force (PMF). A powerful strategy is to initiate simulations from different states (e.g., bound and unbound for a protein-ligand system). The PMF is considered converged when the results from these different starting points agree within statistical error [117]. The use of replica exchange between windows significantly improves convergence by preventing trajectories from becoming trapped in local minima [117].
Table 2: Statistical Assessment Methods for Reliable MD Simulations
| Method | Protocol | Key Outcome | Application Example |
|---|---|---|---|
| Multiple Independent Simulations | Run ≥ 3 replicates from different initial velocities/coordinates [96] | Quantifies uncertainty and distinguishes systematic effects from random sampling noise | 35 replicates of calmodulin revealed mutation effects on dynamics obscured in single runs [55] |
| Statistical Hypothesis Testing | Apply t-tests, F-tests, or non-parametric equivalents to data from multiple replicates [55] | Provides quantitative, statistically rigorous comparison between simulation conditions (e.g., wild-type vs. mutant) | Testing the hypothesis that radius of gyration is the same for wild-type and mutant calmodulin [55] |
| Time-series Block Analysis | Divide a long trajectory into sequential blocks and calculate block averages of key properties [112] | Assesses stability of property estimates; fluctuations between blocks should be small relative to the mean | Determining if a multi-microsecond trajectory has sampled a stable average RMSD [112] |
| Forward/Backward Convergence | For PMF calculations, run simulations starting from both end-states (e.g., bound & unbound) [117] | Strong evidence for convergence when both directions produce the same free energy profile | Calculating the protein-lipid binding PMF for ANT/cardiolipin [117] |
Practical Protocols and Workflows
General Equilibration Protocol
A typical MD workflow begins with an equilibration phase to relax the system from its initial, often non-equilibrium, coordinates (e.g., from a crystal structure). A robust protocol includes:
- Energy minimization: Removes steric clashes.
- Solvent equilibration: With protein heavy atoms restrained, allowing solvent and ions to relax.
- Full system equilibration: Gradually releasing restraints while heating the system to the target temperature (e.g., 310 K) and pressurizing it (e.g., 1 bar).
- Unrestrained production simulation: The data from this final phase is used for analysis. The initial portion of the production run, during which properties may still be drifting, should be discarded as additional equilibration [112] [117].
Convergence should be checked by monitoring the stability of multiple properties (see Table 1) over time, not just RMSD.
Protocol for Converged Potential of Mean Force (PMF) Calculations
To obtain a reliable PMF for processes like protein-ligand binding or membrane protein association, the following protocol is recommended, as applied in studies of glycophorin A transmembrane helix dimerization [117]:
- Choose an optimal collective variable (CV): A poor CV (e.g., center-of-mass distance for helix dimerization) leads to poor convergence. An optimal CV (e.g., a distance matrix RMSD for glycophorin A) accurately describes the transition.
- Employ replica exchange umbrella sampling (REUS): This enhances sampling by allowing exchanges between adjacent umbrella windows along the CV.
- Initialize from multiple states: Perform two independent REUS calculations, one starting from the "bound" state and another from the "unbound" state.
- Check for agreement: The PMF is considered converged when the results from both independent calculations agree within statistical uncertainty.
The following workflow diagram summarizes the key steps in a robust convergence assessment strategy:
Special Considerations for Intrinsically Disordered Proteins (IDRs)
Sampling the conformational ensemble of IDRs is exceptionally challenging due to the vast, flat energy landscape. Recommended protocols include:
- Using enhanced sampling methods like Replica Exchange Solute Tempering (REST2) to improve conformational sampling [115].
- Validating against experimental data such as NMR chemical shifts, J-couplings, and SAXS profiles to ensure the simulated ensemble is physically realistic [115].
- Employing Markov State Models (MSMs) to extract a statistically representative ensemble from a large number of fast, short simulations [115].
