tRNA Duplication: An Evolutionary Blueprint for Genetic Code Expansion and Therapeutic Development
This article explores the cutting-edge field of genetic code expansion (GCE) through the lens of tRNA gene duplication, an evolutionary mechanism now being harnessed for synthetic biology.
tRNA Duplication: An Evolutionary Blueprint for Genetic Code Expansion and Therapeutic Development
Abstract
This article explores the cutting-edge field of genetic code expansion (GCE) through the lens of tRNA gene duplication, an evolutionary mechanism now being harnessed for synthetic biology. Tailored for researchers, scientists, and drug development professionals, it provides a comprehensive analysis spanning from the foundational principles of tRNA biology and conservation to advanced methodologies for engineering orthogonal translation systems. We delve into critical troubleshooting strategies for optimizing incorporation efficiency and orthogonality, and present rigorous validation frameworks for assessing therapeutic potential. By synthesizing insights from evolutionary biology and modern engineering approaches, this review serves as a strategic guide for leveraging tRNA duplication to overcome the constraints of the canonical genetic code and develop novel biomedical tools and therapeutics.
The Evolutionary Foundation: How Natural tRNA Duplication Informs Synthetic Biology
Transfer RNA (tRNA) represents one of the most ancient biological molecules, often described as a "living fossil" that preserves primordial genetic coding mechanisms across all domains of life [1]. As an evolutionary ancient molecule, tRNA exhibits remarkable conservation in sequence and structure while maintaining its fundamental role in protein synthesis. The concept of tRNA as a living fossil reflects its persistence since the RNA world hypothesis, with contemporary tRNA-like structures providing clues to early evolutionary processes [2]. This conservation makes tRNA an invaluable subject for studying deep evolutionary relationships and developing tools for genetic code expansion.
The structural conservation of tRNA is particularly striking. All canonical tRNAs fold into a relatively rigid three-dimensional L-shaped structure through the formation of two orthogonal helices, consisting of the acceptor and anticodon domains [3]. This conserved tertiary organization arises through intramolecular interactions between the D- and T-arms, maintaining functional integrity across billions of years of evolution. Regardless of sequence variability, this architectural blueprint remains consistent, supporting tRNA's canonical function in translation while enabling its recruitment for non-canonical biological functions.
Quantitative Analysis of tRNA Conservation
Genome-Wide Conservation Patterns
Recent comparative genomics studies reveal profound conservation of tRNA genes across diverse species. A comprehensive analysis of 50 plant species identified 28,262 high-confidence tRNA genes encompassing eight divisions within the plant kingdom, demonstrating consistent patterns in gene length, intron distribution, and GC content [1]. The structural parameters of these tRNA genes show remarkable stability despite vast evolutionary distances between species.
Table 1: Conservation of tRNA Genes Across 50 Plant Species
| Genomic Feature | Conservation Range | Representative Examples | Evolutionary Significance |
|---|---|---|---|
| tRNA Gene Length | 62-98 bp (peaking at 72 bp and 82 bp) | All angiosperms, bryophytes, chlorophytes | Highly constrained length distribution suggests structural optimization |
| GC Content | Variable but patterned | Consistent GC distribution patterns across species | Maintains structural stability and transcriptional efficiency |
| Intron Distribution | Ubiquitous in all species | tRNAMetCAT and tRNATyrGTC most abundant | Splicing mechanisms conserved across plant kingdom |
| Tandem Duplications | 578 identical tandemly duplicated pairs | Proline tRNA pairs in 33 species | Important evolutionary mechanism for tRNA gene expansion |
The abundance of tRNA genes shows surprising variation, ranging from just 56 in red algae (Pum) to 1,451 in Camelina sativa, with analysis revealing no significant correlation between tRNA gene number and genome size (r = 0.18, p = 0.21) [1]. This lack of correlation suggests that tRNA gene copy number is regulated by functional constraints rather than genome size dynamics, highlighting the specialized evolutionary pressures on this essential gene family.
Deep Evolutionary Conservation
The conservation of tRNA extends beyond plants to encompass all eukaryotic kingdoms. Evidence from mitochondrial genomes reveals that animal mtDNAs typically contain 22 tRNA genes as part of the conserved set of 37 mitochondrial genes [4]. This consistent gene complement in mitochondrial genomes, which are much diminished from their bacterial ancestors, underscores the essential nature of tRNA for organellar function.
The promoter architecture of tRNA genes reveals intriguing evolutionary patterns. Plant tRNA genes exhibit a highly conserved TATA motif followed by a CAA motif in their upstream regions, while animal tRNA upstream regions are highly heterogeneous and lack a common conserved sequence signature [5]. This fundamental difference in transcriptional regulation suggests divergent evolutionary paths in how tRNA gene expression is controlled across kingdoms, despite conservation of the genes themselves.
Table 2: Comparative Analysis of tRNA Features Across Kingdoms
| Molecular Feature | Plant-Specific Patterns | Animal-Specific Patterns | Universal Conservation |
|---|---|---|---|
| Upstream Promoter | Conserved TATA + CAA motif | Heterogeneous, anticodon-dependent motifs | Internal A and B box promoters |
| Tandem Duplications | Widespread (e.g., 27 tRNAPro in Arabidopsis) | Less common, more dispersed | Duplication as evolutionary mechanism |
| Isoacceptor Diversity | 49 distinct types for 22 amino acids | Similar diversity with tissue-specific expression | Consistent recognition of genetic code |
| tRNA-derived Fragments | Stress-responsive tsRNAs | Tissue-specific regulatory roles | Conservation of cleavage pathways |
Experimental Protocols for Studying tRNA Conservation and Duplication
Genome-Wide tRNA Gene Identification and Analysis
Protocol 1: Identification and Characterization of tRNA Genes
This protocol enables comprehensive annotation of tRNA genes across any genome, facilitating comparative analysis of conservation patterns.
Materials and Reagents:
- Nuclear genome sequence data in FASTA format
- tRNAscan-SE software (version 2.0.12 or higher)
- RNAFold for Minimum Free Energy (MFE) calculations
- VARNA GUI for secondary structure visualization
- R scripting environment with ggplot2 package
Methodology:
- Data Acquisition: Download nuclear genome sequences, coding sequences, and protein sequences from appropriate databases (e.g., Phytozome for plants)
- tRNA Gene Annotation: Execute tRNAscan-SE with eukaryotic parameters (
-Hand-yflags) followed by filtration for high-confidence sets using EukHighConfidenceFilter [1] - Structural Analysis: Calculate Minimum Fold Energy (MFE) for each identified tRNA gene using RNAFold to assess structural stability
- Sequence Alignment: Perform multiple sequence alignment of identical-sequence, intron-containing tRNA genes using multialin or similar tools
- GC Content Analysis: Calculate GC content using a sliding window approach (5 bp window, 1 bp step) with custom R scripts, normalizing against total tRNA gene length
- Phylogenetic Analysis: Cluster tRNA sequences using MMseqs2 with minimum sequence identity of 0.9 and coverage of 0.8, followed by phylogenetic tree construction with IQ-TREE 2 using best-fit models
Applications: This protocol successfully identified 28,262 tRNA genes across 50 plant species, revealing conservation in gene length (62-98 bp) and the presence of intron-containing genes in all species studied [1].
Identification of Tandem Duplication Events
Protocol 2: Analysis of Tandem tRNA Gene Duplications
Tandem duplication represents a fundamental evolutionary mechanism for tRNA gene expansion. This protocol details computational identification and characterization of these events.
Materials and Reagents:
- Genomic coordinates of annotated tRNA genes
- Custom scripts for genomic interval analysis (Python/R)
- Sequence alignment software (ClustalO)
- KaKs_Calculator 3.0 for evolutionary analysis
Methodology:
- Initial Identification: Scan genomic coordinates to identify tRNA gene pairs and clusters located on the same chromosome with physical distance less than 1 kb [1]
- Sequence Similarity Assessment: For clusters with sequence similarity below 100%, use unique tRNA gene sequences for further screening
- Cluster Definition: Define tandem repeats as clusters where different combinations of tRNA genes recur, and where tRNA genes sharing the same anticodon exhibit identical sequences
- Evolutionary Analysis: Calculate Kn/Ks ratios using KaKs_Calculator 3.0 to assess selective pressure on duplicated genes
- Classification: Categorize duplication types (double-, triple-, or quintuple-tRNA genes) and determine repetition frequency
Applications: Application of this protocol revealed 578 identical tandemly duplicated tRNA gene pairs grouped into 410 clusters across plant species, with proline tRNA pairs widely distributed in 33 species including both lower and higher plants [1].
Figure 1: Experimental workflow for analyzing tRNA conservation and tandem duplications across genomes.
tRNA Engineering for Genetic Code Expansion
Structural Principles for tRNA Engineering
The highly conserved structure of tRNA provides both opportunities and challenges for genetic code expansion (GCE). Engineering tRNAs for GCE requires balancing orthogonality to host cell systems with cooperativity with translational machinery [6]. Successful engineering strategies focus on specific structural domains:
Acceptor Stem Engineering: Modifications to the acceptor stem (particularly positions 1-7 and 66-72) can enhance orthogonality by preventing recognition by endogenous aminoacyl-tRNA synthetases (AARS). The discriminator base (position 73) serves as a key identity element for many AARS [6].
Anticodon Loop Modifications: Engineered alterations to the anticodon enable reassignment of stop codons or quadruplet codons to unnatural amino acids. Except for SerRS, AlaRS, LeuRS, and PylRS, most AARS utilize anticodon recognition [6].
Variable Loop Optimization: The variable loop exhibits significant length and composition variation across species, providing an engineering target for creating orthogonal tRNA/AARS pairs, particularly for seryl, phenylalanyl, and tyrosyl tRNAs [6].
Practical tRNA Engineering Protocol
Protocol 3: Engineering Orthogonal tRNA Systems for Genetic Code Expansion
This protocol details the development of orthogonal tRNA systems for incorporating unnatural amino acids into proteins.
Materials and Reagents:
- Host organism cell lines (E. coli, yeast, or mammalian)
- Orthogonal AARS/tRNA pairs from divergent organisms
- Plasmid vectors for tRNA expression
- Unnatural amino acids for incorporation
- Antibiotics for selection
- Western blot reagents for detection
Methodology:
- Orthogonal Pair Selection: Select AARS/tRNA pairs from organisms of different phyla than the host to maximize orthogonality (e.g., archaeal pairs in eukaryotic hosts) [6]
- tRNA Library Construction: Create mutant tRNA libraries focusing on acceptor stem, anticodon loop, and variable loop regions
- Screening for Orthogonality: Transform host cells with tRNA library and screen for absence of mis-incorporation of natural amino acids
- Efficiency Optimization: Select variants that maintain high charging efficiency by the cognate AARS while rejecting recognition by endogenous AARS
- Functional Validation: Assess incorporation efficiency of unnatural amino acids at amber stop codons or other reassigned codons
- Specificity Testing: Verify specific incorporation at target sites without global proteomic disruption
Applications: Engineered tRNA systems have enabled incorporation of over 150 unnatural amino acids with diverse chemical properties, expanding the functional repertoire of recombinant proteins for therapeutic and research applications [6].
Figure 2: tRNA engineering workflow for genetic code expansion applications, highlighting iterative optimization of orthogonality and efficiency.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for tRNA Conservation and Engineering Studies
| Reagent/Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Bioinformatics Tools | tRNAscan-SE, RNAFold, MMseqs2 | tRNA gene identification, structural prediction, sequence clustering | Specialized algorithms for non-coding RNA features |
| Evolutionary Analysis | KaKs_Calculator, IQ-TREE 2 | Selection pressure analysis, phylogenetic reconstruction | Handles specific evolutionary patterns of structural RNAs |
| Structural Analysis | VARNA GUI, PyMOL | Visualization of secondary and tertiary structures | Molecular graphics optimized for nucleic acids |
| Orthogonal Systems | Archaeal tRNA/AARS pairs, Pyrrolysyl system | Genetic code expansion foundation | Cross-kingdom incompatibility enables orthogonality |
| Expression Vectors | Amber suppressor tRNA plasmids, Inducible promoters | Controlled tRNA expression in host systems | Regulated expression critical for toxic variants |
| Detection Reagents | Northern blot probes, Antibodies against epitope tags | Validation of tRNA expression and aminoacylation | Specific detection challenging for mature tRNAs |
Evolutionary Perspectives and Emerging Applications
Deep Evolutionary Origins
The origin of tRNA predates the advent of templated protein synthesis, with evidence suggesting tRNA-like structures first functioned as "genomic tags" in RNA world replication [2]. These primordial tRNA ancestors marked the 3' ends of ancient RNA genomes for replication by RNA enzymes, solving both specificity and telomere maintenance problems. This evolutionary history explains the conserved involvement of contemporary tRNA-like structures in viral replication, such as in bacteriophage Qβ and brome mosaic virus [2].
The modular structure of modern tRNA supports this evolutionary model. The simplest early tRNA tags may have been predecessors of the "top half" of modern tRNA, consisting of a coaxial stack of the TΨC arm on the acceptor stem [2]. This evolutionary perspective informs engineering strategies that treat tRNA as a modular scaffold with evolutionarily distinct domains that can be independently optimized.
tRNA-Derived Fragments in Stress Response
Beyond their canonical role in translation, tRNAs serve as precursors for regulatory small RNAs known as tRNA-derived fragments (tRFs) or tRNA-derived small RNAs (tsRNAs) [7]. These molecules represent a novel category of gene expression regulators that function at both transcriptional and post-transcriptional levels.
In plants, specific tsRNAs show altered expression under diverse stress conditions including salt, drought, temperature extremes, and pathogen infection [7]. The biogenesis of these fragments involves cleavage by specific ribonucleases, with RNase T2 family proteins playing crucial roles in generating tRNA halves in Arabidopsis [7]. This emerging field reveals another dimension of tRNA evolutionary conservation, with the same ancient molecular scaffold being repurposed for regulatory functions across diverse lineages.
The deep conservation of tRNA as a living fossil provides both constraints and opportunities for genetic code expansion research. The highly conserved structural core enables predictive engineering based on evolutionary principles, while lineage-specific variations offer templates for developing orthogonal systems. The documented patterns of tRNA gene duplication and conservation across plants and animals [1] [4] inform strategies for optimizing tRNA copy number and expression in engineered systems.
Future directions include mining the expanding genomic resources from diverse species, particularly "living fossil" organisms like gymnosperms [8], to identify novel tRNA variants with unique properties. The evolutionary perspective on tRNA origins [2] suggests that engineering minimal tRNA scaffolds may yield efficient systems unburdened by evolutionary constraints of modern translational apparatus. Similarly, insights into how essential tRNA synthetases can evolve new functions [9] provide paradigms for directed evolution of orthogonal pairs.
The study of tRNA as a living fossil continues to yield fundamental insights into molecular evolution while providing practical tools for synthetic biology. By leveraging deep conservation patterns and understanding the exceptions to these patterns, researchers can develop increasingly sophisticated genetic code expansion systems with applications in therapeutic protein production, basic biological research, and understanding the fundamental constraints on the evolution of biological information processing.
Application Notes
Tandem duplication serves as a fundamental evolutionary mechanism driving genome plasticity and adaptation across plant species. This process, which generates tandem arrays of identical or similar sequences in close genomic proximity, occurs through unequal chromosomal recombination and represents a widespread phenomenon in plant genomes [10] [11]. Unlike whole-genome duplication events that affect all genes simultaneously, tandem duplication operates at a finer scale, producing significant gene copy number and allelic variation within populations [12]. Recent research has revealed that tandem duplication contributes substantially to the expansion of gene families, with approximately 4.74% to 14% of genes in various plant species classified as tandem duplicated genes (TDGs) [10] [12].
The evolutionary significance of TDGs is particularly evident in their functional bias toward environmental adaptation. Genes involved in stress responses show an elevated probability of retention following tandem duplication, suggesting these duplicates play crucial roles in adaptive evolution to rapidly changing environments [12]. This adaptive mechanism enables plants to develop enhanced resistance to both biotic and abiotic stressors, including pathogen attacks, salinity, and other environmental challenges [10] [13]. The lineage-specific nature of tandem duplication events further contributes to the diversification of plant species by creating genetic innovations that may be selectively advantageous in particular ecological niches.
Key Quantitative Findings from Cross-Species Analysis
Comprehensive analysis across multiple plant species has revealed striking patterns in TDG distribution and abundance. Table 1 summarizes the quantitative findings from genome-wide studies of tandem duplication events, highlighting species-specific variations that underscore the dynamic nature of plant genome evolution.
Table 1: Genome-Wide Tandem Duplication Patterns Across Plant Species
| Species | Genome Size | Total Genes | TDG Number | TDG Percentage | Key Enriched Functions |
|---|---|---|---|---|---|
| Seashore Paspalum (Paspalum vaginatum) | 517.98 Mb | 28,712 | 2,542 | 8.85% | Ion transmembrane transport, ABC transport [10] |
| Pigeonpea (Cajanus cajan) | 833 Mb | 48,680 | 3,837 | 7.88% | Stress resistance pathways, retrotransposons [13] |
| Arabidopsis (Arabidopsis thaliana) | 125 Mb | 35,386 | 3,503 | 9.90% | Environmental stress response, membrane functions [11] [12] |
| Rice (Oryza sativa) | ~400 Mb | Not specified | ~7.78% | ~7.78% | Stress tolerance, membrane functions [10] [11] |
| Maize (Zea mays) | ~2,400 Mb | Not specified | ~4.74% | ~4.74% | Stress tolerance, membrane functions [10] [11] |
| Foxtail Millet (Setaria italica) | ~490 Mb | Not specified | ~11.55% | ~11.55% | Stress tolerance, membrane functions [10] |
| Sorghum (Sorghum bicolor) | ~730 Mb | Not specified | ~10.82% | ~10.82% | Stress tolerance, membrane functions [10] |
Analysis of 50 plant species spanning eight divisions within the plant kingdom (Angiospermae, Bryophyta, Chlorophyta, Lycopodiophyta, Marchantiophyta, Pinophyta, Pteridophyta, and Rhodophyta) has identified 28,262 high-confidence tRNA-coding genes, with abundance ranging from 56 in red algae (Pum) to 1,451 in Camelina sativa [1]. This substantial variation in tRNA gene number shows no significant correlation with genome size (r = 0.18, p = 0.21), suggesting specific evolutionary pressures rather than random expansion mechanisms [1].
A critical finding across studies is the functional enrichment of TDGs in stress response pathways. In seashore paspalum, TDGs show significant enrichment in Gene Ontology terms including "ion transmembrane transporter activity," "anion transmembrane transporter activity," and "cation transmembrane transport," along with KEGG pathways such as "ABC transport" [10]. Similarly, pigeonpea TDGs are significantly enriched in resistance-related pathways, indicating that stress resistance in this species may be ascribed to these pathways originating from tandem duplications [13].
tRNA Gene Duplication and Genetic Code Expansion
The conservation and tandem duplication of tRNA genes represents a particularly insightful model for understanding evolutionary mechanisms in plant genomes. Plant tRNA genes demonstrate remarkable conservation in terms of gene length (ranging from 62 to 98 bp), intron length, GC content, and sequence identity [1]. This conservation highlights the structural and functional constraints on these essential components of the translation machinery while allowing for evolutionary innovation through duplication events.
A comprehensive study identified 578 identical tandemly duplicated tRNA gene pairs grouped into 410 clusters, with some clusters containing up to 26 identical tRNA genes [1]. Different duplication patterns were observed, including double-, triple-, and quintuple-tRNA genes repeated for varying numbers of times. Notably, tandemly located tRNA gene pairs with anticodons to proline were widely distributed across 33 plant species, including both lower and higher plants, suggesting an evolutionarily conserved duplication mechanism with potential adaptive significance [1].
Table 2: tRNA Gene Duplication Patterns Across Plant Species
| Duplication Feature | Findings | Evolutionary Significance |
|---|---|---|
| Total tRNA Genes | 28,262 across 50 plant species | Essential translation components with high conservation [1] |
| Gene Length Range | 62-98 bp (peaking at 72 bp and 82 bp) | Structural constraints in secondary structure formation [1] |
| Tandem Duplication Events | 578 identical tandemly duplicated tRNA gene pairs grouped into 410 clusters | Mechanism for increasing dosage of specific tRNAs [1] |
| Maximum Cluster Size | Up to 26 identical tRNA genes | Potential for substantial changes in translation efficiency [1] |
| Conserved Anticodon Duplication | Proline anticodon tandems widespread in 33 species | Lineage-specific adaptation in translation machinery [1] |
| Duplication Types | Double-, triple-, and quintuple-tRNA gene repeats | Diverse evolutionary trajectories in different lineages [1] |
The expansion of tRNA genes through tandem duplication provides a mechanism for genetic code flexibility and potential expansion. According to the evolutionary trajectory hypothesis, the genetic code sectorized from a glycine code to 4 amino acid codes, then to 8 amino acid codes, then to 16 amino acid codes, and finally to the standard 20 amino acid codes with stops [1]. Tandem duplication of tRNA genes may represent a contemporary mechanism supporting this evolutionary trajectory, potentially enabling the incorporation of novel amino acids or the refinement of translation efficiency under specific environmental conditions.
Protocols
Genome-Wide Identification of Tandem Duplicated Genes
Principle: This protocol enables systematic identification and characterization of tandem duplicated genes (TDGs) from plant genome sequences using a combination of sequence similarity search and genomic location analysis [10] [11].
Materials:
- Plant genome sequence in FASTA format
- Genome annotation in GFF/GTF format
- Computing infrastructure with adequate storage and memory
- BLAST+ suite (v2.0 or higher)
- MCScanX software
- Perl and Python scripting environments
- R statistical platform with clusterProfiler package
Procedure:
Data Preparation
- Obtain protein sequence files and General Feature Format (GFF) files for the target species [10].
- For genes with multiple transcripts, select the longest transcript for subsequent analysis to ensure consistent comparison [10].
- Format the genome sequences and create BLAST databases using makeblastdb command.
Homologous Gene Identification
- Perform an all-against-all BLASTP search using protein sequences with an E-value cutoff of 1e-10 and retain the first 10 matches [10].
- Execute the following command:
Tandem Duplication Detection
Evolutionary Analysis
- Calculate non-synonymous (Ka) and synonymous (Ks) substitution rates for identified TDG pairs using ParaAT software [10].
- Compute approximate dates of duplication events using the formula T = Ks/2λ, where λ represents the clocklike rate of synonymous substitutions (typically 1.5 × 10⁻⁸ for plants) [10].
- Determine selection pressure by calculating Ka/Ks ratios for each gene pair.
Functional Enrichment Analysis
- Perform Gene Ontology (GO) and KEGG pathway enrichment analysis using the clusterProfiler R package [10].
- Calculate the "rich factor" as the ratio of the number of differentially expressed genes annotated in a term to the number of all genes annotated in that term.
- Apply false discovery rate (FDR) correction for multiple testing, with FDR ≤ 0.05 considered statistically significant.
Troubleshooting Tips:
- If MCScanX produces insufficient TDG calls, adjust the BLASTP E-value threshold to 1e-5 to capture more distant homologs.
- For large genomes, consider parallelizing BLAST searches by chromosome.
- Verify ambiguous TDG calls by manually inspecting genomic coordinates in a genome browser.
Identification and Analysis of Tandemly Duplicated tRNA Genes
Principle: This protocol enables comprehensive identification and characterization of tandemly duplicated tRNA genes using specialized tRNA detection software and phylogenetic analysis [1].
Materials:
- Nuclear genome sequences of target plant species
- tRNAscan-SE software (v2.0.12 or higher)
- RNAfold software
- MMseqs2 software
- IQ-TREE (v2.0 or higher)
- KaKs_Calculator (v3.0)
- R software with ggplot2 and ComplexHeatmap packages
Procedure:
tRNA Gene Identification
Sequence and Structural Analysis
- Calculate GC content using a sliding window approach (5 bp window, 1 bp step) and normalize against total tRNA gene length [1].
- Generate fitting curves and confidence intervals for average GC content using the 'loess' method in ggplot2.
- Visualize secondary structures of representative tRNA genes using VARNA GUI [1].
Tandem Duplication Identification
- Identify tRNA gene pairs and clusters located on the same chromosome or scaffold with physical distance less than 1 kb [1].
- Define clusters where different combinations of tRNA genes recur, and where tRNA genes sharing the same anticodon exhibit identical sequences, as tandem repeats [1].
- Classify tandem arrays by repetition pattern (double-, triple-, or quintuple-tRNA genes).
Phylogenetic and Evolutionary Analysis
- Create a database of all tRNA genes using MMseqs2 createdb function [1].
- Cluster sequences with minimum sequence identity of 0.9 and coverage of 0.8.
- Perform multiple sequence alignment for tRNA genes with specific anticodons using clustalo.
- Identify best substitution models using ModelFinder in IQ-TREE.
- Construct phylogenetic trees with 1000 bootstrap replicates using the best-fit models [1].
- Calculate Kn/Ks ratios using KaKs_Calculator 3.0 with default parameters [1].
Comparative Genomics
- Statistically analyze the number of tRNA-coding genes with specific anticodons across species.
- Visualize results using heatmaps generated with ComplexHeatmap [1].
- Correlate tRNA abundance with genomic features such as genome size and codon usage bias.
Validation Methods:
- Verify tandem clusters by PCR amplification and Sanger sequencing of selected loci.
- Validate expression of duplicated tRNA genes using Northern blotting or RT-qPCR.
- Confirm structural predictions using chemical mapping or enzymatic probing for representative tRNAs.
Expression Analysis of Tandem Duplicated Genes Under Stress Conditions
Principle: This protocol assesses the expression patterns of TDGs in response to environmental stressors using RNA sequencing and differential expression analysis [10].
Materials:
- Plant materials subjected to stress treatments and controls
- RNA extraction kit with DNase treatment
- RNA quality assessment equipment (e.g., Bioanalyzer)
- Library preparation kit for RNA-seq
- High-throughput sequencing platform (e.g., Illumina HiSeq 2000)
- HISAT2 alignment software
- DESeq2 R package
- Computer with adequate RAM for processing large datasets
Procedure:
Experimental Design and Stress Treatment
- Grow plants under controlled conditions for two months [10].
- Apply stress treatment (e.g., 400 mM NaCl for salt stress) or mock treatment as control for varying durations (8, 12, 24, 48 hours, or 5 days) [10].
- Harvest tissues of interest with at least three biological replicates for each condition and time point.
RNA Sequencing
- Extract total RNA using quality-controlled methods ensuring RIN > 8.0.
- Prepare paired-end cDNA libraries using standard protocols.
- Sequence libraries on an appropriate platform (e.g., HiSeq 2000) to generate at least 20 million reads per sample [10].
Expression Quantification
- Align clean reads to the reference genome using HISAT2 with default parameters [10].
- Quantify gene expression levels using FPKM (fragments per kilobase per million mapped reads) or TPM (transcripts per million) values [10].
- Compile expression values into a count matrix for differential expression analysis.
Differential Expression Analysis
- Identify differentially expressed genes using DESeq2 with FDR ≤ 0.05 and |log2FC| ≥ 1 as significance thresholds [10].
- Classify tissue-specific expression patterns based on the following criteria:
Integration with TDG Data
- Overlap differentially expressed genes with previously identified TDGs.
- Perform functional enrichment analysis specifically on stress-responsive TDGs.
- Visualize expression patterns using heatmaps and cluster analysis.
Quality Control Measures:
- Monitor sequencing quality using FastQC.
- Remove low-quality reads and adapters using Trimmomatic or similar tools.
- Verify sample correlation through PCA and clustering analysis.
- Validate key findings using RT-qPCR on independent biological samples.
Visualization
Workflow for Genome-Wide Analysis of Tandem Duplicated Genes
Diagram 1: Comprehensive workflow for genome-wide identification and analysis of tandem duplicated genes in plant species.
Evolutionary Trajectory of Tandem Duplication in Plant Genomes
Diagram 2: Evolutionary pathways and fate of tandem duplicated genes in plant genomes under selective pressures.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for TDG Analysis
| Category | Tool/Reagent | Specific Function | Application Context |
|---|---|---|---|
| Genome Analysis Software | MCScanX | Detection and classification of tandem duplicated genes | Identifying TDGs from genomic sequences [10] |
| Sequence Alignment | BLAST+ Suite | Homology search and sequence similarity analysis | Identifying homologous gene pairs for TDG detection [10] |
| Evolutionary Analysis | ParaAT | Calculation of Ka/Ks ratios and divergence times | Estimating selection pressure and duplication timing [10] |
| tRNA Specialized Tools | tRNAscan-SE | Annotation of tRNA genes in genomic sequences | Identifying tRNA-coding genes and their locations [1] |
| Phylogenetic Analysis | IQ-TREE | Phylogenetic tree construction with model selection | Inferring evolutionary relationships of duplicated genes [1] |
| Expression Analysis | DESeq2 | Differential expression analysis of RNA-seq data | Identifying stress-responsive tandem duplicated genes [10] |
| Functional Enrichment | clusterProfiler | GO and KEGG pathway enrichment analysis | Determining functional biases in TDGs [10] |
| Sequence Clustering | MMseqs2 | Rapid clustering of large sequence datasets | Grouping related tRNA genes for duplication analysis [1] |
| Database Resources | PTGBase | Plant Tandem Duplicated Genes Database | Comparative analysis of TDGs across species [11] |
| Visualization | VARNA GUI | Visualization of RNA secondary structures | Examining structural features of duplicated tRNA genes [1] |
Within the broader framework of genetic code expansion (GCE) research, the duplication of transfer RNA (tRNA) genes presents a fundamental pathway for the evolution of novel translational components. Duplicated tRNA genes can serve as raw material for the development of orthogonal tRNA partners, which are crucial for incorporating unnatural amino acids (UAAs) into proteins [14]. The functional fate of duplicated genes is diverse; copies may be retained through subfunctionalization or neofunctionalization, or they may be lost [15]. A key conjecture in this field, the "least diverged ortholog" (LDO) conjecture, posits that following duplication, the copy that undergoes less sequence divergence is more likely to retain the ancestral function, while the more diverged copy (MDO) may acquire new, specialized roles [15]. Understanding the structural hallmarks of these duplicated genes—specifically their gene length, intron patterns, and GC content—is therefore not merely a descriptive exercise but a critical endeavor for rationally selecting and engineering tRNA duplicates for GCE applications. This Application Note provides detailed methodologies for the quantitative analysis of these structural features, equipping researchers with the tools to characterize and exploit tRNA gene duplications systematically.
Quantitative Analysis of Structural Hallmarks
The accurate quantification of tRNA pools, including duplicated genes, is hampered by technical challenges such as pervasive RNA modifications that block reverse transcription and the high sequence similarity among tRNA genes [16] [17]. The following methodologies are designed to overcome these hurdles and provide high-resolution data on tRNA abundance and sequence features.
Table 1: Key Methodologies for tRNA Abundance and Modification Profiling
| Method Name | Core Principle | Key Applications | Advantages | Limitations |
|---|---|---|---|---|
| mim-tRNAseq [16] | Uses a thermostable group II intron reverse transcriptase (TGIRT) under optimized conditions for efficient readthrough of modified sites, capturing misincorporation signatures. | - Quantifying tRNA abundance- Profiling tRNA modification status- Assessing aminoacylation levels | - Applicable to any organism with a known genome- Captures abundance and modification data in one reaction- Full-length cDNA sequences | - Requires a comprehensive computational toolkit for analysis |
| Nano-tRNAseq [17] | Direct sequencing of native tRNA molecules using nanopore technology, with 5' and 3' adapter ligation to improve capture. | - Simultaneous quantification of tRNA abundance and modifications- Analysis of modification dynamics and crosstalk | - No reverse transcription or PCR bias- Detects modifications directly from native RNA- Single-molecule resolution | - Default sequencing settings can discard tRNA reads, requiring custom data reprocessing- Lower throughput compared to NGS |
Table 2: Key Research Reagent Solutions for tRNA Analysis
| Reagent / Tool | Function / Application | Key Features / Considerations |
|---|---|---|
| TGIRT Enzyme [16] | Reverse transcriptase for mim-tRNAseq; enables readthrough of many Watson-Crick face tRNA modifications. | - High processivity- Template-switching capability- Optimal performance in low-salt buffers at 42°C |
| Barcoded DNA Adapters [16] | Ligation to tRNA 3' ends for library preparation; enables sample multiplexing. | - Designed to minimize co-folding with structured tRNAs- High ligation efficiency (89%–95%) |
| Orthogonal AARS/tRNA Pairs [14] | Core components for genetic code expansion; enable incorporation of unnatural amino acids. | - Must be orthogonal to host AARSs- Must function cooperatively with host translational machinery |
| tRNA Engineering Techniques [14] | Directed evolution and rational design to optimize tRNA orthogonality and efficiency in GCE. | - Targets interactions with AARS, EF-Tu, and the ribosome- Can alter identity elements and binding sites |
Experimental Protocols
Protocol for mim-tRNAseq Library Construction and Analysis
This protocol is adapted from Behrens et al. (2021) for high-resolution quantitation of tRNA abundance and modification status, which is essential for characterizing duplicated genes [16].
I. tRNA Purification and 3' Adapter Ligation
- Purification: Isolate mature tRNA pools from total RNA by gel size selection for RNAs between 60–100 nucleotides.
- Adapter Ligation: Ligate barcoded DNA adapters to the 3' end of deacylated tRNAs using T4 RNA ligase 2. The adapter design should limit potential secondary structure formation with tRNA.
- Efficiency Check: Confirm ligation efficiency (typically 89-95%) by analytical gel electrophoresis.
II. Reverse Transcription with TGIRT
- Pooling: Pool samples ligated with different barcoded adapters.
- Primer Annealing: Anneal a primer complementary to the 3' adapter to the pooled tRNA.
- cDNA Synthesis: Perform reverse transcription using TGIRT enzyme in a low-salt buffer at 42°C for an extended reaction time (e.g., 3 hours) to nearly eliminate premature RT stops at modified nucleosides.
III. Library Completion and Sequencing
- cDNA Circularization: Circularize the synthesized cDNA. The primer for cDNA synthesis should contain a 5′ RN dinucleotide to facilitate this step.
- Amplification and Sequencing: Amplify the library and sequence using an Illumina platform.
IV. Computational Analysis
- Use the dedicated mim-tRNAseq computational toolkit for read alignment, which accounts for modification-induced misincorporations to accurately assign reads to highly similar tRNA genes, including duplicates.
- Analyze output data for tRNA abundance, modification sites (from misincorporation patterns), and isodecoder dynamics.
Protocol for Functional Interrogation of Duplicated tRNA Genes
This protocol outlines a computational and experimental workflow to determine the functional fate of duplicated tRNA genes, based on the LDO conjecture [15].
I. Sequence Divergence Analysis
- Identification: Identify tRNA gene duplicates from genomic databases (e.g., using tRNAscan-SE).
- Alignment: Perform multiple sequence alignment of the duplicated tRNA genes.
- Ancestral Reconstruction: Reconstruct the ancestral sequence and calculate the evolutionary rates (number of substitutions per site) for each duplicate.
- Classification: Classify duplicates as "Least Diverged" (LDO) or "Most Diverged" (MDO) based on their branch lengths from the ancestral node [15].
II. Structural and Expression Profiling
- Gene Architecture: Analyze the LDO and MDO for differences in:
- Gene Length: Note variations in the length of the variable arm.
- Intron Patterns: Identify the presence or absence of introns and their sequences.
- GC Content: Calculate the GC content, particularly in the acceptor stem and anticodon loop, as it can influence tRNA stability and interactions with the ribosome and elongation factors.
- Expression Correlation: Integrate the structural data with expression profiles (e.g., from mim-tRNAseq or Nano-tRNAseq) to determine if structural divergence correlates with functional specialization.
The Scientist's Toolkit: Visualization and Workflow
A core component of analyzing duplicated tRNA genes is a clear experimental workflow that integrates computational predictions with empirical validation. The diagram below outlines this logical pathway.
Diagram 1: Analysis of Duplicated tRNA Genes
The strategic analysis of structural hallmarks in duplicated tRNA genes—gene length, intron patterns, and GC content—provides a powerful foundation for advancing genetic code expansion research. By applying the detailed protocols for mim-tRNAseq and functional interrogation outlined in this document, researchers can move beyond simple sequence identification to a deeper understanding of the evolutionary forces shaping the tRNA repertoire. This enables the rational selection and engineering of specialized tRNAs from duplicated pairs, particularly the neofunctionalized MDOs, for developing highly efficient orthogonal translation systems. The integration of robust quantitative profiling, computational evolutionary analysis, and a clear understanding of tRNA structure-function relationships, as detailed in the provided toolkit and workflows, will accelerate the design of novel biologics and therapeutic agents through the site-specific incorporation of unnatural amino acids.
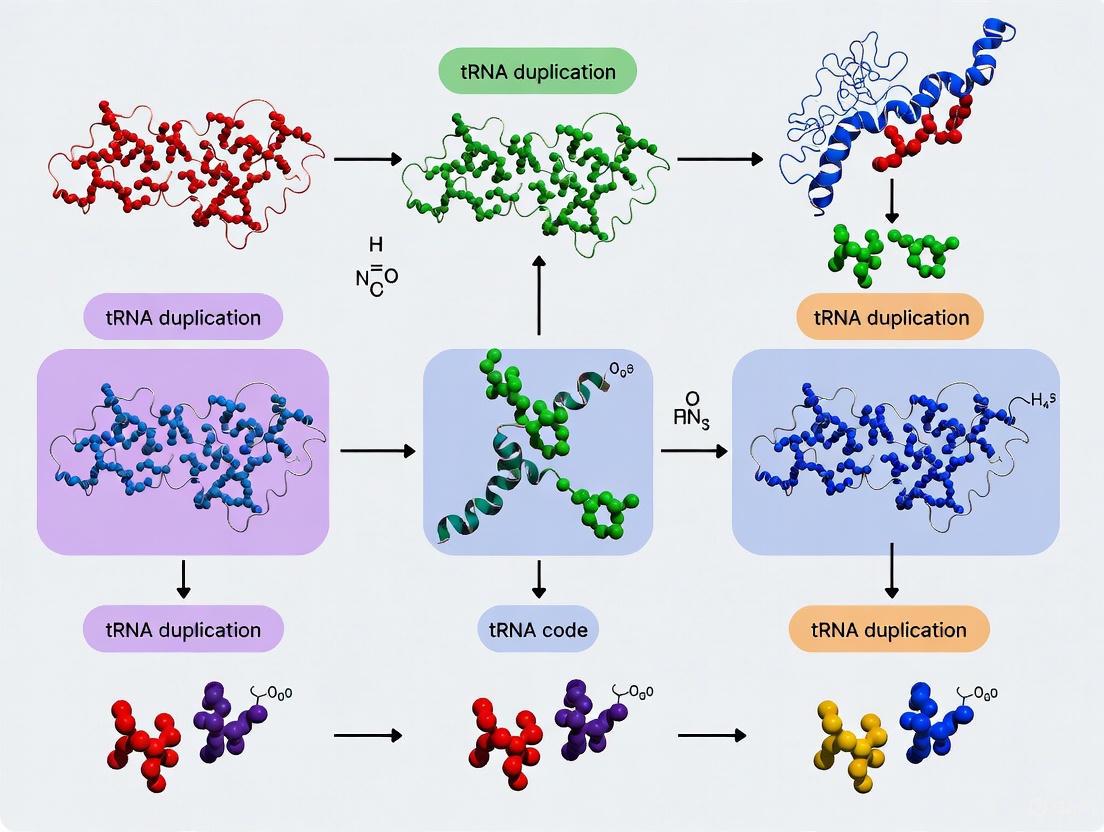
The evolution of the genetic code represents one of biology's most fundamental transitions, yet its origins remain actively debated. Contemporary research has undergone a paradigm shift from an mRNA-centric to a tRNA-centric model of code evolution, which posits that cloverleaf tRNA served as the molecular archetype around which translation systems evolved [18]. This framework suggests that the genetic code is a triplet code specifically because the structure of the tRNA anticodon loop forces a triplet register for two adjacent tRNAs paired to mRNA in the ribosome's decoding center [18]. The evolutionary trajectory proceeded from a primitive system utilizing a limited amino acid repertoire toward the complex modern code through mechanisms including tRNA gene duplication, anticodon modification, and functional specialization.
This application note situates the evolutionary trajectory of tRNA within the context of modern genetic code expansion research, providing researchers with both theoretical frameworks and practical methodologies for investigating and manipulating tRNA-based coding systems. We present quantitative analyses of tRNA gene conservation and duplication patterns across species, detailed protocols for experimental tRNA evolution, and visualization of key evolutionary and engineering concepts to facilitate research in synthetic biology and therapeutic development.
Evolutionary Foundations: From Proto-tRNA to Modern Diversity
The Proto-tRNA Hypothesis and Code Expansion
The polyglycine hypothesis proposes that the initial product of the genetic code may have been short-chain polyglycine synthesized to stabilize protocells [18]. Under this model, archaeal tRNAGly appears closest to the root of the tRNA evolutionary tree, suggesting that a primordial cloverleaf tRNA (tRNAPri) most strongly resembling tRNAGly diversified by mutation to include all permitted anticodons [18]. The initial 3-nucleotide code may have functioned primarily to synthesize short polyglycine chains (typically ~5 residues in length for structural stabilization), with translational processivity limited by primitive machinery [18].
Code expansion followed a sectoring-degeneracy hypothesis, whereby the code sectors from a 1→4→8→∼16 letter code [18]. At initial stages, strong positive selection existed for wobble base ambiguity, supporting convergence to 4-codon sectors and approximately 16 letters. Subsequently, approximately 5-6 additional letters, including stops, were added through innovation at the anticodon wobble position [18]. The initial expansion was physically constrained by negative selection against adenine in the tRNA wobble position, limiting the primordial code to approximately 48 anticodons rather than the full 64 potential codons [18].