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Essential Software and Tools for Convergence Analysis
| Tool Name | Type | Primary Function in Convergence | Reference/Resource |
|---|---|---|---|
| GROMACS | MD Simulation Package | High-performance engine for running simulations. | [113] [116] |
| AMBER | MD Simulation Package | Suite for running simulations and analyzing trajectories. | [113] |
| CHARMM | MD Simulation Package | Suite for running simulations and analyzing trajectories. | [113] |
| NAMD | MD Simulation Package | Parallel MD code for running simulations. | [113] |
| PLUMED | Plugin Library | Defines collective variables and performs enhanced sampling (e.g., Umbrella Sampling, Metadynamics). | [117] |
| DynDen | Analysis Tool | Assesses convergence in systems with interfaces/surfaces via linear partial density correlation. | [114] |
| MDAnalysis | Python Library | Analyzes trajectories, calculates RMSD, Rg, and other metrics. | [117] [114] |
| PyTorch | Machine Learning Library | Enables AI-driven potentials (MLIPs) for faster, accurate simulations via interfaces like ML-IAP-Kokkos. | [9] |
| LAMMPS | MD Simulation Package | Simulates materials with various force fields, including MLIPs. | [9] |
The following diagram illustrates the workflow for a convergence-checked enhanced sampling study, as used in PMF calculations:
Assessing the convergence and sampling of molecular dynamics simulations is not a mere formality but a fundamental requirement for producing statistically reliable and scientifically valid results. Relying on intuitive, single-metric checks like RMSD visualization is insufficient and potentially misleading. Instead, researchers must adopt a rigorous, multi-faceted strategy that includes:
- Monitoring multiple complementary metrics beyond RMSD.
- Performing multiple independent simulation replicates.
- Quantifying uncertainty for all reported properties.
- Using statistical tests for objective comparisons.
- Implementing enhanced sampling protocols with robust convergence checks for complex transformations.
By integrating these practices into the standard MD workflow, researchers can significantly enhance the credibility and reproducibility of their computational findings, ensuring that molecular dynamics simulations continue to provide powerful and reliable insights into the molecular world.
Conclusion
A robust molecular dynamics simulation workflow integrates a solid understanding of physical principles, meticulous system setup, efficient execution, and rigorous validation. As demonstrated, success hinges not only on choosing appropriate force fields and software but also on proactively troubleshooting common errors and systematically benchmarking results against experimental data. The integration of automated analysis tools and machine learning interatomic potentials is poised to further transform the field, enabling larger and longer simulations with enhanced accuracy. For biomedical research, these advancing capabilities in MD simulation promise to deepen our understanding of disease mechanisms at an atomic level, accelerate structure-based drug design by precisely characterizing binding interactions, and ultimately contribute to the development of novel therapeutics with greater efficiency and predictive power.
References
- 1. Classical and reactive molecular dynamics: Principles and ... [https://www.sciencedirect.com/science/article/pii/S036012852300014X]
- 2. Molecular Dynamics: From Basics to Application [Article v1.0] [https://livecomsjournal.org/index.php/livecoms/article/view/v6i1e3797]
- 3. Supplemental: Overview of the Common Force Fields [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/S01-Force_Fields_Overview/index.html]
- 4. OPLS4 - Schrödinger [https://www.schrodinger.com/platform/products/ms-opls4/]
- 5. End-to-end differentiable construction of molecular mechanics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9600499/]
- 6. Quantitative analysis of protein dynamics using a deep ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10941960/]
- 7. A workflow for molecular dynamics simulation of casting ... [https://chemrxiv.org/engage/chemrxiv/article-details/68b9eaf8728bf9025e616d13]
- 8. MDCrow: Automating Molecular Dynamics Workflows with ... [https://arxiv.org/html/2502.09565v1]
- 9. Enabling Scalable AI-Driven Molecular Dynamics ... [https://developer.nvidia.com/blog/enabling-scalable-ai-driven-molecular-dynamics-simulations/]
- 10. Molecular dynamics — AMS 2025.1 documentation - SCM [https://www.scm.com/doc/AMS/Tasks/Molecular_Dynamics.html]