Conserved Structural Transitions in tRNA Evolution
The evolutionary trajectory from proto-tRNA to modern diversity maintained remarkable structural conservation while permitting functional diversification. The cloverleaf tRNA structure is proposed to have evolved through a gradual, Fibonacci process-like elongation from a primordial coding triplet and 5'DCCA3' quadruplet to the eventual 76-90 base cloverleaf [19]. The conserved L-shaped tertiary structure comprises two functional branches: the acceptor branch (acceptor stem and T arm) where amino acids are charged, and the anticodon branch (D arm and anticodon arm) responsible for mRNA decoding [14].
Table 1: Evolutionary Trajectory of Genetic Code Expansion
| Evolutionary Phase | Amino Acid Diversity | tRNA Complexity | Key Mechanisms |
|---|---|---|---|
| Initial Glycine Phase | 1 amino acid (Glycine) | Single proto-tRNA species | Non-specific charging, polyglycine synthesis |
| Early Sectoring | 4 amino acids | Limited anticodon diversity | Wobble position ambiguity, initial duplication |
| Intermediate Expansion | 8-16 amino acids | Specialized isoacceptors | Anticodon modification, sectoring degeneracy |
| Modern Code | 20+ amino acids | Full isoacceptor/isodecoder families | tRNA gene duplication, synthetase coevolution |
Analysis of RNA secondary structures reveals an evolutionary axis from tRNA-like to rRNA-like configurations, with tRNA-like structures representing more primitive forms characterized by short RNAs with high proportions of external loops topping stems [20]. The relative similarity of tRNAs to this primitive structural class correlates with genetic code inclusion orders of tRNA cognate amino acids, confirming the biological relevance of this evolutionary axis [20].
Quantitative Analysis of tRNA Gene Evolution
Conservation and Duplication Patterns Across Species
Systematic analysis of tRNA genes across 50 plant species encompassing eight divisions within the plant kingdom reveals profound evolutionary conservation alongside dynamic duplication mechanisms [21]. A total of 28,262 high-confidence tRNA genes identified across these species demonstrate that tRNA gene abundance exhibits no significant correlation with genome size (r = 0.18, p = 0.21), indicating specific evolutionary pressures shaping tRNA copy number independent of general genome expansion [21].
Table 2: tRNA Gene Conservation and Duplication Across Phylogenetic Divisions
| Phylogenetic Division | Number of Species Analyzed | Total tRNA Genes Identified | Gene Length Range (bp) | Tandem Duplication Prevalence |
|---|---|---|---|---|
| Angiospermae | 36 | 14,827 | 62-98 | High (Proline anticodon clusters widespread) |
| Bryophyta | 4 | 3,215 | 70-92 | Moderate |
| Chlorophyta | 4 | 298 | 65-88 | Low |
| Lycopodiophyta | 2 | 537 | 68-90 | Moderate |
| Marchantiophyta | 1 | 824 | 71-95 | High |
| Pinophyta | 1 | 387 | 69-89 | Moderate |
| Pteridophyta | 1 | 483 | 67-91 | Moderate |
| Rhodophyta | 1 | 56 | 62-79 | Minimal |
Identical tandemly duplicated tRNA gene pairs are abundant across plant species, with 578 identified pairs grouped into 410 clusters containing up to 26 identical tRNA genes [21]. Different duplication types include double-, triple-, and quintuple-tRNA genes repeated variably, with tandemly located tRNA gene pairs with anticodons to proline widespread in 33 plant species across both lower and higher plants [21].
Experimental Evolution of tRNA Genes
Landmark experimental evolution studies in Saccharomyces cerevisiae demonstrate the rapid adaptive capacity of tRNA genes when faced with novel translational demands [22]. Deletion of the single-copy tRNA gene decoding the AGG arginine codon initially reduced fitness, but evolved populations recovered wild-type growth rates after ~200 generations through a strategic mutation that changed the anticodon of another tRNA gene (normally decoding AGA arginine) to match the deleted AGG anticodon [22].
This anticodon switching mechanism represents a fundamental evolutionary strategy for adapting the tRNA pool to meet novel translational demands. Computational analysis of hundreds of genomes confirms that anticodon mutations occur throughout the tree of life, indicating this represents a general adaptive mechanism rather than a laboratory-specific phenomenon [22]. Beyond meeting translational demand, the evolution of tRNA pools is also constrained by the need to properly couple translation to protein folding, maintaining deliberately suboptimal "slow codons" at domain boundaries to facilitate proper cotranslational folding [22].
Experimental Protocols for tRNA Evolution Studies
Protocol: Experimental Evolution of tRNA Gene Function
This protocol adapts methodology from Yona et al. (2013) for investigating tRNA gene evolution in response to gene deletions or novel translational demands [22].
Materials and Reagents
- Saccharomyces cerevisiae strain with deletion of specific tRNA gene (e.g., ΔtRNA-AGG-Arg)
- Appropriate rich and selective media (YPD, SC)
- Chemostat or serial transfer apparatus
- DNA extraction kit
- PCR reagents
- tRNA-specific sequencing primers
- Computational resources for genome analysis
Procedure
- Strain Construction: Delete target tRNA gene (e.g., tRNA-AGG-Arg) from S. cerevisiae genome using standard gene replacement techniques.
- Evolution Initiation: Inoculate mutant strain into appropriate medium and initiate evolution under either:
- Chemostat conditions: Maintain continuous culture for 200+ generations
- Serial transfer: Dilute 1:100 into fresh medium daily for 200+ generations
- Fitness Monitoring: Sample populations every 20 generations to assess growth rates relative to wild-type
- Genomic Analysis: Extract genomic DNA from evolved populations showing fitness recovery
- tRNA Gene Sequencing: Amplify and sequence all tRNA genes with anticodons cognate to deleted tRNA
- Variant Identification: Identify anticodon mutations and validate causative mutations through reconstruction
Applications and Limitations This approach directly demonstrates how tRNA gene families evolve to meet translational demands but requires specialized expertise in microbial evolution and may produce strain-specific findings.
Protocol: Identification and Analysis of Tandem tRNA Duplications
This protocol describes bioinformatic identification and analysis of tandem tRNA gene duplications from genomic data, based on methods from plant tRNA genomics studies [21].
Materials and Reagents
- Genomic sequences in FASTA format
- High-performance computing cluster
- tRNAscan-SE software (v2.0.12)
- MMseqs2 for sequence clustering
- R scripting environment with ggplot2, ComplexHeatmap
- IQ-TREE for phylogenetic analysis
Procedure
- tRNA Gene Identification:
- Annotate tRNA genes using tRNAscan-SE with eukaryotic parameters:
tRNAscan-SE -H -y genome.fasta - Filter for high-confidence sets using EukHighConfidenceFilter
- Annotate tRNA genes using tRNAscan-SE with eukaryotic parameters:
Tandem Duplication Identification:
- Identify tRNA genes located on same chromosome with <1 kb intergenic distance
- Calculate sequence identity for proximal genes using Needle alignment
- Define tandem duplicates as genes with >90% sequence identity and >80% coverage
Sequence and Evolutionary Analysis:
- Calculate GC content using 5bp sliding windows normalized against total tRNA length
- Perform multiple sequence alignment using clustalo
- Construct phylogenetic trees using IQ-TREE with best-fit models identified by ModelFinder
- Calculate Kn/Ks ratios using KaKs_Calculator 3.0 with default parameters
Visualization and Interpretation:
- Generate heatmaps of tRNA abundance by anticodon across species
- Plot GC content variation across tRNA structures
- Visualize phylogenetic relationships of tandem duplicates
Applications and Limitations This protocol enables systematic comparison of tRNA gene evolution across species but requires quality genome assemblies and may miss evolutionarily recent duplicates under annotation thresholds.
Visualization of Evolutionary and Engineering Concepts
Evolutionary Trajectory of tRNA Gene Function
Evolution of tRNA Gene Function
tRNA Engineering Workflow
tRNA Engineering Workflow
Research Reagent Solutions for Genetic Code Expansion
Table 3: Essential Research Reagents for tRNA and Genetic Code Expansion Studies
| Reagent/Category | Function/Application | Key Characteristics | Experimental Considerations |
|---|---|---|---|
| Orthogonal tRNA/synthetase Pairs | Genetic code expansion with unnatural amino acids | Species-cross reactive, non-immunogenic to host AARS | Requires directed evolution for orthogonality and efficiency |
| tRNA Gene Deletion Strains | Studying tRNA evolution and essentiality | Single-copy tRNA gene deletions in model organisms | Fitness defects often observed; enables experimental evolution |
| tRNAscan-SE Software | Bioinformatics annotation of tRNA genes | Covariance model-based prediction, cloverleaf scoring | Standard for genomic tRNA identification; requires parameter optimization |
| Directed Evolution Systems | tRNA engineering for improved function | Library generation, orthogonality selection | Critical for optimizing tRNA efficiency in non-native hosts |
| Aminoacyl-tRNA Synthetase Libraries | Expanding substrate specificity | Mutant libraries for novel amino acid incorporation | Enables genetic code expansion to non-canonical amino acids |
The evolutionary trajectory from proto-tRNA to modern diversity reveals fundamental principles governing genetic code expansion and adaptability. The documented mechanisms of tandem gene duplication, anticodon switching, and structural conservation provide both explanatory power for natural code evolution and engineering strategies for synthetic biology applications. The experimental and computational protocols presented here enable researchers to directly investigate tRNA evolution and harness these principles for genetic code expansion.
For drug development professionals, these insights facilitate engineering of novel tRNA-based therapeutics and optimization of heterologous protein expression systems. The conservation of tRNA duplication mechanisms across the tree of life suggests generalizable approaches to manipulating translational systems for industrial and therapeutic applications, including readthrough of disease-causing nonsense mutations and incorporation of novel amino acids for biologics engineering.
Transfer RNA (tRNA) serves as the fundamental molecular bridge that translates genetic code into functional proteins. The conserved functional modules of tRNA—specifically the acceptor stem and anticodon loop—work in concert with specific identity elements to ensure the fidelity and efficiency of protein synthesis. Within the context of genetic code expansion (GCE), these modules provide both a framework of natural constraints and a platform for engineering. GCE technologies aim to incorporate non-canonical amino acids (ncAAs) into proteins, requiring the development of orthogonal translation systems (OTSs) that function outside the natural machinery while adhering to its core principles [23]. Research into tRNA gene duplication events reveals an evolutionary pathway that has diversified the tRNA repertoire while conserving these critical functional modules, offering valuable insights for synthetic biology [21]. This application note provides a detailed analysis of these modules, supported by quantitative data and experimental protocols, to facilitate advanced research in genetic code expansion.
Quantitative Analysis of Conserved tRNA Modules
Genome-Wide tRNA Gene Conservation
A comprehensive analysis of 50 plant species identified 28,262 high-confidence tRNA genes, revealing significant conservation across the plant kingdom. The abundance of tRNA genes showed a weak, non-significant correlation with genome size (r = 0.18, p > 0.05), indicating that factors beyond genome scale govern tRNA gene copy number [21]. The study also documented 578 identical tandemly duplicated tRNA gene pairs, grouped into 410 clusters, with some clusters containing up to 26 repeated tRNA genes. These duplication events were observed across both lower and higher plants, suggesting tandem duplication serves as a fundamental evolutionary mechanism for tRNA gene family expansion [21].
Table 1: Conservation of Intron-Containing tRNA Genes and Tandem Duplication Events in Plants
| Analysis Category | Findings | Significance |
|---|---|---|
| Total tRNA Genes Identified | 28,262 genes across 50 plant species | Demonstrates widespread presence and conservation [21] |
| Gene Length Conservation | Ranged from 62 to 98 bp, peaking at 72 bp and 82 bp | Indicates strong structural conservation [21] |
| Abundant Intron-Containing tRNAs | tRNAMet_CAT and tRNATyr_GTC were most abundant | Specific tRNA families are consistently intron-containing [21] |
| Tandem Duplication Events | 578 identical tandemly duplicated tRNA gene pairs (410 clusters) | Tandem duplication is a key evolutionary driver [21] |
| Widespread Tandem Duplication | tRNAPro anticodon pairs found in 33 species | Highlights a conserved duplication event [21] |
Functional Coding of Acceptor Stems and Anticodon Loops
The acceptor stem and anticodon loop encode distinct physicochemical properties of amino acids, implementing a dual-level proofreading system. Research demonstrates that the anticodon primarily encodes the hydrophobicity of the amino acid side-chain, represented by its water-to-cyclohexane distribution coefficient (ΔGw>c) [24]. In contrast, the acceptor stem codes preferentially for the size or surface area of the side-chain, as represented by its vapor-to-cyclohexane distribution coefficient (ΔGv>c) [24]. These orthogonal properties are both necessary to satisfactorily account for the exposed surface area of amino acids in folded proteins. Furthermore, the acceptor stem correctly codes for β-branched and carboxylic acid side-chains, while the anticodon codes for a wider range of properties but not for size or β-branching [24].
Table 2: Functional Coding Properties of tRNA Modules
| tRNA Module | Encoded Amino Acid Property | Experimental Measure | Contribution to Protein Folding |
|---|---|---|---|
| Acceptor Stem | Side-chain size / Surface area | Vapor-to-cyclohexane transfer equilibrium (ΔGv>c) | Determines van der Waals contacts in folded state [24] |
| Anticodon Loop | Side-chain hydrophobicity / Polarity | Water-to-cyclohexane transfer equilibrium (ΔGw>c) | Governs hydrophilic character and solvent interaction [24] |
Experimental Protocols
Protocol 1: Genome-Wide Identification and Analysis of tRNA Genes
Objective: To identify tRNA genes, characterize their structural features, and detect duplication events in genomic sequences.
Materials:
- Nuclear genome sequences in FASTA format
- tRNAscan-SE software (v2.0.12 or higher)
- RNAFold software
- Computing environment with R and ggplot2 package
- MMseqs2 software for sequence clustering
Procedure:
- Data Acquisition: Download nuclear genome sequences, coding sequences, and protein sequences for target species from databases such as Phytozome [21].
- tRNA Gene Identification: Annotate tRNA genes using tRNAscan-SE with parameters "-H" and "-y" optimized for eukaryotic tRNAs. Filter results for high-confidence sets using EukHighConfidenceFilter [21].
- Structural Characterization:
- Sequence Conservation Analysis:
- Perform multiple sequence alignment of identical-sequence tRNA genes using Multialin or similar tools.
- Calculate sequence identity between tRNA gene pairs using global alignment tools such as Needle [21].
- Estimate non-synonymous (Kn) and synonymous (Ks) substitution rates using KaKs_Calculator 3.0 to evaluate evolutionary pressure [21].
- Phylogenetic Analysis:
- Cluster tRNA sequences using MMseqs2 with a minimum sequence identity of 0.9 and coverage of 0.8.
- Construct phylogenetic trees using IQ-TREE 2 with appropriate substitution models and 1000 bootstrap replicates [21].
- Tandem Duplication Detection:
- Identify tRNA gene pairs and clusters located on the same chromosome with a physical distance of less than 1 kb.
- Define tandem repeats as clusters where tRNA genes with the same anticodon exhibit identical sequences or where different combinations of tRNA genes recur [21].
Protocol 2: Analyzing Identity Elements and Editing Mechanisms
Objective: To characterize tRNA identity elements and their role in aminoacylation fidelity and editing.
Materials:
- Purified aminoacyl-tRNA synthetases (ARSs) and editing enzymes
- In vitro transcription system for tRNA variants
- Radiolabeled or fluorescent-labeled amino acids
- HPLC system with appropriate columns
- Stop-flow quenching instruments for kinetic assays
Procedure:
- tRNA Variant Design: Design tRNA mutants with systematic alterations at known identity element positions (e.g., positions 1, 72, 73 in the acceptor stem; positions 34, 35, 36 in the anticodon loop) [25].
- In Vitro Aminoacylation Assays:
- Charge wild-type and mutant tRNAs with cognate and non-cognate amino acids using purified ARSs.
- Quantify aminoacylation efficiency and mischarging rates using radiolabeled amino acids or other detection methods [25].
- Editing Assay Setup:
- Incubate mischarged tRNAs with cis-editing domains of ARSs or trans-editing enzymes.
- Monitor deacylation kinetics using stop-flow quenching techniques or HPLC to separate aminoacyl-tRNAs from free amino acids [25].
- Specificity Determination:
- Compare editing rates for cognate versus non-cognate aa-tRNAs.
- Identify specific tRNA identity elements recognized by editing domains through kinetic analysis of mutants [25].
- Data Analysis: Calculate kinetic parameters (kcat, KM) for both aminoacylation and editing reactions to determine how identity elements contribute to overall fidelity.
Protocol 3: tRNA Engineering for Genetic Code Expansion
Objective: To engineer orthogonal tRNA/synthetase pairs for incorporation of non-canonical amino acids.
Materials:
- Library of tRNA variants (e.g., with mutations in anticodon stem-loop)
- Orthogonal aminoacyl-tRNA synthetase (aaRS) library
- Non-canonical amino acid of interest
- Reporter plasmid with target codon (e.g., amber stop codon UAG)
- Host cells (E. coli, yeast, or mammalian cells)
- Fluorescence-activated cell sorting (FACS) equipment
Procedure:
- tRNA Library Construction: Create a diverse library of orthogonal tRNA variants (e.g., pyrrolysyl-tRNA) with randomized mutations in the anticodon stem-loop region to recognize non-standard codons [26].
- Selection System Setup: Co-express the tRNA library with a cognate aaRS library and a reporter gene containing the target codon (e.g., Ψ-modified stop codon for RCE systems) [26].
- High-Throughput Screening:
- For efficiency screening: Use fluorescent reporter genes (e.g., GFP) whose full-length production depends on successful ncAA incorporation at the target codon. Sort high-efficiency clones via FACS [23].
- For orthogonality screening: Use negative selection markers (e.g., toxin genes) to eliminate clones with cross-reactivity to endogenous amino acids or tRNAs [23].
- Characterization of Engineered Pairs:
- Measure ncAA incorporation efficiency and fidelity using western blotting and mass spectrometry.
- Assess protein yields and incorporation accuracy at multi-site locations [6].
- System Optimization: Iteratively evolve both tRNA and synthetase components to improve orthogonality, efficiency, and specificity for the desired ncAA [6].
Visualizing tRNA Modules and Experimental Workflows
Diagram 1: Integrated workflow for analyzing conserved tRNA modules and engineering for genetic code expansion.
Diagram 2: Functional modules of tRNA and their interaction with cellular machinery, highlighting key identity elements.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for tRNA and Genetic Code Expansion Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Bioinformatics Tools | tRNAscan-SE, RNAFold, MMseqs2, KaKs_Calculator, IQ-TREE 2 | tRNA gene identification, structural prediction, evolutionary analysis [21] |
| Orthogonal tRNA/synthetase Pairs | Pyrrolysyl-tRNA/synthetase pair, Engineered M. jannaschii tyrosyl pair | Core components for genetic code expansion and ncAA incorporation [23] [26] |
| Non-Canonical Amino Acids | Photo-crosslinkers, Bio-orthogonal handles (azides, alkynes), Fluorescent analogs | Expanding chemical functionality of synthesized proteins [23] |
| Selection & Screening Systems | GFP-based reporters, Toxin counter-selection, FACS, Compartmentalized partnered replication | High-throughput identification of efficient orthogonal systems [23] |
| In Vitro Translation Systems | PURE system (Reconstituted E. coli components), Cell lysates | Flexible genetic code reprogramming without cellular constraints [23] |
| Analytical Techniques | HPLC-MS for modified nucleosides, NMR, X-ray crystallography | Quantifying tRNA modifications and determining 3D structures [27] [25] |
Engineering the Code: Methodologies for Harnessing tRNA Duplication in GCE
Genetic Code Expansion (GCE) technology enables the site-specific incorporation of non-canonical amino acids (ncAAs) into proteins, thereby overcoming the limitations of the standard genetic code and creating novel protein functions and properties [6] [14]. This technique has matured into a versatile tool with applications across protein science, therapeutic engineering, and synthetic biology [28]. At the heart of every GCE system lies the orthogonal aminoacyl-tRNA synthetase/tRNA pair (aaRS/tRNA), a fundamental component that acts as an autonomous translation system within the host organism [28] [29]. For successful GCE, this pair must function without cross-reacting with the host's native translational machinery; the orthogonal aaRS must specifically charge the ncAA onto its cognate orthogonal tRNA, which in turn must be aminoacylated only by its orthogonal partner and not by any endogenous host aaRSs [6] [30]. The tRNA must also specifically recognize a "blank" codon—most commonly the amber stop codon (UAG)—that is not assigned to a canonical amino acid [28]. The strategic sourcing and engineering of these pairs from diverse biological origins are therefore critical for expanding the scope and efficiency of GCE.
Sourcing Orthogonal Pairs from Nature
Orthogonal aaRS/tRNA pairs are typically sourced from organisms across different phylogenetic domains to ensure they do not cross-talk with the host's native translation systems [6] [14]. The underlying principle is that aaRS/tRNA pairs from distantly related species (e.g., archaea transplanted into bacteria) have evolved distinct identity elements, making them functionally independent in the new host environment [31] [29].
Table 1: Naturally Sourced Orthogonal aaRS/tRNA Pairs and Their Applications
| Orthogonal Pair | Organism of Origin | Common Hosts | Key Features and Applications | References |
|---|---|---|---|---|
| Pyrolysyl-tRNA Synthetase/tRNAPyl (PylRS/tRNAPyl) | Methanosarcina species (e.g., barkeri, mazei) | E. coli, Yeast, Mammalian Cells | - Naturally incorporates pyrrolysine [29].- Extremely versatile substrate specificity [28].- Used to incorporate >200 distinct ncAAs [28]. | [28] [29] [30] |
| Tyrosyl-tRNA Synthetase/tRNATyr (TyrRS/tRNATyr) | Methanocaldococcus jannaschii (Mj) | E. coli | - One of the first pairs developed for GCE [29].- Used to incorporate various phenylalanine and tyrosine analogs [29]. | [31] [29] |
| Tyrosyl-tRNA Synthetase/tRNATyr (TyrRS/tRNATyr) | E. coli | Yeast, Mammalian Cells | - Demonstrates orthogonality in eukaryotic hosts [29].- Bacterial identity elements differ from eukaryotic counterparts. | [29] |
| Tyrosyl-tRNA Synthetase/tRNATyr (TyrRS/tRNATyr) | Methanosaeta concilii (Mc) | E. coli | - A newly developed pair for incorporating para-azido-L-phenylalanine (AzF) [30].- Broadens the pool of available orthogonal pairs. | [30] |
The PylRS/tRNAPyl pair is exceptionally prominent in GCE due to its unique natural function and remarkable plasticity. It was originally discovered in methanogenic archaea, where it charges the rare amino acid pyrrolysine into proteins in response to an in-frame amber codon [29]. Its versatility and high orthogonality across diverse hosts, from bacteria to mammalian cells, have made it a cornerstone for incorporating a vast range of ncAAs [28] [29].
Experimental Protocol: Establishing and Validating Orthogonality
This protocol outlines the key steps for establishing a new orthogonal aaRS/tRNA pair in a host organism, such as E. coli, and validating its functionality and orthogonality.
Computational Identification and Library Construction
- Step 1: In Silico Screening. Screen millions of tRNA sequences from genomic databases (e.g., from bacteria, archaea, bacteriophages) to identify candidate tRNAs with low similarity to the host's identity elements. A scoring system based on known identity elements for host aaRSs can predict orthogonality; tRNAs scoring below a threshold (e.g., +0.5) for all host synthetases are strong candidates [31].
- Step 2: Library Construction. For the candidate aaRS, create mutant libraries. This can involve:
- Site-Specific Mutagenesis: Target residues in the aaRS active site known to be critical for substrate recognition based on homologous pairs (e.g., Tyr32, Asp158, and Leu162 in M. jannaschii TyrRS) to disrupt binding to canonical amino acids [30].
- Random Mutagenesis: Use error-prone PCR or an orthogonal DNA replication system (e.g., OrthoRep in yeast) to generate diverse aaRS variants [28] [30]. OrthoRep continuously mutates the aaRS gene at a high rate (∼10⁻⁵ substitutions per base), enabling rapid, open-ended evolution [28].
Experimental Validation of Orthogonality and Function
- Step 3: tRNA Orthogonality Assessment. Use the tRNA Extension (tREX) method to determine the in vivo aminoacylation status of candidate tRNAs [31].
- Principle: Fluorescent DNA probes are designed to selectively invade the acceptor stem of the target tRNA and anneal to its 3'-end. The presence of an amino acid on the tRNA blocks this annealing, allowing differentiation between charged and uncharged tRNA [31].
- Procedure: Extract total RNA from host cells expressing the candidate tRNA. Incubate the RNA with Cy5-labelled DNA probes. Analyze the samples using gel electrophoresis. A charged tRNA will show no shift, while an uncharged tRNA will form a probe-tRNA complex with a mobility shift. Orthogonal tRNAs should remain uncharged in the host unless their cognate aaRS is present [31].
- Step 4: Selection for Functional aaRS/tRNA Pairs. Use a dual positive/negative selection system in the host organism to isolate aaRS variants that charge the ncAA onto the orthogonal tRNA.
- Reporter System: A common method uses a fluorescent reporter gene (e.g., super-folder GFP, sfGFP) containing an amber codon at a permissive site [28] [30].
- Positive Selection: Grow cells in the presence of the ncAA. Cells with a functional aaRS/tRNA pair will incorporate the ncAA, produce full-length fluorescent protein, and can be isolated via Fluorescence-Activated Cell Sorting (FACS) [28] [30].
- Negative Selection: Grow sorted cells in the absence of the ncAA. Cells where the aaRS charges the orthogonal tRNA with a canonical amino acid will read through amber codons in essential genes, leading to cell death or reduced fitness. Only cells with highly specific aaRSs survive [28].
Diagram 1: Directed evolution workflow for orthogonal aaRS using dual selection.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of GCE requires a suite of specialized reagents and molecular tools.
Table 2: Key Research Reagents for GCE Experiments
| Reagent / Tool | Function in GCE | Specific Examples |
|---|---|---|
| Orthogonal aaRS/tRNA Pair | The core engine for ncAA incorporation; must be orthogonal and efficient in the host. | PylRS/tRNAPyl from M. barkeri; TyrRS/tRNATyr from M. jannaschii; E. coli TyrRS/tRNA pair in eukaryotes [29] [30]. |
| Reporter Plasmid | Reports on the efficiency and fidelity of ncAA incorporation, enabling selection and screening. | Plasmids encoding sfGFP with an amber codon [30]; Ratiometric RFP-GFP (RXG) reporters for quantifying readthrough efficiency [28]. |
| Selection System | Enriches for functional, specific aaRS variants from large libraries. | FACS for fluorescent reporters [28] [30]; growth-based selection with antibiotic resistance genes containing amber codons. |
| Hypermutation System | Accelerates directed evolution by introducing targeted mutations into the aaRS gene. | OrthoRep (an orthogonal error-prone DNA polymerase system in yeast) [28]. |
| tRNA Analysis Tool | Directly measures the aminoacylation status of tRNAs to confirm orthogonality. | tRNA Extension (tREX) method with fluorescent DNA probes [31]. |
Engineering and Optimization Strategies
Sourcing pairs from nature is only the first step. Extensive engineering is often required to enhance orthogonality, improve ncAA incorporation efficiency, and adapt the pair for new hosts or ncAAs.
tRNA Engineering for Enhanced Performance
While early GCE efforts focused predominantly on aaRS engineering, optimizing the tRNA is equally critical for high efficiency [6] [14]. tRNA engineering focuses on two conflicting demands: maintaining orthogonality to host aaRSs while ensuring efficient cooperation with the host's transcriptional and translational machinery [14].
- Modifying Identity Elements: Identity elements are specific nucleotides within a tRNA that are recognized by its cognate aaRS [31] [14]. Mutating these elements in the orthogonal tRNA can reduce its mis-aminoacylation by host aaRSs, thereby improving orthogonality [29]. For example, mutating the tRNA's acceptor stem and anticodon loop can enhance its exclusive recognition by its engineered aaRS [14].
- Optimizing for Host Machinery: The orthogonal tRNA must be efficiently transcribed, processed, and interact with host elongation factors (EF-Tu in bacteria) and the ribosome. Engineering the tRNA's promoter, 5' and 3' sequences, and bases that interact with EF-Tu (e.g., positions in the acceptor stem and T stem) can significantly boost the yield of full-length protein [6] [14].
Diagram 2: Key targets for engineering functional orthogonal tRNAs.
aaRS Engineering for Novel Specificity
The aaRS active site must be redesigned to accommodate a specific ncAA, which is typically achieved through directed evolution [29] [30]. The general workflow involves:
- Creating a large library of aaRS mutants.
- Using a selection system (as described in Section 3.2) to isolate variants that enable ncAA-dependent protein synthesis.
- Iteratively cycling through positive and negative selection to evolve aaRS mutants that are both highly active and specific for the target ncAA [28] [30].
Advanced platforms like OrthoRep streamline this process by enabling continuous in vivo mutagenesis and selection, leading to the rapid discovery of highly efficient aaRSs that can rival the performance of natural translation systems [28].
The deliberate sourcing and sophisticated engineering of orthogonal aaRS/tRNA pairs are foundational to the power and success of Genetic Code Expansion. By strategically selecting pairs from disparate branches of the tree of life and refining them through state-of-the-art directed evolution and engineering protocols, researchers can reliably create custom translation systems. These systems serve as programmable engines for installing novel chemical functionalities directly into proteins, thereby pushing the boundaries of synthetic biology, therapeutic development, and fundamental biological research.
The field of genetic code expansion (GCE) leverages engineered cellular machinery to incorporate unnatural amino acids (UAAs) into proteins, enabling the creation of polypeptides with novel chemical, structural, and functional properties that surpass the constraints of the canonical 20 amino acids [14] [6]. While early GCE efforts predominantly focused on engineering aminoacyl-tRNA synthetases (AARSs), recent advances have underscored the pivotal role of transfer RNA (tRNA) itself as a critical component for enhancing UAA incorporation efficiency and system orthogonality [14]. The core of this technology relies on orthogonal AARS/tRNA pairs—where the AARS specifically charges the desired UAA only onto its cognate orthogonal tRNA, and this tRNA is not recognized by the host's endogenous AARSs [14] [23].
An emerging and powerful substrate for this engineering is the duplicated tRNA scaffold. Genomic analyses reveal that tandem duplication is a fundamental evolutionary force driving tRNA gene family expansion across diverse plant species [21]. These duplicated genes provide a rich natural reservoir of sequence variation and a template for engineered tRNA sets. This Application Note details a comprehensive toolkit of experimental and computational protocols for the directed evolution and rational design of duplicated tRNA scaffolds, providing researchers with methodologies to advance GCE for basic science and therapeutic development.
tRNA Structure and Function: A Primer for Engineering
A foundational understanding of tRNA architecture and its molecular interactions is a prerequisite for effective engineering. The canonical tRNA structure is an L-shaped molecule, historically represented by a cloverleaf secondary structure comprising the acceptor stem, D arm, anticodon arm, variable arm, and T arm, culminating in a 3′ CCA sequence for aminoacylation [14] [6]. This structure folds into a three-dimensional L-form, highly conserved across life [14].
The functionality of tRNA is defined by its precise interactions with key binding partners during translation, each engaging distinct structural elements:
- Aminoacyl-tRNA Synthetases (AARS): AARS enzymes charge the cognate amino acid onto the 3′ end of the tRNA. Recognition depends on "identity elements," which are often located in the anticodon loop and the discriminator base at position 73, though this varies by AARS type [14] [6].
- Elongation Factor Tu (EF-Tu): In prokaryotes, EF-Tu delivers the aminoacylated tRNA to the ribosome. Critical interaction sites are localized to base pairs in the acceptor stem and T stem (e.g., 51:63, 50:64, 49:65, and 7:66) [14].
- The Ribosome: The ribosome has three tRNA binding sites (A, P, and E). The anticodon loop, elbow region, and 3′ end interact at the A and P sites, while the acceptor stem facilitates dissociation at the E site [14].
Table 1: Key tRNA Binding Partners and Their Interaction Sites
| Binding Partner | Primary Interaction Sites on tRNA | Functional Consequence of Engineering |
|---|---|---|
| Aminoacyl-tRNA Synthetase (AARS) | Anticodon loop, discriminator base (position 73), acceptor stem, variable loop [14] | Alters orthogonality and charging efficiency with UAAs [14] |
| Elongation Factor (EF-Tu/EF-1α) | Acceptor stem, T stem (e.g., pairs 51:63, 50:64) [14] | Influences ternary complex stability and delivery kinetics to the ribosome [14] |
| Ribosome (A/P Sites) | Anticodon loop, elbow region, 3' acceptor end [14] | Affects decoding accuracy, translocation efficiency, and susceptibility to ribosome-based quality control |
This intricate network of interactions means that any engineering strategy must balance the introduction of orthogonality against the preservation of cooperativity with the host's native translational machinery [14] [6].
Quantitative Analysis of tRNA Duplication and Diversity
The conservation and tandem duplication of tRNA genes is a widespread phenomenon. A comprehensive analysis of 50 plant species identified 28,262 high-confidence tRNA genes, revealing that tandem duplication is a major driver of tRNA gene evolution and abundance, with no significant correlation between tRNA gene number and genome size [21]. The study identified 578 identical tandemly duplicated tRNA gene pairs, grouped into 410 clusters. Notable examples include a cluster of 27 tandemly duplicated tRNAPro genes in Arabidopsis thaliana and a repeat of 28 tRNAIle genes in Zea mays [21]. This natural duplication and divergence provide a blueprint for creating engineered tRNA libraries.
Table 2: Experimentally Determined Parameters for Engineered tRNA Distributions
| tRNA Abundance Distribution Type | Correlation with Codon Usage (Slope) | Average Expected Elongation Latency (ms, Mean ± SD) | Relative Performance vs. Wild-Type |
|---|---|---|---|
| Wild-Type E. coli | Positive correlation | 193 ± 5.5 | Baseline |
| Uniform Distribution | Low positive (0.1) | 214 ± 1.4 | ~11% slower |
| Stepwise Correlated | Positive | 194 ± 5.0 | Comparable |
| Stepwise Anticorrelated | Negative | 232 ± 3.8 | ~20% slower |
| Codon-Weighted | Strong positive (1.26) | 185 ± 6.7 | ~4% faster |
| CAD-Optimized (Fast) | Strong positive (1.51) | 175 ± 8.6 | ~10% faster |
| CAD-Optimized (Slow) | Strong negative | 244 ± 2.9 | ~25% slower |
These quantitative metrics, derived from colloidal dynamics simulations, provide a framework for predicting the functional outcomes of engineering tRNA abundances and sequences [32]. The data demonstrate that strategic manipulation of tRNA pools can significantly modulate translation kinetics.
Core tRNA Engineering Strategies
Strategy 1: Directed Evolution of tRNA Scaffolds
Directed evolution applies iterative cycles of diversification and selection to engineer tRNAs with enhanced properties for GCE.
Protocol 1: Directed Evolution of Orthogonal tRNA for Improved UAA Incorporation [14] [23]
- Library Construction: Generate a diverse library of tRNA variants based on a duplicated parental scaffold (e.g., a prokaryotic tRNA scaffold for use in a eukaryotic host). Focus mutagenesis on key regions such as the anticodon loop (for codon reassignment), the acceptor stem, and the T-stem (to modulate EF-Tu binding and orthogonality) [14].
- Selection System:
- Positive Selection: Use a reporter gene (e.g., GFP or an antibiotic resistance marker) wherein its successful expression depends on the suppression of a nonsense (e.g., amber) or quadruplet codon by the aminoacylated orthogonal tRNA. Grow cells in the presence of the UAA and the selective agent (e.g., antibiotic) [23].
- Negative Selection: Use a reporter gene expressing a toxic protein (e.g., barnase) under the control of the same codon to be suppressed. In the absence of the UAA, cells with tRNAs that are mis-acylated by endogenous amino acids will not survive. This counterselection eliminates non-orthogonal tRNAs [23].
- High-Throughput Screening: For higher throughput, use fluorescence-activated cell sorting (FACS) to isolate cells based on the expression of a fluorescent reporter protein (e.g., GFP) that requires successful UAA incorporation [23].
- Iteration and Validation: Isolate plasmid DNA from selected clones, transform into fresh cells, and repeat the selection cycle to enrich for superior performers. Finally, sequence the evolved tRNA genes and characterize their performance in UAA incorporation assays.
Strategy 2: Rational Design of tRNA Sequences and Abundances
Rational design leverages structural knowledge and computational tools to predictively engineer tRNA components.
Protocol 2: Rational Design of a tRNA Pool for Optimized Translation Kinetics [32]
- Define Objective: Specify the desired translational output (e.g., maximum speed for a specific set of codons, or slowed translation for protein folding studies).
- Utilize Computer-Aided Design (CAD) Tool:
- Employ the Colloidal Dynamics CAD (CD-CAD) tool, which uses a genetic algorithm (GA) to optimize a population of tRNA abundance distributions [32].
- The GA initializes with random distributions, then applies rounds of computational mutation and selection based on performance metrics (e.g., simulated elongation latency) calculated by a first-principles colloidal dynamics simulator [32].
- Constrain the algorithm to keep individual tRNA abundances within natural observed bounds (e.g., 0.15% to 8.5% of total tRNA pool) and total tRNA concentration constant (~225 µM) to ensure biological feasibility [32].
- Implementation with Synthesized tRNAs (TINA):
- Once an optimal abundance distribution is computed, implement it using the Tunable Implementation of Nucleic Acids (TINA) method [32].
- Directly synthesize the 21 required tRNA surrogates via in vitro transcription (IVT) in the precise abundances specified by the CD-CAD output [32].
- Assemble the synthetic cell-free system (e.g., PURE system) using this engineered tRNA pool [32].
Strategy 3:In SituSynthesis of Engineered tRNA Sets
This strategy focuses on generating a complete, functional set of tRNAs directly within a cell-free system, a critical step toward self-regenerating synthetic cells.
Protocol 3: Simultaneous Synthesis of a Complete tRNA Set via the tRNA Array Method [33] [34]
- Template Design and Construction:
- Clone genes encoding all 21 minimal tRNAs required for translation into a single plasmid DNA template. For non-guanylated tRNAs, optimize promoter sequences (e.g., using T7 class II promoter φ2.5 for A-initiated tRNAs) or introduce point mutations at the 5' end (e.g., C-to-G for tRNAfMet) to enhance transcription yields by T7 RNA polymerase without needing 5' processing enzymes [33].
- Incorporate self-cleaving ribozyme sequences (e.g., HDV ribozyme) and RNase P recognition sites between tRNA genes on the plasmid to ensure proper processing into individual, mature tRNAs post-transcription [33] [34].
- In Situ Transcription and Processing:
- Use a transcription-translation system (e.g., tRNA-omitted PURE system) containing the plasmid template, T7 RNAP, NTPs, and necessary processing enzymes.
- Co-express the tRNA array and a desired reporter gene (e.g., sfGFP) from its own template in the same reaction vessel.
- The tRNAs are transcribed, self-processed, and directly utilized by the translation machinery to synthesize the target protein [33].
- Continuous Operation:
- To achieve sustained synthesis, run the PURE reaction in a microfluidic chemostat device that allows for the continuous replenishment of substrates (NTPs, amino acids) and removal of reaction byproducts, supporting steady-state protein expression for over 20 hours [33].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for tRNA Engineering and Application
| Reagent / Tool Name | Function and Application in tRNA Engineering |
|---|---|
| PURE System | A defined, reconstituted cell-free transcription-translation system; ideal for testing engineered tRNA pools without complex cellular background [32] [33]. |
| CD-CAD (Colloidal Dynamics Computer-Aided Design) | A physics-based software tool that designs optimal tRNA abundance distributions to achieve user-specified protein synthesis rates [32]. |
| tRNA Array Plasmid | A single plasmid template encoding all 21 tRNAs, often with ribozyme sequences, for the simultaneous in situ production of a complete tRNA set [33] [34]. |
| T7 RNA Polymerase Mutants | Engineered polymerases with reduced sequence bias for more uniform in vitro transcription of diverse tRNA sequences from DNA templates [33]. |
| tRNA-guanine transglycosylase (TGT) | An RNA-modifying enzyme used as a tool for the post-transcriptional introduction of non-natural bases into tRNA molecules for labeling or cross-linking studies [35]. |
Workflow and Pathway Visualizations
Diagram 1: Integrated tRNA Engineering Workflow. This diagram outlines the core decision points and parallel methodologies for developing engineered tRNAs, from goal definition to final validation.
Diagram 2: Mechanistic Pathway of tRNA Engineering. This diagram illustrates the logical relationship between specific engineering interventions on the tRNA molecule, their effects on key molecular interactions, and the resulting functional improvements for Genetic Code Expansion.
The fundamental process of protein synthesis is governed by the genetic code, a set of rules that maps nucleotide triplets (codons) to specific amino acids. While nature primarily uses 64 codons to encode 20 canonical amino acids and translation termination, this system inherently limits the chemical diversity of proteins [23] [36]. Codon reassignment technologies overcome this limitation by reprogramming the translation apparatus to incorporate noncanonical amino acids (ncAAs) with novel chemical, physical, and biological properties [23] [37]. These strategies form the cornerstone of genetic code expansion (GCE), a powerful synthetic biology approach that enables the precise installation of ncAAs into proteins at specified positions [38].