- 11. Software in the Real World: 5 Uses... Molecular Dynamics Simulation [https://www.linkedin.com/pulse/molecular-dynamics-simulation-software-real-6faqf]
- 12. mdciao: Accessible Analysis and Visualization of Molecular ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1012837]
- 13. Optimize Your Simulation Workflow [https://www.digitalengineering247.com/article/optimize-your-simulation-workflow]
- 14. Construction of a specialized integrated simulation platform ... [https://www.nature.com/articles/s41598-023-42913-5]
- 15. Machine learning analysis of molecular dynamics ... [https://www.nature.com/articles/s41598-025-11392-1]
- 16. Structural Optimization for Simulation-Driven Design [https://www.simscale.com/blog/structural-optimization-for-simulation-driven-design/]
- 17. FastMDAnalysis: Software for Automated Analysis of ... [https://chemrxiv.org/engage/chemrxiv/article-details/69097c5aef936fb4a23023b0]
- 18. [2502.09565] MDCrow: Automating Molecular ... Dynamics Workflows [https://arxiv.org/abs/2502.09565]
- 19. AMBER vs GROMACS [https://diphyx.com/stories/amber-vs-gromacs/]
- 20. Which MD software is preferable for protein simulation? [https://mattermodeling.stackexchange.com/questions/11701/which-md-software-is-preferable-for-protein-simulation]
- 21. Choosing the best tool for MD simulations: GROMACS ... [https://www.linkedin.com/posts/omarariasgaguancela_gromacs-vs-amber-vs-namd-which-one-do-you-activity-7339451262129606656-xgBd]
- 22. Best CPU, GPU, RAM for Molecular Dynamics in 2024 ... [https://bizon-tech.com/blog/best-cpu-gpu-ram-for-molecular-dynamics?srsltid=AfmBOopqNU_s-U4bBRsiL3nhejO0URrSDG23baUx8qTz62_CbQvUCXpJ]
- 23. Comparison of software for molecular mechanics modeling [https://en.wikipedia.org/wiki/Comparison_of_software_for_molecular_mechanics_modeling]
- 24. Molecular Dynamics Simulations for Materials and ... [https://matlantis.com/en/resources/blog/md-intro/]
- 25. Implementing reactivity in molecular dynamics simulations ... [https://www.nature.com/articles/s41467-024-50793-0]
- 26. Accurate evaluation of molecular potential energy functions ... [https://www.nature.com/articles/s41598-025-20295-0]
- 27. An empirical formulation of accelerated molecular ... [https://www.sciencedirect.com/science/article/abs/pii/S0010465525004448]
- 28. MDAnalysisData: MD data for testing, learning, and ... [https://www.mdanalysis.org/MDAnalysisData/]
- 29. T020 · Analyzing molecular dynamics simulations [https://projects.volkamerlab.org/teachopencadd/talktorials/T020_md_analysis.html]
- 30. System preparation - GROMACS 2025.4 documentation [https://manual.gromacs.org/current/user-guide/system-preparation.html]
- 31. Molecular dynamics simulation for all - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6209097/]
- 32. the GROMOS force-field parameter sets 53A5 and 53A6 [https://pubmed.ncbi.nlm.nih.gov/15264259/]
- 33. Solvation Models - NWChem [https://nwchemgit.github.io/Solvation-Models.html]
- 34. Preparing Your System for Molecular Dynamics (MD) [https://ctlee.github.io/BioChemCoRe-2018/system-prep/]
- 35. Solvation structures and ion dynamics of CaCl2 aqueous ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261425001253]
- 36. Automating MD simulations for Proteins using Large ... [https://arxiv.org/html/2507.07887v1]
- 37. Computational BioPhysics Tutorials - 2025/2026 [https://cbp-unitn.gitlab.io/QCB/tutorial2_gromacs.html]
- 38. High Throughput Molecular Dynamics and Analysis [https://training.galaxyproject.org/training-material/topics/computational-chemistry/tutorials/htmd-analysis/tutorial.html]
- 39. Free energy techniques [https://cgmartini.nl/docs/tutorials/Martini3/Free_Energy_Techniques/]
- 40. Practical guidelines for optimising free energy calculations ... [https://www.sciencedirect.com/science/article/pii/S0009261425005378]
- 41. leveraging AI-agents for customized research workflows [https://pubs.rsc.org/en/content/articlehtml/2025/me/d5me00062a]
- 42. Fast Analysis of Molecular Dynamics Trajectories with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3085256/]
- 43. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 44. Frontier advances in molecular dynamics simulations for ... [https://pubs.rsc.org/en/content/articlelanding/2025/ra/d5ra00059a]
- 45. Molecular dynamics simulation approach for discovering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8916543/]
- 46. How to Analyze Results from Molecular Dynamics Simulations [https://www.iaanalysis.com/how-to-analyze-molecular-dynamics-simulation-results.html]
- 47. Analysis of molecular dynamics simulations [https://training.galaxyproject.org/training-material/topics/computational-chemistry/tutorials/analysis-md-simulations/tutorial.html]
- 48. 2.2.3: Hydrogen Bonding [https://bio.libretexts.org/Bookshelves/Microbiology/Microbiology_(Boundless)/02%3A_Chemistry/2.02%3A_Chemical_Bonds/2.2.03%3A_Hydrogen_Bonding]
- 49. Carbon-Oxygen Hydrogen Bonding in Biological Structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3516709/]
- 50. A combination of virtual screening, molecular dynamics ... [https://www.nature.com/articles/s41598-024-58485-x]
- 51. The subtle strength of hydrogen bonds [https://www.asbmb.org/asbmb-today/opinions/102224/the-subtle-strength-of-hydrogen-bonds]
- 52. Introduction to Principal Component Analysis [https://amberhub.chpc.utah.edu/introduction-to-principal-component-analysis/]
- 53. PCAViz: An Open-Source Python/JavaScript Toolkit for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6849643/]
- 54. From complex data to clear insights: visualizing molecular ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2024.1356659/full]
- 55. A statistical approach to the interpretation of molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2253239/]
- 56. pyPcazip: A PCA-based toolkit for compression and ... [https://www.sciencedirect.com/science/article/pii/S2352711016300036]
- 57. Visualization Software - GROMACS 2025.3 documentation [https://manual.gromacs.org/current/how-to/visualize.html]
- 58. SAMSON Connect | Home [https://www.samson-connect.net/]
- 59. Nanome: Virtual Reality for Drug Design and Molecular ... [https://nanome.ai/]
- 60. MDAKits and MDA-based tools [https://www.mdanalysis.org/pages/mdakits/]
- 61. Combining Experiment and Simulation in Protein Folding: Closing the Gap for Small Model Systems - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2291534/]
- 62. Graph Theory Approaches for Molecular Dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11853848/]
- 63. MDTraj: A Modern Open Library for the Analysis of ... [https://www.sciencedirect.com/science/article/pii/S0006349515008267]
- 64. AI-Enhanced Molecular Dynamics Simulations for Protein Folding and Drug Binding Prediction | American Journal of Botany and Bioengineering [https://biojournals.us/index.php/AJBB/article/view/1076]
- 65. Forces have not converged during energy minimization - User discussions - GROMACS forums [https://gromacs.bioexcel.eu/t/forces-have-not-converged-during-energy-minimization/12105]
- 66. Energy minimization error - User discussions - GROMACS forums [https://gromacs.bioexcel.eu/t/energy-minimization-error/478]
- 67. Minimization - General Methodology [https://www.uoxray.uoregon.edu/local/manuals/biosym/discovery/General/Minimization/Gen_Method.html]
- 68. Energy Minimization - GROMACS 2025.3 documentation [https://manual.gromacs.org/documentation/2025.3/reference-manual/algorithms/energy-minimization.html]
- 69. Common errors when using GROMACS — GROMACS 2019-rc1 documentation [https://manual.gromacs.org/documentation/2019-rc1/user-guide/run-time-errors.html]
- 70. Removing friction in Structure-Based Drug Design through ... [https://www.desertsci.com/2025/03/19/removing-friction-in-structure-based-drug-design-through-advanced-molecular-dynamics/]
- 71. Top Drug Discovery Software Solutions to Watch in 2025 [https://www.deepmirror.ai/post/top-drug-discovery-software-solutions-to-watch-in-2025]
- 72. Constraint algorithms - GROMACS 2025.4 documentation [https://manual.gromacs.org/documentation/current/reference-manual/algorithms/constraint-algorithms.html]
- 73. Molecular Dynamics Simulations and Drug Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10705576/]
- 74. Enriching stabilizing mutations through automated analysis ... [https://pubmed.ncbi.nlm.nih.gov/41074875/]
- 75. Understanding LINCS Warnings in GROMACS [https://parssilico.com/blogs/97-lincs-warnings-in-gromacs]
- 76. LINCS Error in production run - GROMACS forums [https://gromacs.bioexcel.eu/t/lincs-error-in-production-run/10434]
- 77. Thermal Stability Ranking of Energetic Crystals via a ... [https://www.sciencedirect.com/science/article/pii/S2667134425000525]
- 78. Practical considerations for Molecular Dynamics - GitHub Pages [https://computecanada.github.io/molmodsim-md-theory-lesson-novice/aio/index.html]
- 79. Metadata practices for simulation workflows | Scientific Data [https://www.nature.com/articles/s41597-025-05126-1]
- 80. Density artefacts at interfaces caused by multiple time-step effects in... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6441880/]