The driving premise for codon reassignment lies in expanding protein functionality beyond natural constraints. By incorporating ncAAs, researchers can equip proteins with unique handles for conjugation, crosslinkable groups for target engagement, post-translational modifications at defined sites, and properties that reduce immune recognition [23] [37]. The implementation of these technologies requires an orthogonal translation system (OTS)—a pair consisting of a tRNA and its cognate aminoacyl-tRNA synthetase (aaRS) that functions independently of the host's native translation machinery [23] [38]. This system must specifically charge the orthogonal tRNA with the desired ncAA and incorporate it in response to a reassigned codon without cross-reacting with endogenous components [38].
This protocol details three primary strategies for codon reassignment: stop codon suppression, quadruplet codon decoding, and unnatural base pair integration. We frame these methods within the context of ongoing tRNA duplication research, which seeks to create new coding capacity through the generation of additional orthogonal tRNA-codon pairs. The ability to reassign multiple codons simultaneously is paramount for synthesizing proteins with multiple distinct ncAAs, enabling the creation of sophisticated biomaterials and therapeutics with custom-tailored functionalities [39] [40].
Stop Codon Suppression
Conceptual Foundation and Applications
Stop codon suppression represents the most widely utilized method for site-specific ncAA incorporation [23] [36]. This approach repurposes one of the three native stop codons—typically the amber codon (UAG)—to encode an ncAA instead of signaling translation termination [23] [37]. The implementation requires an orthogonal aaRS/tRNA pair where the tRNA contains an anticodon (CUA) complementary to the UAG codon [36]. When an in-frame UAG codon is encountered in an mRNA, the orthogonal tRNA delivers the charged ncAA to the ribosome, allowing incorporation into the growing polypeptide chain [37].
A significant advantage of this system is its ability to create precise point mutations with ncAAs that minimally disrupt overall protein structure [23]. However, a primary challenge is competition between the orthogonal suppressor tRNA and release factor 1 (RF1), which naturally recognizes UAG codons to terminate translation [36]. This competition can limit incorporation efficiency, necessitating engineering solutions to improve system performance [36] [39].
Table 1: Comparison of Stop Codon Suppression Platforms
| Orthogonal Pair | Organism of Origin | Host Organisms | ncAA Examples | Key Features |
|---|---|---|---|---|
| MjTyrRS/tRNATyr | Methanocaldococcus jannaschii | E. coli | Aromatic ncAAs with azide, ketone, alkyne groups [37] | First GCE system developed; highly efficient for aromatic ncAAs [37] |
| PylRS/tRNAPyl | Methanosarcina species | E. coli, Mammalian cells [37] | Pyrrolysine analogs, lysine derivatives with diverse side chains [36] [37] [41] | Naturally orthogonal; works in prokaryotes and eukaryotes; permissive substrate range [37] |
| EcTyrRS/tRNATyr | Escherichia coli | Yeast, Mammalian cells [37] | Phe derivatives with benzophenone, azide, ketone groups [37] | Engineered for use in eukaryotic systems [37] |
Protocol: Amber Stop Codon Suppression in E. coli
This protocol describes the incorporation of an ncAA in response to the UAG codon using the PylRS/tRNAPyl orthogonal pair in E. coli, with options for adaptation to RF1-deficient strains to enhance efficiency.
Reagents and Equipment:
- Orthogonal plasmid containing PylRS gene and tRNAPyl with CUA anticodon under appropriate promoters
- Target protein plasmid with UAG codon at desired position
- Chemically competent E. coli cells (standard or RF1-deficient strains like C321.ΔA)
- ncAA stock solution (e.g., Nε-(tert-butyloxycarbonyl)-L-lysine, Boc-Lys)
- LB broth and agar plates with appropriate antibiotics
- IPTG for induction
- Equipment for protein expression and analysis (incubator, centrifuge, SDS-PAGE, mass spectrometry)
Procedure:
- Strain Preparation: Co-transform the orthogonal plasmid and target protein plasmid into your selected E. coli host strain. Plate on LB agar containing the appropriate antibiotics and incubate overnight at 37°C.
- Culture Inoculation: Pick a single colony and inoculate 5 mL of LB medium with antibiotics. Grow overnight at 37°C with shaking.
- Protein Expression: Dilute the overnight culture 1:100 into fresh LB medium with antibiotics. Grow at 37°C until OD600 reaches 0.6-0.8.
- Induction and Incorporation: Add the ncAA to a final concentration of 1-5 mM. Immediately add IPTG to a final concentration of 0.1-1 mM to induce protein expression.
- Expression and Harvest: Incubate the culture for 12-16 hours at 30°C or 3-4 hours at 37°C with shaking. Harvest cells by centrifugation.
- Analysis: Analyze protein expression by SDS-PAGE. Verify ncAA incorporation and fidelity through mass spectrometry (e.g., ESI-MS or MALDI-TOF) and functional assays.
Troubleshooting Notes:
- Low Incorporation Efficiency: Use RF1-deficient strains (e.g., C321.ΔA) or the PURE in vitro translation system, which omits RF1, to eliminate competition [36] [39].
- Cellular Toxicity: Titrate the concentration of ncAA and IPTG. Consider using a different ncAA or an orthogonal pair with higher specificity.
- Misincorporation: Ensure the orthogonal aaRS is highly specific for the ncAA over canonical amino acids. Evolve the aaRS for enhanced specificity if necessary [23].
Diagram 1: Stop codon suppression workflow. The orthogonal aaRS charges tRNAᶜᵁᴬ with the ncAA. This complex competes with RF1 at the UAG codon to incorporate the ncAA into the protein.
Quadruplet Codon Decoding
Conceptual Foundation and Applications
Quadruplet codon decoding expands the genetic code by using four-base codons (e.g., AGGA) instead of traditional triplet codons [42] [41]. This approach effectively creates 65 or more codons available for translation, providing new orthogonal channels for ncAA incorporation that do not compete with endogenous triplet codons [42]. Since quadruplet codons induce a frameshift during translation, their decoding requires engineered tRNAs with complementary four-base anticodons [41].
This strategy is particularly valuable for incorporating multiple distinct ncAAs into a single protein. When combined with stop codon suppression, quadruplet codons enable the biosynthesis of proteins with two different unnatural functionalities, significantly expanding the chemical space accessible to protein engineers [42]. A key challenge, however, is the typically low efficiency of quadruplet codon suppression, which often necessitates engineering both the tRNA and the ribosomal machinery for improved performance [41].
Research by Neumann et al. demonstrated the feasibility of this approach by evolving a ribosome capable of efficiently decoding quadruplet codons [42]. Subsequent work has focused on optimizing tRNA structure to enhance four-base codon recognition without compromising orthogonality or efficiency [41].
Protocol: tRNA Evolution for Enhanced Quadruplet Codon Suppression
This protocol outlines a directed evolution approach to engineer tRNAs with enhanced efficiency for quadruplet codon suppression, based on methodology successfully applied to the pyrrolysyl-tRNA (tRNAPyl) system [41].
Reagents and Equipment:
- tRNA library with randomized positions in the anticodon loop and stem
- Selection plasmid with a reporter gene (e.g., chloramphenicol acetyltransferase) containing a quadruplet codon at a permissive site
- Orthogonal aaRS specific for the target ncAA (e.g., PylRS variant)
- Appropriate E. coli host strain
- Selection antibiotic (e.g., chloramphenicol) at varying concentrations
- ncAA of interest (e.g., Boc-Lys)
- Equipment for library transformation, colony screening, and DNA sequencing
Procedure:
- Library Design: Create a tRNA library targeting the quadruplet codon of interest. Begin by randomizing four bases of the anticodon loop (positions 29, 30, 34, 35 using standard numbering) to generate an initial library [41].
- Positive Selection: Transform the tRNA library and the orthogonal aaRS plasmid into E. coli containing the selection plasmid. Plate cells on media containing the ncAA and a low concentration of antibiotic (e.g., 34 μg/mL chloramphenicol). Incubate until colonies appear.
- Dependency Screening: Replate survivors on media with and without the ncAA, containing the same antibiotic concentration. Select clones that only grow in the presence of the ncAA, indicating dependent quadruplet suppression.
- Secondary Evolution: Use selected hits as templates for secondary libraries with randomized anticodon stem positions (25-28 and 36-39) [41]. Repeat selection with increasing antibiotic concentrations (50-150 μg/mL) to identify enhanced mutants.
- Characterization: Sequence validated clones and characterize suppression efficiency in vivo using a fluorescent reporter (e.g., GFP) containing the quadruplet codon. Confirm ncAA incorporation fidelity via mass spectrometry.
Troubleshooting Notes:
- Poor Library Coverage: Ensure transformation efficiency exceeds library diversity. Use electrocompetent cells for high efficiency.
- Limited Selection Stringency: Gradually increase antibiotic concentration across selection rounds to identify progressively better performers.
- Context Effects: Test evolved tRNAs with different reporter genes and at different positions to assess codon suppression robustness.
Table 2: Evolved tRNAPyl Mutants for AGGA Quadruplet Codon Suppression
| Mutant | Mutations | Anticodon Loop Sequence (29,30,34,35) | Relative Efficiency | Key Structural Features |
|---|---|---|---|---|
| Wild-type | - | C U A A | 1.0x (baseline) | Standard anticodon loop [41] |
| M1 | C29A, A35C | A U A C | ~2-3x improvement | Improved base stacking [41] |
| M2 | A35U | C U A U | ~2-3x improvement | Alternative loop conformation [41] |
| M4 | C29A, A35C, A28G, U36C, others | A U A C (with stem mutations) | ~5x improvement | Strengthened anticodon stem with G28-C36 pair [41] |
| M7 | A35U, A28G, U36C, others | C U A U (with stem mutations) | ~5x improvement | Strengthened anticodon stem with mismatched positions 26-38 [41] |
Diagram 2: Directed evolution of tRNAs for quadruplet decoding. The process involves iterative library selection to identify mutants with enhanced four-base codon recognition.
Unnatural Base Pairs (UBPs)
Conceptual Foundation and Applications
Unnatural base pair (UBP) technology represents the most radical approach to genetic code expansion by creating entirely new nucleotides that function alongside natural A-T and G-C pairs [36] [43]. These synthetic nucleobases form a third, orthogonal base pair that can be incorporated into DNA and RNA through replication and transcription, ultimately creating novel codons for ncAA incorporation [36].
The power of UBPs lies in their ability to generate truly orthogonal codons that have no counterpart in natural systems. A six-letter genetic alphabet (A, T, G, C, X, Y) could theoretically produce 216 novel codons (6×6×6) in addition to the natural 64, dramatically expanding the potential for incorporating multiple ncAAs [36]. This complete orthogonality eliminates competition with endogenous translation factors, potentially enabling higher fidelity incorporation of multiple ncAAs compared to other reassignment strategies.
Several UBP systems have been developed and optimized for in vitro and in vivo applications. The Ds-Px and NaM-TPT3 pairs are among the most advanced, showing good efficiency in replication, transcription, and translation [36]. These systems require the engineering of polymerases that can recognize and incorporate the unnatural triphosphates, as well as orthogonal aaRS/tRNA pairs that recognize the novel codons containing unnatural bases.
Protocol: Incorporating Unnatural Base Pairs for ncAA Incorporation
This protocol describes the use of UBPs to create novel codons for ncAA incorporation, focusing on in vitro transcription and translation systems as a starting point for implementation.
Reagents and Equipment:
- Unnatural nucleobases (e.g., Ds, Px, NaM, TPT3) and their triphosphate forms
- Engineered DNA polymerase capable of synthesizing DNA containing UBPs
- T7 RNA polymerase mutant for transcribing RNA containing unnatural bases
- PURE in vitro translation system or reconstituted translation machinery
- Orthogonal aaRS/tRNA pair engineered to recognize the unnatural codon
- Plasmid template containing the UBP at desired position
- ncAA of interest
- Equipment for HPLC purification, in vitro transcription/translation
Procedure:
- DNA Template Preparation: Synthesize a DNA template containing the UBP at the desired codon position using an engineered DNA polymerase and the appropriate dNTPs plus unnatural dXTPs. Purify the template using HPLC or affinity purification.
- mRNA Transcription: Transcribe the DNA template using an engineered T7 RNA polymerase and a mixture of natural NTPs plus the unnatural rXTP. Verify transcription efficiency and mRNA integrity by gel electrophoresis.
- tRNA Engineering: Engineer an orthogonal tRNA with an anticodon complementary to the novel codon containing the unnatural base. Ensure the tRNA is efficiently charged by its cognate aaRS and functions with the translation machinery.
- In Vitro Translation: Use the PURE system or reconstituted translation system supplemented with the engineered aaRS/tRNA pair, ncAA, and unnatural base-containing mRNA. Incubate according to standard protocols.
- Analysis: Purify the expressed protein and verify incorporation of the ncAA at the specified position using mass spectrometry. Confirm fidelity by mutational analysis and functional assays.
Troubleshooting Notes:
- Poor Transcription Efficiency: Optimize the ratio of natural to unnatural NTPs. Engineer RNA polymerase variants with enhanced acceptance of unnatural substrates.
- Inefficient Translation: Ensure the unnatural base-containing mRNA is properly recognized by the ribosome. Engineer the ribosomal decoding center if necessary.
- Cellular Toxicity (for in vivo applications): For in vivo implementation, consider using engineered GROs with enhanced membrane permeability to unnatural triphosphates or implement synthetic auxotrophy systems.
Diagram 3: Unnatural base pair expansion system. UBPs create novel codons outside the natural code, enabling fully orthogonal encoding of ncAAs.
Advanced Integrated Systems and Genomically Recoded Organisms
Conceptual Foundation and Applications
The ultimate expression of codon reassignment involves creating genomically recoded organisms (GROs) in which multiple codons have been systematically replaced throughout the entire genome, freeing them for reassignment to ncAAs [39] [40]. These engineered organisms represent integrated platforms that combine multiple reassignment strategies to achieve unprecedented expansion of the genetic code.
GROs offer several transformative advantages. They provide complete resistance to viral infection by deleting tRNAs essential for reading reassigned codons, making them unable to produce functional viral proteins [39]. They enable multi-site incorporation of distinct ncAAs into single proteins with high fidelity by eliminating competition with endogenous translation factors [39] [40]. Additionally, they serve as robust platforms for biocontainment by creating organisms that depend on synthetic nutrients (ncAAs) not found in natural environments [40].
The construction of the Syn61Δ3 strain exemplifies this approach, where TCG, TCA, and TAG codons were replaced throughout the E. coli genome, followed by deletion of the corresponding tRNAs (tRNASer CGA, tRNASer UGA) and RF1 [39]. This strain exhibited complete resistance to a broad cocktail of bacteriophages and enabled the reassignment of all three freed codons to incorporate ncAAs [39]. More recently, the Ochre strain was engineered to use UAA as the sole stop codon, reassigning both UAG and UGA for multi-site incorporation of two distinct ncAAs with >99% accuracy [40].
Protocol: Utilizing GROs for Multi-ncAA Incorporation
This protocol describes the use of advanced GROs like Syn61Δ3(ev5) or Ochre for incorporating multiple distinct ncAAs into a single protein through sense and stop codon reassignment.
Reagents and Equipment:
- GRO strain (e.g., Syn61Δ3(ev5) or Ochre)
- Multiple orthogonal aaRS/tRNA pairs with complementary anticodons to reassigned codons
- Target protein plasmid with reassigned codons at desired positions
- Distinct ncAAs for each reassigned codon
- Appropriate growth media and antibiotics
- Expression induction reagents
- Mass spectrometry equipment for verification
Procedure:
- Strain Verification: Confirm the GRO genotype by PCR and sequencing of key recoded regions. Verify absence of target tRNAs/RF1 and orthogonality of reassigned codons.
- Plasmid Design: Engineer expression plasmids for your target protein with selected reassigned codons (e.g., TCG, TCA, TAG) at desired positions. Include genes for complementary orthogonal aaRS/tRNA pairs with appropriate anticodons.
- Transformation: Co-transform the target plasmid and orthogonal pair plasmids into the GRO. Select on appropriate antibiotic media.
- Multi-ncAA Expression: Grow transformed cells to mid-log phase in media supplemented with all required ncAAs (typically 1-5 mM each). Induce protein expression with IPTG or other inducer.
- Analysis: Purify the expressed protein and verify multi-ncAA incorporation using high-resolution mass spectrometry. Confirm incorporation fidelity at each position through tandem MS/MS and functional protein assays.
Troubleshooting Notes:
- Poor Protein Yield: Optimize ncAA concentrations and growth conditions. Consider using evolved GRO strains with improved fitness (e.g., Syn61Δ3(ev5)).
- Misincorporation at Reassigned Codons: Ensure orthogonal pairs are highly specific for their cognate codons. Engineer tRNAs to minimize wobble pairing.
- Cellular Fitness Issues: Use adaptive laboratory evolution to improve growth characteristics while maintaining codon reassignment fidelity.
Table 3: Advanced Genomically Recoded Organisms and Their Applications
| GRO Strain | Recoded Features | Freed Codons | Key Applications | Performance Metrics |
|---|---|---|---|---|
| C321.ΔA | All 321 TAG codons replaced with TAA; RF1 deleted [36] [40] | TAG (amber) | Single ncAA incorporation; Phage resistance [36] | Improved ncAA incorporation efficiency; Some viral resistance [36] |
| Syn61Δ3 | TCG, TCA, TAG replaced; serT, serU, prfA deleted [39] | TCG, TCA, TAG | Multi-ncAA incorporation; Complete viral resistance [39] | Resistant to phage cocktail; Three orthogonal coding channels [39] |
| Ochre | 1,195 TGA codons replaced with TAA; RF2 and tRNATrp engineered [40] | TAG, TGA (with UAA sole stop) | Dual ncAA incorporation; Non-degenerate code [40] | >99% accuracy dual incorporation; Single stop codon [40] |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for Codon Reassignment Studies
| Reagent Category | Specific Examples | Function and Utility | Implementation Notes |
|---|---|---|---|
| Orthogonal Pairs | MjTyrRS/tRNATyr, PylRS/tRNAPyl, EcTyrRS/tRNATyr [37] | Charge ncAAs and deliver to ribosome | Species-specific orthogonality; PylRS most versatile across domains [37] |
| Engineered Strains | RF1-deficient E. coli (C321.ΔA), Syn61Δ3(ev5), Ochre [39] [40] | Enhance incorporation efficiency; Enable multi-ncAA incorporation | GROs provide complete resistance to viral contamination [39] |
| Noncanonical Amino Acids | p-Acetylphenylalanine (pAcF), Nε-Boc-L-lysine (BocK), Azidohomoalanine [37] [41] | Provide novel chemical functionalities | Consider cell permeability, stability, and metabolic fate [38] |
| In Vitro Systems | PURE System [36] | Controlled translation environment | Omits RF1; Enhanced UAG suppression [36] |
| Unnatural Base Pairs | Ds-Px, NaM-TPT3 [36] | Create novel orthogonal codons | Require engineered polymerases for replication/transcription [36] |
| Selection Systems | Chloramphenicol resistance with in-frame reassigned codons [41] | Directed evolution of orthogonal components | Use increasing antibiotic concentrations for progressive evolution [41] |
Genetic code expansion represents a revolutionary approach in synthetic biology, enabling the biosynthesis of proteins with novel properties and functions. Central to this field are two principal methodologies for incorporating non-canonical amino acids (ncAAs) into proteins: residue-specific incorporation and site-specific incorporation [23] [44]. These techniques have transformed protein engineering by moving beyond the constraints imposed by the twenty canonical amino acids, allowing researchers to create proteins with enhanced or entirely new chemical properties [23].
The fundamental distinction between these approaches lies in their scope and precision. Residue-specific incorporation enables the global replacement of a canonical amino acid with its ncAA counterpart throughout the entire proteome, while site-specific incorporation allows for the precise installation of a ncAA at a single, predetermined location within a target protein [23] [44]. Both methods rely on the cellular translation machinery but manipulate it in different ways to achieve their distinct outcomes.
Understanding the mechanistic basis, applications, and limitations of each approach is crucial for researchers aiming to utilize genetic code expansion in their work. This article provides a comprehensive comparative analysis of these two foundational techniques, supported by experimental protocols and practical implementation guidelines for scientific researchers and drug development professionals.
Fundamental Principles and Mechanisms
Residue-Specific Incorporation
Residue-specific incorporation operates through the global replacement of a specific canonical amino acid with a structurally similar ncAA across all proteins being synthesized [44]. This method typically requires an auxotrophic host organism that cannot synthesize the canonical amino acid being replaced [23]. When this auxotroph is grown in medium containing the ncAA instead of the canonical amino acid, the native translation machinery, including aminoacyl-tRNA synthetases (aaRSs) and tRNAs, accepts the ncAA as a substrate and incorporates it at every position normally occupied by the canonical amino acid [23] [44].
A key advantage of this approach is its technical simplicity, as it often does not require engineering of the translation machinery, particularly when using ncAAs that are close structural analogs of canonical amino acids [23]. The resulting proteins contain ncAAs at multiple sites, which can significantly alter their overall physical and chemical properties [44]. This global modification approach is particularly valuable for applications requiring proteome-wide labeling or fundamental alterations to protein characteristics [44].
Site-Specific Incorporation
In contrast, site-specific incorporation (also termed genetic code expansion) enables the precise installation of a ncAA at a defined position in a target protein without replacing canonical amino acids [23]. This precision is achieved through the introduction of an orthogonal translation system (OTS) - a pair consisting of an orthogonal aaRS and its cognate tRNA that do not cross-react with the host's native translation components [23].
The most common implementation involves repurposing a stop codon, typically the amber stop codon (UAG), to encode the ncAA [23]. The orthogonal tRNA is engineered to recognize this codon, while the orthogonal aaRS is specifically designed or evolved to charge the tRNA exclusively with the desired ncAA [45]. When the engineered system is introduced into a host cell along with a target gene containing the repurposed codon at the desired position, the ncAA is incorporated specifically at that site during translation [23] [45].
This approach maintains the rest of the protein's sequence intact, allowing for minimal structural perturbation while introducing unique chemical functionalities at precise locations [23]. The primary challenge lies in developing highly specific and efficient orthogonal pairs for each ncAA of interest [45].
Table 1: Comparative Analysis of Incorporation Approaches
| Characteristic | Residue-Specific Incorporation | Site-Site-Specific Incorporation |
|---|---|---|
| Genetic Basis | Reinterpretation of sense codons [23] | Repurposing of blank codons (e.g., stop codons) [23] |
| Translation Machinery | Native aaRS/tRNA pairs [44] | Engineered orthogonal aaRS/tRNA pairs [23] |
| Incorporation Pattern | Multiple sites throughout proteome [44] | Single predetermined site [23] |
| Structural Perturbation | Global modification of protein properties [44] | Minimal disruption to protein structure [44] |
| Technical Complexity | Lower - often uses auxotrophic strains [23] | Higher - requires orthogonal pair engineering [45] |
| Primary Applications | Proteomics, biomaterials, global property alteration [44] | Biophysical probes, mechanistic studies, protein engineering [23] |
Experimental Design and Implementation
Workflow Comparison
The following diagram illustrates the fundamental mechanistic differences between residue-specific and site-specific incorporation approaches:
Protocol for Residue-Specific Incorporation
Objective: Global replacement of methionine with azidohomoalanine (Aha) in Escherichia coli proteins for subsequent bioorthogonal labeling [44] [46].
Materials:
- Methionine auxotroph E. coli strain (e.g., ΔmetE)
- Minimal medium lacking methionine
- Azidohomoalanine (Aha) stock solution (100 mM in DMSO)
- Isopropyl β-d-1-thiogalactopyranoside (IPTG) for induction
- Phosphate-buffered saline (PBS) for washing
Procedure:
- Inoculate a single colony of methionine auxotroph E. coli into minimal medium containing 0.5 mM methionine and grow overnight at 37°C with shaking.
- Subculture the overnight culture 1:100 into fresh minimal medium containing 0.5 mM Aha instead of methionine.
- Grow the culture to mid-log phase (OD600 ≈ 0.5-0.6) at 37°C with shaking.
- Induce protein expression with appropriate inducer (e.g., 0.5 mM IPTG for lac-based promoters) for 2-4 hours.
- Harvest cells by centrifugation (5,000 × g, 10 min, 4°C) and wash twice with PBS.
- Process cells for downstream applications including:
- Click chemistry conjugation with alkyne-functionalized probes
- Affinity purification using alkyne-functionalized beads
- Protein analysis by SDS-PAGE and western blotting
Technical Notes: For efficient replacement, ensure complete methionine starvation by washing cells with methionine-free medium before adding Aha-containing medium. The extent of incorporation can be verified by mass spectrometry or through detection of the incorporated bioorthogonal handle [44].
Protocol for Site-Specific Incorporation
Objective: Precise incorporation of 3-iodo-L-tyrosine at an amber stop codon position in a target protein expressed in mammalian cells [45].
Materials:
- Mammalian cell line (e.g., HEK293T, CHO)
- Expression plasmid encoding target protein with TAG codon at desired position
- Orthogonal plasmid system: B. stearothermophilus suppressor tRNATyr and E. coli TyrRS(V37C195) [45]
- 3-iodo-L-tyrosine stock solution (100 mM in NaOH)
- Transfection reagent (e.g., Lipofectamine 2000)
- Standard cell culture medium and supplements
Procedure:
- Culture mammalian cells in appropriate medium (e.g., DMEM with 10% FBS) at 37°C, 5% CO₂ to 70-80% confluence.
- Co-transfect cells with:
- Plasmid encoding target protein with TAG codon at desired position
- Plasmid encoding orthogonal B. stearothermophilus suppressor tRNATyr
- Plasmid encoding engineered E. coli TyrRS(V37C195)
- Add 1 mM 3-iodo-L-tyrosine to culture medium immediately after transfection.
- Incubate cells for 24-48 hours to allow protein expression.
- Harvest cells and lyse using appropriate lysis buffer.
- Purify target protein using affinity tags (e.g., FLAG-tag) and verify incorporation efficiency via:
- Mass spectrometry analysis
- Western blotting with anti-FLAG antibodies
- Functional assays specific to the target protein
Technical Notes: For enhanced suppression efficiency, use a tRNA expression vector with multiple tandem copies of the suppressor tRNA gene [45]. Optimization of tRNA:aaRS ratios may be necessary for different ncAAs or target proteins. Always include controls without ncAA to assess readthrough by endogenous machinery.
Research Reagent Solutions
Successful implementation of genetic code expansion requires carefully selected reagents and tools. The following table outlines essential components for both incorporation strategies:
Table 2: Essential Research Reagents for Genetic Code Expansion
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Non-Canonical Amino Acids | Azidohomoalanine (Aha) [44], Homopropargylglycine (Hpg) [44], 3-iodo-L-tyrosine [45], Se-allyl selenocysteine [46] | Provide unique chemical handles (azides, alkynes, halogens, photocaged groups) for conjugation, crosslinking, or spectroscopic studies |
| Orthogonal aaRS/tRNA Pairs | E. coli TyrRS/tRNA pair [45], M. jannaschii TyrRS/tRNA pair [23], Engineered MetRS variants [44] | Enable specific charging of tRNAs with ncAAs for site-specific incorporation; engineered for orthogonality in host systems |
| Specialized Cell Strains | Amino acid auxotrophs [23] [44], Genomically recoded organisms (GROs) [23] [43] | Provide cellular environment permissive for ncAA incorporation by eliminating competing pathways or creating blank codons |
| Bioorthogonal Chemistry Reagents | Copper(I) catalysts, cyclooctynes, tetrazines [46] | Enable selective conjugation to incorporated ncAAs for detection, purification, or immobilization applications |
| Analytical Tools | Nano-tRNAseq [17], LC-MS/MS systems [17], Modification-specific antibodies | Characterize incorporation efficiency, tRNA abundance, and modification status using advanced analytical techniques |
Applications and Future Directions
Current Research Applications
The complementary strengths of residue-specific and site-specific incorporation have enabled diverse applications across chemical biology and protein engineering:
Residue-Specific Applications:
- BONCAT (Bioorthogonal Non-canonical Amino Acid Tagging): Pulse-chase labeling of newly synthesized proteins for proteomic analysis using methionine analogs like Aha and Hpg [44] [46]
- FUNCAT (Fluorescent Non-canonical Amino Acid Tagging): Visualization of newly synthesized proteins in live cells through click chemistry with fluorescent dyes [44]
- Biomaterial Engineering: Creation of protein-based materials with enhanced stability or novel properties through global amino acid replacement [44]
Site-Specific Applications:
- Biophysical Probes: Incorporation of spectroscopic labels (NMR, EPR, fluorescent) for studying protein structure and dynamics [23]
- Post-Translational Modification Mimicry: Installation of phosphomimetics, acetyllysine, or other modified amino acids to study signaling pathways [23]
- Therapeutic Protein Engineering: Development of antibody-drug conjugates and optimized biologics with improved pharmacokinetic properties [23]
Emerging Methodologies and Future Outlook
The field of genetic code expansion continues to evolve with several promising directions:
Enhanced Orthogonality and Efficiency: Ongoing efforts focus on improving the orthogonality of aaRS/tRNA pairs through protein engineering and creating optimized expression systems [43]. Computational design and machine learning approaches are increasingly being employed to predict and enhance orthogonal pair functionality [23].
Genome Recoding: Creation of genomically recoded organisms (GROs) with reassigned codons provides blank codons for expanded genetic code manipulation without competition from endogenous factors [23] [43].
Novel Codon Systems: Development of unnatural base pairs and quadruplet codons further expands the number of available blank codons for simultaneous incorporation of multiple ncAAs [43].
Advanced Screening Technologies: High-throughput methods like yeast display, phage display, and compartmentalized partnered replication enable rapid evolution of orthogonal translation systems with enhanced specificity and efficiency [23].
The integration of these advanced methodologies with both residue-specific and site-specific incorporation approaches will continue to push the boundaries of genetic code expansion, enabling increasingly sophisticated manipulation of protein structure and function for basic research and therapeutic applications.
Genetic code expansion technologies have emerged as transformative approaches for incorporating non-canonical amino acids (ncAAs) into biosynthesized proteins, thereby overcoming the structural and functional limitations imposed by the twenty canonical amino acids. The development of orthogonal translation systems (OTSs)—comprising engineered aminoacyl-tRNA synthetases (aaRSs) and their cognate tRNAs—is fundamental to these efforts. However, engineering high-performing OTS requires sophisticated screening methods capable of evaluating immense molecular diversity to identify rare variants with desired specificity and efficiency [23].
High-throughput screening platforms provide the critical infrastructure needed to optimize these complex biomolecular systems. As outlined in Table 1, several powerful screening methodologies are employed in OTS development, each offering distinct advantages in terms of host system, library diversity, and selectable phenotypes. This application note details three key platforms—Yeast Display, mRNA Display, and Continuous Evolution—providing experimental protocols and contextual data to facilitate their implementation in genetic code expansion research, particularly within the framework of tRNA duplication and engineering studies.
Table 1: High-Throughput Screening Methods for OTS Development
| Screening Method | Common Engineering Targets | Phenotype | Host System | Typical Library Diversity |
|---|---|---|---|---|
| Yeast Display | Antibodies, enzymes, peptides, aaRS | Fluorescence | S. cerevisiae | 108–109 [23] |
| mRNA Display | Peptides | DNA amplification | In vitro | 1013–1014 [23] |
| Continuous Evolution | aaRS/tRNA | Phage propagation; Luminescence | Phage, E. coli | Experiment-dependent [23] |
| Live/Dead Selections | aaRS/tRNA | Growth | E. coli; S. cerevisiae | 106–109 [23] |
| Fluorescent Reporters | aaRS/tRNA | Fluorescence | E. coli; S. cerevisiae | 106–108 [23] |
Platform 1: Yeast Display for Orthogonal Peptide and Protein Engineering
Principle and Applications
Yeast display couples the phenotype (displayed peptide or protein) with the genotype (enclosed plasmid) on the surface of Saccharomyces cerevisiae. This platform is particularly valuable for screening combinatorial libraries of macrocyclic peptides and engineering aaRSs, as it enables real-time monitoring of screening processes using quantitative flow cytometry. This allows for precise control over selection stringency and direct affinity ranking of binders [47] [23].
A key application in OTS development involves using yeast display to screen libraries of macrocyclic peptides. Recent work demonstrates the generation of structurally diverse disulfide-cyclized peptide libraries displayed on yeast surfaces via a cysteine-free glycosylphosphatidylinositol (GPI) anchor system. This system minimizes undesirable intermolecular disulfide bonds and offers flexibility in yeast strain selection [47]. Quantitative flow cytometry facilitates the screening of millions of individual macrocyclic peptides against protein targets, enabling the identification of high-affinity ligands.
Detailed Protocol: Yeast Surface Display of Macrocyclic Peptide Libraries
Objective: To screen a yeast-displayed macrocyclic peptide library for high-affinity binders against a target protein.
Workflow Diagram: Yeast Display Screening
Materials:
- pYDS Vector: Episomal plasmid containing cysteine-free GPI anchor, (G4S)3 linker, and HA tag [47].
- Yeast Strain: Saccharomyces cerevisiae (e.g., EBY100).
- Library Oligonucleotides: Designed for formats like CX7C (one ring) or CX3CX9C (two rings), where X represents random amino acids encoded by NNK codons [47].
- Target Protein: Biotinylated or fluorescently labeled.
- Antibodies: Anti-HA tag antibody (fluorescently conjugated), streptavidin-conjugated fluorophore if using biotinylated target.
- Growth Media: SDCAA and SGCAA media.
- Flow Cytometer: Equipped for fluorescence-activated cell sorting (FACS).
Procedure:
- Library Generation:
- Clone the oligonucleotide library into the pYDS vector, downstream of the secretion signal and upstream of the (G4S)3 linker and GPI anchor sequence. The theoretical library diversity can reach up to 2x109 transformants [47].
- Electroporate the library DNA into competent yeast cells and culture in SDCAA to ensure library representation.
Induction:
- Harvest log-phase cells and resuspend in SGCAA to induce peptide expression. Incubate for 24-48 hours at 20-30°C with shaking.
Staining and FACS:
- Harvest induced yeast cells and wash.
- Co-stain cells with the target protein (e.g., 10-100 nM) and anti-HA antibody, each conjugated to distinct fluorophores, for 30-60 minutes on ice.
- Analyze and sort cells using FACS. Gate for cells with high HA signal (good expression) and high target signal (high binding). The ratio of target to HA signal allows for affinity ranking normalized for expression [47].
Recovery and Iteration:
- Culture sorted cells in SDCAA. After recovery, subject the population to 2-4 additional rounds of induction and sorting under increasing stringency (e.g., reduced target concentration).
- After the final sort, plate cells to isolate single clones for binding validation and sequencing.
Platform 2: mRNA Display for In Vitro Selection of OTS Components
Principle and Applications
mRNA display is a completely in vitro platform that creates a physical covalent linkage between a peptide (phenotype) and its encoding mRNA (genotype) via a puromycin linker. This method supports the highest library diversity among common screening platforms (Table 1), enabling the discovery of high-affinity peptides and optimized tRNAs without cellular constraints [23].
This platform is exceptionally suited for screening under conditions that would be toxic to cells and for incorporating ncAAs via genetic code reprogramming. The ultra-high diversity allows for deep sampling of sequence space, which is crucial for isolating rare, highly active OTS components from large random libraries.
Detailed Protocol: In Vitro Selection of tRNA-Binding Motifs via mRNA Display
Objective: To isolate RNA-binding peptides that specifically bind engineered tRNAs.
Workflow Diagram: mRNA Display Selection
Materials:
- DNA Library: A linear DNA template library encoding random peptides (e.g., 20-40 aa), flanked by constant regions for T7 promoter, reverse transcription, and PCR. The 3' end must be conjugated to puromycin.
- Puronucleotide: Puromycin-linked oligonucleotide.
- Translation System: Reconstituted E. coli or rabbit reticulocyte lysate, or PURE system.
- Immobilization Matrix: Streptavidin-coated beads and biotinylated target tRNA.
- Enzymes: T7 RNA polymerase, reverse transcriptase, Taq DNA polymerase.
Procedure:
- Library Preparation:
- Transcribe the DNA library in vitro to produce mRNA. Ligate the puromycin linker to the 3' end of the mRNA.
In Vitro Translation and Fusion:
- Incubate the mRNA-puromycin construct in the chosen translation system. The ribosome translates the mRNA and puromycin enters the ribosome's A-site, forming a covalent mRNA-peptide fusion.
- Purify the fusion molecules from the translation mixture by oligo(dT) chromatography or other methods.
Selection Panning:
- Incubate the purified fusion library with the immobilized target (biotinylated tRNA bound to streptavidin beads). Use a negative selection step with immobilized non-target tRNA to remove non-specific binders.
- Wash extensively to remove unbound and weakly bound fusions.
Recovery and Amplification:
- Elute specifically bound fusions (e.g., using denaturing conditions or competitive elution).
- Reverse transcribe the mRNA component into cDNA.
- Amplify the cDNA by PCR to generate a new DNA library for the next round.
Iteration and Analysis:
- Subject the library to 5-10 rounds of selection, increasing wash stringency in later rounds.
- Clone the final PCR product and sequence individual clones to identify enriched peptide sequences.
Platform 3: Continuous Evolution for OTS Optimization
Principle and Applications
Continuous evolution systems directly link a desired molecular function, such as the efficiency of an OTS, to the replication of a genetic element (e.g., a bacteriophage). This enables the autonomous and parallel evolution of millions of variants over many generations without manual intervention, allowing for the accumulation of beneficial mutations that might be missed in stepwise screens [23].
A common implementation is Phage-Assisted Continuous Evolution (PACE), where the gene of interest (e.g., an aaRS variant) is encoded on a plasmid. Its activity is coupled to the expression of a phage protein essential for propagation. Only host cells containing functional aaRS variants support phage replication, leading to the continuous enrichment of improved OTS components over time.
Detailed Protocol: Continuous Evolution of aaRS Using PACE
Objective: To evolve an aaRS with enhanced charging efficiency for a specific ncAA.
Workflow Diagram: Continuous Evolution Setup
Materials:
- Apparatus: PACE system with a chemostat (turbidostat) and a series of "lagoon" vessels for continuous dilution.
- Host Cells: E. coli cells harboring two plasmids: 1) the "accessory plasmid" containing the gene for the mutagenesis system, and 2) the "selection plasmid" carrying the aaRS mutant library and a reporter gene (e.g., gIII for phage propagation) whose expression is dependent on successful ncAA incorporation [23].
- M13 Bacteriophage: An engineered M13 phage whose genome lacks the gene III (gIII), making its propagation dependent on gIII expression from the host cell's selection plasmid.
- Growth Media: Including the ncAA of interest.
Procedure:
- System Setup:
- Inoculate the host E. coli strain (containing both the accessory and selection plasmids) into the lagoon and start continuous media flow from the chemostat.
- Introduce the engineered M13 phage into the lagoon.
Evolution Run:
- As host cells flow into the lagoon, they are infected by the phage. Only host cells containing an aaRS variant that efficiently charges the orthogonal tRNA with the ncAA will express the full-length reporter protein (gIII).
- This allows for the packaging and release of new, infectious phage particles, which flow out of the lagoon to infect fresh host cells entering from the chemostat.
- The mutagenesis plasmid continuously introduces random mutations into the aaRS gene in the host cells, generating new diversity.
Monitoring and Harvesting:
- Monitor phage titer in the lagoon effluent over time. A rising titer indicates successful evolution.
- The evolution run can continue for dozens to hundreds of hours. Sample phage from the effluent periodically.
- Isolate the selection plasmid from the phage particles to characterize the evolved aaRS variants.
The Scientist's Toolkit: Essential Research Reagent Solutions
The successful implementation of high-throughput screening platforms relies on specialized reagents and genetic tools. The following table details key solutions for OTS development and screening.
Table 2: Key Research Reagent Solutions for OTS Screening
| Reagent / Solution | Composition / Key Feature | Function in Screening |
|---|---|---|
| Cysteine-free GPI Anchor System [47] | Episomal plasmid, (G4S)3 linker, HA tag | Yeast surface display of disulfide-cyclized peptides without unwanted inter-chain bonds. |
| ACE-tRNA Expression Cassette Libraries [48] | >1800 unique variants of 5'-UCE, tRNA body, flanking sequences | Optimizes sup-tRNA transcription, processing, stability, and translational efficiency for nonsense suppression. |
| Nonsense Reporter HTCS Plasmid [48] | pNanoRePorter 2.0 (Nanoluciferase with PTC, UbC-Fluc2 normalization) | High-throughput quantification of nonsense suppression efficiency in a normalized system. |
| Orthogonal tRNA–Ψ Codon Pairs [26] | Engineered tRNA with mutated anticodon stem-loop, Ψ-modified stop codon on mRNA | Enables specific incorporation of ncAAs via RNA codon-expansion (RCE) with reduced crosstalk. |
| Prime Editing Installed sup-tRNA [49] | PE machinery, optimized sup-tRNA sequence, endogenous genomic locus | Converts a dispensable endogenous tRNA into a potent, genomically integrated suppressor tRNA for PTC readthrough. |
The integration of yeast display, mRNA display, and continuous evolution provides a powerful, multi-faceted toolkit for advancing genetic code expansion research. By enabling the screening of vast molecular libraries, these platforms accelerate the development of optimized orthogonal translation systems and novel therapeutic agents. The detailed protocols and reagent solutions outlined herein offer a practical foundation for researchers to implement these cutting-edge screening methodologies in their own investigations of tRNA biology and ncAA incorporation.