- 81. [1710.00941] Communication: Truncated non-bonded potentials can... [https://arxiv.org/abs/1710.00941]
- 82. Molecular dynamics-driven drug discovery [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp00380f]
- 83. A Review of Global Optimization Methods for Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12537999/]
- 84. Learning the action for long-time-step simulations of ... [https://arxiv.org/html/2508.01068v1]
- 85. Molecular Dynamics - GROMACS 2025.4 documentation [https://manual.gromacs.org/documentation/current/reference-manual/algorithms/molecular-dynamics.html]
- 86. EMFF-2025: a general neural network potential for ... [https://www.nature.com/articles/s41524-025-01809-w]
- 87. Accelerating Molecular Dynamics Simulations with ... [https://arxiv.org/html/2510.06562v2]
- 88. Which Optimizer Should You Use With NNPs? | Rowan [https://www.rowansci.com/blog/which-optimizer-should-you-use-with-nnps]
- 89. Refining coarse-grained molecular topologies: a Bayesian ... [https://www.nature.com/articles/s41524-025-01729-9]
- 90. GōMartini 3: From large conformational changes in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12043922/]
- 91. A Quick Guide to Discrete Color Palettes in Molecular ... [https://blog.samson-connect.net/a-quick-guide-to-discrete-color-palettes-in-molecular-visualization]
- 92. Validating Molecular Dynamics Simulations Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420231/]
- 93. Structure-Based Experimental Datasets for Benchmarking ... [https://livecomsjournal.org/index.php/livecoms/article/view/v6i1e3871]
- 94. Integrating experimental data with molecular simulations to ... [https://arxiv.org/html/2207.08622v4]
- 95. Integrating experimental data with molecular simulations to ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X22001828]
- 96. Reliability and reproducibility checklist for molecular ... [https://www.nature.com/articles/s42003-023-04653-0]
- 97. Optimized protein-water interactions and torsional ... [https://www.nature.com/articles/s41467-025-65603-4]
- 98. Structure-Based Experimental Datasets for Benchmarking ... [https://pubmed.ncbi.nlm.nih.gov/40196146/]
- 99. Comparison of protein force fields for molecular dynamics ... [https://pubmed.ncbi.nlm.nih.gov/18446282/]
- 100. Comparative Study of Molecular Mechanics Force Fields for β ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10302482/]
- 101. Comparison of the AMBER, CHARMM, COMPASS ... [https://www.sciencedirect.com/science/article/abs/pii/S0378381206003323]
- 102. Evaluation of nine condensed-phase force fields ... [https://pubs.rsc.org/en/content/articlelanding/2021/cp/d1cp00215e]
- 103. Force field - GROMACS 2025.3 documentation [https://manual.gromacs.org/documentation/2025.3/reference-manual/functions/force-field.html]
- 104. Force field comparison for molecular dynamics simulations ... [https://www.sciencedirect.com/science/article/abs/pii/S0167732224024061]
- 105. Benchmarking biomolecular force fields for molecular ... [https://www.sciencedirect.com/science/article/pii/S2405844025019644]
- 106. Accelerating structural dynamics through integrated research ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12313326/]
- 107. Shedding Light on the Dark Matter of Molecular Dynamics ... [https://elifesciences.org/reviewed-preprints/90061v1]
- 108. Highly accurate protein structure prediction with AlphaFold [https://www.nature.com/articles/s41586-021-03819-2]
- 109. AI Models For Protein Structure Prediction [https://frontlinegenomics.com/ai-models-for-protein-structure-prediction/]
- 110. BioEmu: AI‐Powered Revolution in Scalable Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12643044/]
- 111. From complex data to clear insights: visualizing molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11043564/]
- 112. Convergence and equilibrium in molecular dynamics ... [https://www.nature.com/articles/s42004-024-01114-5]
- 113. Is an Intuitive Convergence Definition of Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3145956/]
- 114. DynDen: Assessing convergence of molecular dynamics ... [https://www.sciencedirect.com/science/article/pii/S0010465521002381]
- 115. Statistical accuracy of molecular dynamics-based methods ... [https://pubs.rsc.org/en/content/articlehtml/2024/cp/d4cp02564d]
- 116. On the importance of statistics in molecular simulations for ... [https://elifesciences.org/articles/57589]
- 117. Convergence and Sampling in Determining Free Energy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5402295/]