Overcoming Hurdles: Strategies for Optimizing Efficiency and Orthogonality
Genetic code expansion (GCE) technology enables the site-specific incorporation of noncanonical amino acids (ncAAs) into proteins, revolutionizing protein engineering for therapeutic and research applications [37]. The core of this technology relies on orthogonal aminoacyl-tRNA synthetase/tRNA pairs (OTSs)—components that must function without interference from the host's native translation machinery [23] [37]. These pairs are typically sourced from organisms of different phyla to minimize cross-reactivity, yet achieving true orthogonality in complex cellular environments remains a significant challenge [6].
A primary obstacle in GCE is cross-talk, where endogenous cellular components mistakenly interact with orthogonal elements. This can manifest as endogenous aminoacyl-tRNA synthetases (aaRSs) charging canonical amino acids onto orthogonal tRNAs, resulting in misincorporation, or orthogonal tRNAs being inefficiently processed by host translational machinery, reducing ncAA incorporation efficiency [6]. As research expands into more complex eukaryotic systems and demands the incorporation of multiple ncAAs simultaneously, the problem of cross-talk becomes increasingly pronounced [50]. This application note details targeted strategies and practical protocols to characterize, quantify, and mitigate cross-talk, ensuring the high-fidelity genetic code expansion required for advanced therapeutic development.
Quantitative Analysis of Orthogonality Challenges
The following table summarizes the primary sources of cross-talk in genetic code expansion systems and their impact on protein synthesis.
Table 1: Common Sources of Cross-Talk in Genetic Code Expansion Systems
| Source of Cross-Talk | Effect on System | Quantitative Impact |
|---|---|---|
| Mis-charging by endogenous aaRSs | Incorporation of canonical amino acids at ncAA sites, reducing product homogeneity [6] | Can exceed 30% mis-incorporation in poorly optimized systems [51] |
| Non-orthogonal tRNA-EF-Tu interaction | Reduced efficiency of ncAA incorporation and potential truncation of target protein [6] | Up to 60% reduction in yield for some engineered tRNAs [51] |
| Wobble base pairing / Poor codon specificity | Mis-reading of synonymous codons, disrupting precise ncAA placement [51] | 5-10% mis-incorporation even with modified tRNAs; reduced to <2% with hyperaccurate ribosomes [51] |
| Competition with release factors | Reduced full-length protein yield during stop codon suppression [23] | Efficiency highly dependent on codon context and system optimization [23] |
Experimental Protocols for Characterizing Cross-Talk
Protocol: Competitive Codon Reading Assay
This assay quantifies the ability of various aminoacyl-tRNAs to compete for a single codon, directly measuring potential wobble-pairing cross-talk [51].
Materials:
- Custom reconstituted PURE translation system [51]
- In vitro transcribed mRNA templates featuring a single codon type of interest
- Individual tRNA isoacceptors, either wild-type (fully modified) or in vitro transcribed (unmodified)
- Leucine isotopologues (e.g., [¹³C₆,¹⁵N]-L-Leucine, [D₁₀]-L-Leucine) or isotopically labeled ncAAs
- MALDI-TOF Mass Spectrometer
Procedure:
- Aminoacylation: Charge each individual tRNA isoacceptor with a unique leucine isotopologue or a distinct isotopically labeled ncAA.
- Competition Translation: Combine the prepared AA-tRNAs in equimolar ratios into the PURE translation reaction containing the mRNA template.
- Product Analysis: Purify the synthesized peptide and analyze by MALDI-TOF mass spectrometry.
- Data Quantification: The relative incorporation of each isotopologue is determined from the peak intensities in the mass spectrum. This creates a quantitative profile of which tRNAs successfully read the target codon under competitive conditions [51].
Interpretation: This assay reveals ambiguous codon reading that leads to cross-talk. A well-behaved, orthogonal system will show strong preference for the cognate tRNA. Significant read-through by non-cognate tRNAs indicates a need for optimization, such as using unmodified tRNAs or hyperaccurate ribosomes [51].
Protocol: In Vivo Orthogonality Assessment via Fluorescence Reporting
This method uses a dual-reporter system in live cells to simultaneously measure suppression efficiency (full-length protein yield) and orthogonality (fidelity of ncAA incorporation).
Materials:
- Engineered host cells (e.g., E. coli, S. cerevisiae) with genomic deletion of the target nonsense codon where feasible [23]
- Two plasmid systems:
- Plasmid 1: pEVOL or similar, expressing the orthogonal aaRS/tRNA pair [23]
- Plasmid 2: Dual-reporter construct expressing:
- Reporter 1: GFP with an amber (TAG) stop codon at a permissive site.
- Reporter 2: mCherry with no amber codon, serving as a transfection/expression control.
- ncAA of interest
- Fluorescence plate reader or flow cytometer
Procedure:
- Transformation: Co-transform the two plasmids into the expression host.
- Culture and Induction: Grow cells in media with and without the ncAA. Induce expression of both the OTS and the reporter construct.
- Measurement: After a suitable expression period, measure GFP and mCherry fluorescence.
- Calculation:
- Suppression Efficiency: Normalize GFP fluorescence of the "+ncAA" sample to the mCherry control. Compare this to the fluorescence from a "no ncAA" control. High GFP signal in the "+ncAA" condition indicates successful suppression.
- Orthogonality/Fidelity: Calculate the signal ratio of GFP(+ncAA) / GFP(-ncAA). A high ratio indicates low mis-incorporation of canonical amino acids in the absence of the ncAA, signifying high orthogonality [23].
Protocol: Assessing tRNA Processing and Maturation
In eukaryotic systems, nuclear processing of tRNAs is a major hurdle. This protocol assesses the efficiency of orthogonal tRNA maturation.
Materials:
- Northern Blot equipment or RNA-seq capabilities
- Probes specific to the orthogonal tRNA's sequence
- Cells expressing the orthogonal tRNA
Procedure:
- Total RNA Extraction: Isolate total RNA from cells expressing the orthogonal tRNA.
- Analysis: Perform Northern Blot analysis using probes against the orthogonal tRNA. Alternatively, use YAMAT-seq, a method for deep-sequencing the mature tRNA pool [52].
- Detection: Distinguish between the unprocessed tRNA precursor and the mature, correctly processed tRNA.
Interpretation: A strong band/signal for the mature tRNA indicates successful adaptation to the host's processing machinery. A predominant precursor band suggests the orthogonal tRNA is not being correctly processed, which will severely limit its function and requires sequence re-engineering [52] [6].
Visualization of Orthogonality Engineering Strategies
Diagram 1: A strategic roadmap for troubleshooting and resolving common sources of cross-talk in Genetic Code Expansion systems, linking specific problems to engineering strategies and validation tools.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Ensuring Orthogonality
| Reagent / Tool | Function & Utility | Key Consideration |
|---|---|---|
| Orthogonal aaRS/tRNA Pairs (e.g., MjTyrRS/tRNA, PylRS/tRNA) | Core components for ncAA incorporation; different pairs offer varying orthogonality across hosts [37] [6]. | MjTyrRS/tRNA is highly orthogonal in E. coli; PylRS/tRNA is orthogonal in both prokaryotes and eukaryotes [37]. |
| Hyperaccurate Ribosomes (e.g., mS12 mutant) | Increases ribosomal discrimination against near-cognate tRNAs, drastically reducing wobble pairing and improving codon orthogonality [51]. | Particularly effective when paired with unmodified, in vitro transcribed tRNAs (t7tRNA) [51]. |
| Unmodified tRNAs (t7tRNA) | tRNAs produced by in vitro transcription lack natural post-transcriptional modifications, which often expand codon recognition; this narrows their codon specificity [51]. | Reduces unwanted "sharing" of codons between different tRNAs, but may require compensatory engineering for efficiency. |
| PURE Translation System | A reconstituted in vitro translation system using purified components. Allows precise control over tRNA and ribosome composition [23] [51]. | Ideal for debugging cross-talk and testing novel OTSs without the complexity of a full cellular environment. |
| Genomically Recoded Organisms | Engineered cells with all occurrences of a particular nonsense codon (e.g., TAG) removed from the genome [23]. | Eliminates competition with native release factors, and prevents mis-incorporation of ncAAs into native proteins. |
Achieving robust orthogonality in complex cellular environments is paramount for advancing genetic code expansion from a research tool to a reliable platform for therapeutic applications such as the development of homogeneous antibody-drug conjugates and novel live-attenuated vaccines [37] [53]. The protocols and strategies outlined here—including the use of competitive codon assays, hyperaccurate ribosomes, and deep sequencing of tRNA pools—provide a structured methodology to identify, quantify, and eliminate sources of cross-talk. As the field progresses toward incorporating multiple ncAAs and operating in more complex eukaryotic systems, a meticulous and systematic approach to engineering orthogonality will be the foundation upon which next-generation protein therapeutics are built.
Within the framework of genetic code expansion (GCE) research, the efficient incorporation of non-canonical amino acids (ncAAs) relies on the optimal performance of orthogonal translation systems. A significant bottleneck in this process often occurs during the delivery and incorporation of aminoacyl-tRNAs (aa-tRNAs) into the ribosomal A-site, a process facilitated by elongation factor Tu (EF-Tu in prokaryotes; eEF1A in eukaryotes). Engineering the interactions between tRNA, EF-Tu, and the ribosome presents a powerful strategy for enhancing translational efficiency, particularly for challenging ncAA substrates. This application note details practical strategies and protocols for engineering these molecular interactions to boost translational output, with direct applicability to GCE initiatives involving tRNA duplication and orthogonalization. The core objective is to create engineered components that maintain orthogonality while achieving superior translation kinetics and fidelity, ultimately expanding the toolkit for synthetic biology and therapeutic development [14] [54].
Key Interaction Sites for Engineering
The tRNA molecule interacts with EF-Tu and the ribosome through specific, well-characterized structural domains. Engineering efforts focused on these sites can dramatically alter the binding affinity, accommodation kinetics, and overall efficiency of translation. The table below summarizes the primary engineering targets on tRNA and their functional roles in translation.
Table 1: Key tRNA Engineering Sites for Enhanced EF-Tu and Ribosome Interaction
| Engineering Target | Structural Location | Function in Translation | Engineering Approach |
|---|---|---|---|
| Acceptor Stem & T-stem | Pairs 51:63, 50:64, 49:65, and 7:66 [14] | Primary binding interface with EF-Tu; critical for ternary complex stability [55]. | Rational design or directed evolution to modulate EF-Tu binding affinity and optimize accommodation kinetics [54]. |
| Elbow Region | Junction of D-loop and T-loop [54] | Interacts with the ribosome's A, P, and E sites; crucial for accommodation dynamics [56]. | Introduce modifications or mutations that facilitate pivoting and navigation of the accommodation corridor. |
| Variable Arm | Between the anticodon and T arms [14] | Impacts tRNA flexibility and can influence interactions with elongation factors [54]. | Engineer length and sequence to fine-tune the dynamics of the accommodation process. |
| Anticodon Stem-Loop | Anticodon loop and flanking nucleotides [14] | Decodes mRNA codon within the ribosomal A-site. | While key for codon recognition, it can be engineered to work with elongated or quadruplet codons in GCE. |
| tRNA Body Modifications | Throughout the molecule, especially elbow and anticodon loop [54] | Stabilizes structure, ensures decoding accuracy, and influences EF-Tu binding [57]. | Co-express with specific modification enzymes or use pre-modified transcripts for in vitro systems. |
Recent structural and computational studies have revealed that the accommodation process in humans requires a distinct ~30° pivoting of the aa-tRNA about the anticodon stem to navigate the accommodation corridor, a step that becomes more constrained due to intersubunit rolling in the eukaryotic ribosome [56]. This finding underscores the importance of the elbow region as a critical engineering target for improving translational efficiency in eukaryotic systems or for facilitating the incorporation of bulky ncAAs.
Experimental Protocols for Engineering and Validation
This section provides detailed methodologies for engineering tRNAs and quantitatively assessing their performance in translation.
Protocol: High-Throughput Directed Evolution of tRNA for Improved EF-Tu Engagement
Objective: To generate tRNA variants with enhanced translational efficiency through iterative selection based on cellular survival or fluorescence.
Materials:
- Library Construction: Oligonucleotide pool for mutagenesis (targeting acceptor/T-stem), PCR reagents, plasmid backbone with a selection gene (e.g., antibiotic resistance or GFP) containing a target codon.
- Selection Host: E. coli strain with a knocked-out cognate tRNA gene or an orthogonal EF-Tu variant.
- Analysis: Next-generation sequencing (NGS) platform, flow cytometer (if using FACS).
Procedure:
- Library Generation: Synthesize a diverse tRNA library by introducing degenerate nucleotides at key positions in the acceptor and T-stem, as defined in Table 1. Clone this library into a plasmid where the tRNA is required for the expression of a selectable marker.
- Transformation and Selection: Transform the library into the selection host and plate on selective media (e.g., containing antibiotic). Only cells with functional tRNA variants will survive.
- Iterative Selection: For higher stringency, use fluorescence-activated cell sorting (FACS) if the selectable marker is a fluorescent protein. Gate the top-performing cells (brightest fluorescence) and collect them for plasmid recovery.
- Variant Analysis: Isolate plasmids from the selected population and subject the tRNA region to NGS to identify enriched mutations. These consensus mutations are strong candidates for improving EF-Tu interaction and translational efficiency [54].
Protocol: Measuring Translational Kinetics Using a tRNA-free PURE System
Objective: To directly quantify the efficiency of engineered tRNA in a controlled, in vitro environment devoid of endogenous tRNA background.
Materials:
- tfPURE System: Commercially available or reconstituted tRNA-free Protein Synthesis Using Recombinant Elements system [58].
- tRNA Templates: DNA templates for in vitro transcription of wild-type and engineered tRNAs.
- Reporter Template: DNA encoding a reporter protein (e.g., luciferase, GFP).
- Detection Instrument: Luminometer or fluorometer.
Procedure:
- System Preparation: Assemble the tfPURE reaction according to manufacturer's or standard protocols. This system contains all necessary translation factors, including EF-Tu, but lacks tRNA [58].
- tRNA Expression: Include DNA templates for the tRNA of interest in the reaction. The PURE system's T7 RNA polymerase will transcribe the tRNAs. For tRNAs that do not start with a 5'-G, use a leader sequence and RNase P for correct 5'-end processing [58].
- Translation Reaction: Add the DNA template for your reporter protein. The efficiency of the engineered tRNA will be directly reflected in the synthesis rate and yield of the reporter.
- Kinetic Analysis: Monitor reporter output (e.g., luminescence for luciferase) over time. Calculate the initial rate of synthesis and total yield. Compare engineered tRNA against wild-type controls to quantify improvement [58].
Table 2: Quantitative Analysis of Engineered tRNA Performance
| tRNA Variant | Luciferase Yield (RLU) | Incorporation Rate (Amino acids/sec) | Proofreading Efficiency (Relative to WT) |
|---|---|---|---|
| Wild-Type tRNA | 1.0 x 10⁶ [58] | 10-20 [56] | 1.0 |
| Engineered tRNA (Variant A) | 3.5 x 10⁶ | ~25 | 0.8 |
| Engineered tRNA (Variant B) | 5.7 x 10⁶ | ~30 | 1.1 |
| Note: RLU = Relative Light Units. The values for engineered tRNAs are illustrative of potential improvements. Proofreading efficiency indicates fidelity maintenance. |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Engineering tRNA Translation
| Reagent / Tool | Function / Application | Example & Notes |
|---|---|---|
| Orthogonal EF-Tu/eEF1A Mutants | To study and evolve specific tRNA-EF interactions; can have altered affinity for ncAAs. | E. coli EF-Tu mutants with expanded binding pockets [54]. |
| tRNA-free PURE System | For in vitro validation of tRNA efficiency without background from endogenous cellular tRNAs [58]. | Reconstituted from individually purified components; allows for precise compositional control. |
| Nanopore tRNA-Seq (RNA004) | To simultaneously monitor tRNA abundance and modification status, which affects EF-Tu binding and function [59]. | Oxford Nanopore RNA004 chemistry; enables direct RNA sequencing without RT-PCR biases. |
| Structure-Based Models (SBM) | For molecular simulations of large-scale conformational changes like aa-tRNA accommodation [56] [60]. | All-atom Gō models; used to simulate accommodation and identify steric bottlenecks. |
| Ribosome Profiling (Ribo-Seq) | To provide a genome-wide snapshot of translation efficiency and ribosome occupancy [54]. | Reveals codon-specific translational bottlenecks in vivo. |
Workflow and Pathway Visualizations
Engineering Workflow
tRNA Selection Pathway
Concluding Remarks
Strategic engineering of the tRNA-EF-Tu-ribosome interface provides a powerful avenue for overcoming efficiency barriers in genetic code expansion. By focusing on key structural elements such as the acceptor stem, T-stem, and elbow region, researchers can tailor translational components for enhanced performance with non-canonical amino acids. The integrated use of directed evolution, rational design, and rigorous in vitro validation, as outlined in this application note, creates a robust pipeline for developing next-generation tools that push the boundaries of synthetic biology and therapeutic protein production.
The expansion of the genetic code through the incorporation of noncanonical amino acids (ncAAs) represents a frontier in synthetic biology, enabling the creation of proteins with novel chemical properties and functions. Central to this technology are orthogonal aminoacyl-tRNA synthetase (aaRS)/tRNA pairs, which must charge ncAAs onto tRNAs with high fidelity to prevent misacylation—the erroneous attachment of incorrect amino acids to tRNAs. Misacylation compromises translational fidelity, leading to statistical proteins and potential cellular toxicity. This application note examines the challenge of misacylation within the context of genetic code expansion via tRNA duplication research. We detail mechanisms of natural aaRS editing, present experimental protocols for assessing charging fidelity, and explore engineering strategies aimed at enhancing the specificity and efficiency of orthogonal translation systems. By providing a framework for exploiting and engineering aaRS editing activities, this work supports the development of more robust and reliable genetic code manipulation tools for therapeutic and biotechnological applications.
Genetic code expansion (GCE) allows for the site-specific incorporation of noncanonical amino acids (ncAAs) into proteins, thereby augmenting the chemical and functional diversity of the proteome. This process relies on the establishment of orthogonal aminoacyl-tRNA synthetase (aaRS)/tRNA pairs that operate without cross-reacting with the host's native translation machinery. A persistent challenge in this field is maintaining high fidelity in the aminoacylation reaction. Misacylation, the mischarging of a tRNA with a non-cognate amino acid (be it a canonical or a noncanonical one), directly subverts the accuracy of protein synthesis. This can lead to the production of "statistical proteins" and potentially activate cellular stress responses, undermining both the efficiency of ncAA incorporation and overall cell fitness.
Naturally, many aaRSs have evolved proofreading (editing) activities to cleave misactivated amino acids (pre-transfer editing) or misacylated tRNAs (post-transfer editing). For example, class II prolyl-tRNA synthetase (ProRS) employs both pre- and post-transfer editing pathways to prevent the stable formation of Ala-tRNA^Pro, a common error due to the similarity between alanine and proline [61]. The editing domains of aaRSs are critical for discriminating against structurally similar amino acids; however, these domains are often absent in the minimalist orthogonal aaRS/tRNA pairs derived from archaeal or bacterial systems (e.g., the widely used PylRS/tRNA^Pyl pair), making them inherently prone to misacylation errors when engineered for new ncAA substrates. Consequently, understanding, measuring, and engineering these editing functions is paramount for advancing GCE technologies, particularly in the context of tRNA duplication research which seeks to create new blank codons and orthogonal pairs.
Analytical Methods: Quantifying aaRS Fidelity and Misacylation
Accurately measuring the fidelity of tRNA aminoacylation is a prerequisite for diagnosing misacylation and validating the success of any engineering intervention. The following protocols describe key methods for this purpose.
Protocol: tRNA Microarray for In Vivo Misacylation Profiling
This protocol, adapted from [62] [63], uses radiolabeling and custom microarrays to quantitatively determine which tRNAs are charged with a specific amino acid in living cells. It is particularly powerful for detecting condition-dependent misacylation, such as that induced by oxidative stress.
- Primary Application: Profiling the aminoacylation status of multiple tRNA isoacceptors simultaneously from a cellular sample.
- Principle: Cells are pulsed with a radiolabeled amino acid (e.g.,
[³⁵S]-Methionine). Total RNA is extracted under acidic conditions to preserve the labile aminoacyl bond. The charged tRNAs are then hybridized to a custom DNA microarray where each probe is the reverse complement of a specific tRNA sequence. Phosphorimaging reveals which tRNA spots are radioactive, indicating they were aminoacylated with the labeled amino acid. Materials and Reagents:
[³⁵S]-Methionine(high specific activity)- Acidic RNA extraction buffer (e.g., 0.3 M sodium acetate, pH 4.5)
- Custom tRNA microarray slides (designed based on Genomic tRNA Database)
- DNA oligonucleotide probes (reverse complements of target tRNAs)
- Hybridization chambers and oven
- Phosphorimager and screens
Procedure:
- Cell Labeling and Lysis: Grow cells to the desired density and pulse with
[³⁵S]-Metfor a short duration (e.g., 5-15 minutes). Immediately harvest cells and lyse them in acidic phenol/RNA extraction buffer to preserve aminoacyl-tRNAs. - tRNA Isolation: Precipitate total RNA and treat with aminopeptidase M to remove any N-terminal amino acids from peptidyl-tRNAs, ensuring the signal derives solely from charged tRNAs [63].
- Microarray Hybridization: Resuspend the RNA sample in hybridization buffer and apply it to the custom tRNA microarray. Hybridize overnight at a controlled temperature (e.g., 50°C).
- Washing and Imaging: Wash the array stringently to remove non-specifically bound RNA. Expose the dried array to a phosphorimager screen and scan.
- Data Analysis: Quantify the signal intensity for each tRNA probe. Normalize signals to account for variations in tRNA abundance and probe efficiency. A significant signal from a non-cognate tRNA probe (e.g., signal for a lysine tRNA in a
[³⁵S]-Metlabeled sample) indicates misacylation.
- Cell Labeling and Lysis: Grow cells to the desired density and pulse with
Protocol: In Vitro Aminoacylation and Editing Assay
This biochemical assay directly measures the aminoacylation kinetics and editing efficiency of a purified aaRS [64] [61]. It allows for the dissection of pre- and post-transfer editing pathways.
- Primary Application: Characterizing the kinetic parameters of an aaRS and quantifying its mischarging and editing efficiency against specific amino acids in a controlled system.
- Principle: The aaRS catalyzes the attachment of an amino acid to its cognate tRNA, which can be monitored in real-time. By using non-cognate amino acids (e.g., alanine for ProRS) and mutant aaRSs lacking post-transfer editing functionality, one can isolate and quantify the contributions of different editing pathways.
Materials and Reagents:
- Purified wild-type and editing-deficient aaRS (e.g.,
ProRS-Δedit) - In vitro transcribed tRNA (cognate and non-cognate)
- ATP,
[³²P]-ATPor[³H]-Amino Acids - Reaction buffer (e.g.,
40 mM HEPES-KOH, pH 7.5, 50 mM KCl, 10 mM MgCl₂, 0.1 mg/mL BSA) - Scintillation counter or TLC equipment
- Purified wild-type and editing-deficient aaRS (e.g.,
Procedure:
- Aminoacylation Reaction: Set up reactions containing aaRS, tRNA, ATP, and a
[³H]-labelednon-cognate amino acid (e.g.,[³H]-Alaninefor ProRS). Incubate at 37°C and withdraw aliquots at time intervals. - Quantification of Misacylated tRNA: Spot aliquots on acid-washed filter pads, which retain charged tRNA but not free amino acids. Measure the radioactivity on the filters to quantify the formation of misacylated tRNA (e.g.,
Ala-tRNA^Pro). - Isolating Pre-transfer Editing: To measure pre-transfer editing (hydrolysis of the misactivated aminoacyl-adenylate), perform the aminoacylation reaction in the absence of tRNA and monitor the production of
AMPinstead of the acylated product. - Kinetic Analysis: Determine steady-state kinetic parameters (
K_Mandk_cat) for both cognate and non-cognate amino acids. A low discrimination factor at the aminoacylation active site necessitates robust editing.
- Aminoacylation Reaction: Set up reactions containing aaRS, tRNA, ATP, and a
Table 1: Key Research Reagent Solutions for Misacylation Studies
| Reagent / Tool | Function / Application | Key Characteristics |
|---|---|---|
| OrthoRep System [28] | Continuous in vivo hypermutation of aaRS genes for directed evolution. | Error-prone orthogonal DNA polymerase; enables 10⁻⁵ mutations/base. |
| tRNA Microarray [62] | High-throughput profiling of tRNA charging fidelity in vivo. | Custom DNA probes; requires radiolabeled amino acids. |
| Ratiometric RXG Reporter [28] | Fluorescence-based selection for amber codon suppression efficiency and fidelity. | RFP-GFP fusion with amber codon; measures relative readthrough efficiency (RRE). |
| Editing-Deficient aaRS Mutants [64] [61] | Biochemical tools to isolate and study specific editing pathways. | e.g., ProRS-Δedit or LeuRS-ΔCP1; ablates post-transfer editing. |
| PylRS/tRNA^Pyl Pair [28] | Versatile orthogonal platform for genetic code expansion. | Naturally polyspecific; often requires engineering for high ncAA fidelity. |
Figure 1: Experimental workflows for analyzing misacylation and aaRS editing. The left panel outlines the tRNA microarray protocol for in vivo profiling. The right panel depicts the competing pathways in an in vitro aminoacylation assay with a non-cognate amino acid.
Engineering Strategies to Combat Misacylation
The ultimate goal in GCE is to design orthogonal systems that are both highly efficient and specific. The following strategies leverage insights from natural editing mechanisms and advanced evolutionary techniques to achieve this.
Harnessing Directed Evolution for Enhanced Fidelity
Traditional aaRS engineering is labor-intensive and often yields suboptimal variants. Continuous in vivo evolution platforms, such as OrthoRep, have emerged as powerful alternatives [28]. In this system, the gene for the aaRS of interest is placed on an orthogonal plasmid replicated by an error-prone DNA polymerase, introducing random mutations at a rate of ~10⁻⁵ substitutions per base.
- Selection Strategy: A ratiometric dual-fluorescence reporter (RFP-GFP with an amber stop codon, or RXG) is used to select for desired aaRS activity.
- Positive Selection: High
GFP/RFPratio in the presence of the target ncAA selects for aaRS variants that efficiently charge the orthogonal tRNA and incorporate the ncAA. - Negative Selection: Low
GFP/RFPratio in the absence of the ncAA selects against aaRS variants that promiscuously charge the tRNA with canonical amino acids, thereby enforcing ncAA dependency and reducing misacylation.
- Positive Selection: High
This approach has successfully evolved aaRSs that incorporate 13 different ncAAs with efficiencies rivaling canonical translation [28]. In one instance, this method even yielded an aaRS that evolved to autoregulate its own expression, further minimizing leakiness.
Exploiting Natural and Engineered Editing Mechanisms
Understanding how natural aaRSs achieve high fidelity provides a blueprint for engineering. For instance, studies on Mycoplasma pathogens reveal that they naturally possess aaRSs with inactivated editing domains (e.g., LeuRS and PheRS), leading to elevated mistranslation rates. This "statistical proteome" is believed to provide antigenic variation to evade host immune systems [64]. This phenomenon underscores the critical role of editing in maintaining proteome integrity and demonstrates that its modulation can have profound biological consequences.
Conversely, some aaRSs enhance editing under stress. Salmonella PheRS is oxidized under oxidative stress, which enlarges its editing pocket and increases its efficiency in clearing misacylated m-Tyr-tRNA^Phe and p-Tyr-tRNA^Phe, thus protecting the proteome from these damaging incorporations [65]. This illustrates that editing activity can be a regulated, inducible cellular defense mechanism.
For orthogonal pairs that lack inherent editing domains, one strategy is to introduce or engineer editing functions de novo. This could involve:
- Domain Swapping: Fusing editing domains from related aaRSs onto orthogonal aaRSs.
- Active Site Engineering: Redesigning the aminoacylation active site to improve initial discrimination against canonical amino acids, reducing the burden on downstream editing.
- tRNA Engineering: Modifying the orthogonal tRNA structure to make it a poorer substrate for misacylation by endogenous host aaRSs, thereby improving orthogonality [23].
Table 2: Comparison of aaRS Editing Behaviors Under Different Conditions
| aaRS / System | Condition / Manipulation | Effect on Editing & Fidelity | Experimental Evidence |
|---|---|---|---|
| ThrRS (E. coli) [65] | Oxidative Stress (H₂O₂) |
Inactivated editing due to oxidation of critical editing-site cysteine (C182); leads to Ser misincorporation at Thr codons. | Mass spectrometry on proteome; reporter assays; growth defects in protease-deficient strains. |
| PheRS (Salmonella) [65] | Oxidative Stress (H₂O₂) |
Enhanced editing via oxidation-induced structural changes; protects against m-Tyr and p-Tyr misincorporation. | Cryo-EM structures showing enlarged editing pocket; in vitro and in vivo fitness assays. |
| ProRS (E. coli) [61] | Genetic ablation of post-transfer editing domain | Severe mischarging with alanine; pre-transfer editing alone is insufficient for fidelity. | In vitro kinetics measuring Ala-tRNA^Pro formation; AMP hydrolysis assays. |
| M. mobile LeuRS [64] | Natural genomic deletion of CP1 editing domain | Constitutive mistranslation (e.g., Val, Met incorporated at Leu codons); generates a statistical proteome. | Mass spectrometry analysis of cellular proteome; heterologous expression in E. coli. |
| OrthoRep-evolved aaRS [28] | Continuous directed evolution with dual selection | High ncAA fidelity and efficiency; emergence of autoregulatory mechanisms to minimize leakiness. | Ratiometric fluorescence reporter (RRE); ncAA-dependent cell growth and protein synthesis. |
Combating misacylation is a central challenge in the maturation of genetic code expansion technologies. The strategies outlined here—employing sensitive analytical methods to quantify fidelity and leveraging powerful directed evolution platforms for engineering—provide a robust toolkit for researchers. The field is moving beyond simply achieving ncAA incorporation towards optimizing the entire system for high efficiency, orthogonality, and compatibility with host cells [23] [43]. Future efforts will likely focus on integrating computational design with high-throughput screening, engineering the broader cellular environment (e.g, ribosomes, elongation factors) to better accommodate orthogonal translation, and developing even more sophisticated in vivo evolution systems. By systematically exploiting and engineering aaRS editing activities, we can unlock the full potential of genetic code expansion for drug development, synthetic biology, and fundamental biological research.
The development of genomically recoded organisms (GROs) represents a paradigm shift in synthetic biology, enabling unprecedented expansion of the genetic code for biotechnology and therapeutic applications. This field leverages system-wide optimization of translational components—including elongation factors, ribosomes, and orthogonal translation systems (OTSs)—to overcome the inherent limitations of the canonical genetic code. By engineering these complex biological systems, researchers can create cellular platforms for producing novel protein chemistries with applications in drug development, biomaterials, and fundamental biological research. The integration of tRNA duplication research provides critical insights into the coordination required for efficient re-assignment of codon function, ensuring high fidelity in the incorporation of non-standard amino acids (nsAAs) into proteins.
Recent breakthroughs have demonstrated that successful genome recoding requires a holistic approach that addresses the interconnected nature of translational components. The ribosome, a complex macromolecular machine, coordinates with elongation factors and tRNAs to maintain the balance between rate and fidelity in protein synthesis, with in vivo synthesis rates of 15–20 amino acids per second and an error rate below ~10⁻⁴ [66]. System-wide optimization must therefore consider the dynamic conformational changes and kinetic proofreading mechanisms that govern translational accuracy while implementing large-scale genomic edits to reassign codon functions.
Engineering Elongation Factors for Enhanced Orthogonal Translation
Functional Roles of Native Elongation Factors
In prokaryotic systems, elongation factors EF-Tu and EF-G play essential, complementary roles in the protein synthesis cycle. EF-Tu forms ternary complexes with aminoacyl-tRNAs (aa-tRNAs) and GTP, delivering these substrates to the ribosomal A-site during translation. Following GTP hydrolysis triggered by correct codon-anticodon recognition, EF-Tu dissociates, allowing aa-tRNA accommodation and peptide bond formation [66] [67]. EF-G then catalyzes the translocation of the mRNA-tRNA complex, resetting the ribosome for the next elongation cycle [67]. These factors operate through sophisticated conformational changes that are tightly coupled to the ribosome's functional state, effectively "sensing" the status of tRNAs during translation [67].
Structural studies reveal that EF-G mimics the shape of the ternary complex, with domains III, IV, and V adopting a configuration that resembles the EF-Tu•tRNA complex [67]. This molecular mimicry enables EF-G to drive translocation by displacing tRNAs from the ribosomal A-site. The GTPase activities of both factors are critically regulated by ribosomal components, with GTP hydrolysis preceding the actual movement of tRNAs and mRNA during translocation [67]. Understanding these native mechanisms provides the foundation for engineering orthogonal translation systems that incorporate non-standard amino acids.
Engineering Elongation Factors for Orthogonal Systems
Orthogonal translation systems require specialized elongation factors that can accommodate the unique structural and chemical properties of non-standard amino acids while maintaining orthogonality to endogenous translation machinery. The development of a phosphoserine incorporation system (pSerOTS) exemplifies this approach, where researchers engineered a modified elongation factor (EF-pSer) specifically designed to accommodate the bulky, negative charge of phosphoserine [68]. This engineered factor enhances the delivery of pSer-tRNA^pSer^ to the ribosome and significantly improves overall OTS efficiency compared to native elongation factors [68].
System-wide analysis of OTS-host interactions has revealed that engineering efforts must address the metabolic burden and cellular stress responses induced by heterologous expression of orthogonal components. Studies monitoring growth lag time, specific growth rate, growth efficiency, and cell size distribution demonstrated that OTS expression can cause a ~2-fold reduction in growth rate and efficiency, with a ~3-fold increase in lag time [68]. These physiological impacts highlight the importance of optimizing elongation factor expression and function within the broader context of cellular physiology.
Table 1: Engineering Strategies for Elongation Factors in Orthogonal Translation Systems
| Engineering Target | Approach | Effect on OTS Performance |
|---|---|---|
| Substrate Binding Pocket | Modify structure to accommodate bulky/charged nsAAs | Enhanced delivery of nsAA-tRNAs to ribosome |
| Expression Level | Optimize using constitutive, low-level promoters | Reduced metabolic burden and cellular stress |
| GTPase Activity | Fine-tune interaction with ribosomal factors | Improved kinetics of nsAA incorporation |
| Orthogonality | Reduce affinity for native tRNAs | Minimized interference with host translation |
Experimental Protocol: Optimization of Orthogonal Elongation Factors
Protocol 1: Engineering Elongation Factors for Non-Standard Amino Acid Incorporation
Materials:
- Plasmid system with tunable promoters (e.g., pSerOTS variants)
- GRO strain (e.g., C321.ΔA or rEcΔ2.ΔA)
- Site-directed mutagenesis kit
- Analytics: HPLC for nsAA incorporation assessment, growth rate monitoring equipment
Procedure:
- Clone gene encoding orthogonal elongation factor (e.g., EF-pSer) into expression vector with tunable promoter system (e.g., glnS promoter)
- Generate variant library through site-directed mutagenesis targeting substrate-binding regions
- Transform variants into appropriate GRO strain (e.g., C321.ΔA for UAG reassignment)
- Assess growth characteristics (lag time, specific growth rate, maximum density) to identify variants with reduced metabolic burden
- Measure nsAA incorporation efficiency and fidelity using reporter assays with internal UAG or UGA codons
- For superior variants, perform kinetic analysis of incorporation efficiency versus native amino acids
- Validate orthogonality by proteomic analysis of host cell proteome for misincorporation events
Troubleshooting:
- If cellular toxicity is observed, reduce expression level using weaker promoters
- If misincorporation occurs, perform additional rounds of mutagenesis to enhance specificity
- If incorporation efficiency is low, optimize expression of corresponding orthogonal tRNA
Ribosome Engineering and tRNA Optimization
Ribosome Dynamics in Translation Fidelity
The ribosome undergoes sophisticated large-scale conformational changes during protein synthesis that are essential for maintaining translational fidelity. Cryo-electron microscopy studies have revealed a ratchet-like rotation between ribosomal subunits that accompanies the transition from classic to hybrid states of tRNA binding, facilitating the coordinated movement of tRNAs through the ribosome [66]. These rearrangements are particularly important during the translocation step, where any error would result in loss of the reading frame. The ribosome employs an induced-fit mechanism to discriminate between cognate and near-cognate tRNAs, with structural changes occurring only when correct codon-anticodon pairing is recognized [66].
The ribosome's kinetic proofreading mechanism involves two discrimination steps—initial selection and proofreading—separated by the irreversible step of GTP hydrolysis [66]. This two-step selection process allows the ribosome to sample the energy landscape twice, significantly enhancing selectivity. Pre-steady-state kinetic studies demonstrate that discrimination relies mainly on differences in GTPase activation (k₃) and tRNA accommodation (k₅) rates between cognate and near-cognate species [66]. These fundamental mechanisms must be considered when engineering ribosomes for expanded genetic codes.
tRNA Engineering and Abundance Analysis
tRNAs serve as the physical link between mRNA codons and their corresponding amino acids, making them essential components for genetic code expansion. Recent advances in tRNA analysis, particularly Nano-tRNAseq, have enabled quantitative assessment of tRNA abundance and modification dynamics in a single experiment [17]. This nanopore-based approach sequences native tRNA populations, providing insights into the complex relationship between tRNA modifications and decoding preferences. tRNA modifications, averaging 13 modifications per tRNA molecule, can significantly impact translational efficiency and fidelity by affecting tRNA stability, aminoacylation capability, and codon-anticodon interactions [17].
The development of orthogonal tRNA systems requires careful consideration of both sequence and modification patterns. Engineering efforts must address translational crosstalk between orthogonal and native systems, which can lead to misincorporation and reduced fidelity. Research has shown that tRNA modifications at position 34 of the anticodon directly influence wobbling capacity, thereby changing the set of "preferred" or "optimal" codons [17]. System-wide optimization requires comprehensive analysis of tRNA populations and their modification states under different growth conditions and stress responses.
Table 2: tRNA Engineering Parameters for Genetic Code Expansion
| Parameter | Impact on Translation | Optimization Strategy |
|---|---|---|
| Anticodon Sequence | Determines codon recognition | Engineer for reassigned codons (UAG, UGA) |
| Modification Profile | Affects decoding accuracy and efficiency | Co-express modification enzymes |
| Cellular Abundance | Influences incorporation efficiency | Tunable expression systems |
| Aminoacylation | Critical for orthogonality | Engineer orthogonal aaRS specificity |
| Wobble Position | Alters codon recognition range | Modify position 34 and corresponding enzymes |
Experimental Protocol: Nano-tRNAseq for tRNA Modification Analysis
Protocol 2: Comprehensive tRNA Characterization Using Nanopore Sequencing
Materials:
- Native tRNA samples (minimum 1μg)
- Nanopore RNA CS (RCS) and RNA DX (RDX) adapters
- T4 DNA ligase
- Nanopolish software package
- Oxford Nanopore sequencer (MinION or PromethION)
Procedure:
- Extract total RNA using phenol-chloroform method with special care to preserve small RNAs
- Ligate 5' RNA adapter to tRNA 3' CCA overhang using T4 DNA ligase
- Ligate 3' DNA adapter complementary to the 5' RNA adapter
- Prepare sequencing library according to ONT direct RNA sequencing protocol with modified settings
- Sequence using MinION or PromethION flow cell with 72-hour run time
- Re-process raw current intensity signals to recover discarded tRNA reads
- Map reads to reference genome using minimap2 with relaxed parameters (-ax map-ont -k5)
- Analyze modification patterns using Nanopolish and custom scripts
- Correlate modification changes with tRNA abundance and codon usage patterns
Troubleshooting:
- If sequencing yields are low, optimize adapter ligation efficiency
- If mapping rates are poor, further relax mapping parameters and use custom tRNA reference databases
- For modification detection, validate with LC-MS/MS on selected tRNA species
Development and Optimization of Genomically Recoded Organisms
Design Principles and Construction of GROs
Genomically recoded organisms are engineered with alternative genetic codes in which redundant codons are reassigned to new functions. The foundational approach involves whole-genome codon replacement followed by deletion of the corresponding translation factors. The first GRO construction replaced all 321 known UAG stop codons in E. coli MG1655 with synonymous UAA codons, enabling deletion of release factor 1 (RF1) and reassignment of UAG translation function [69]. This pioneering work demonstrated that GROs exhibit improved properties for incorporating non-standard amino acids and increased resistance to bacteriophage infection [69].
Recent advances have produced more extensively recoded organisms. The "Ochre" GRO represents a landmark achievement, compressing redundant codon functionality into a single codon through replacement of 1,195 TGA stop codons with TAA in a ΔTAG E. coli background [40]. This engineering feat required multi-phase genome editing using multiplex automated genome engineering (MAGE) and conjugative assembly genome engineering (CAGE) to implement thousands of precise genomic changes [40]. The resulting organism utilizes UAA as the sole stop codon, with UGG encoding tryptophan and both UAG and UGA reassigned for incorporation of distinct non-standard amino acids with greater than 99% accuracy [40] [70].
System-Wide Optimization in GROs
Successful GRO development requires system-wide optimization to address the complex interactions between orthogonal components and native cellular processes. Studies of orthogonal translation systems in GRO backgrounds have revealed significant OTS-mediated cytotoxicity resulting from off-target interactions with host translational machinery [68]. System-level analysis of host proteomes in response to OTS expression shows dysregulation of stress response pathways and global metabolic burden caused by elements of episomal vectors [68].
Optimization strategies include modifying plasmid copy number through origin of replication selection, tuning expression levels using constitutive promoters, and engineering OTS components for enhanced orthogonality [68]. Research demonstrates that OTS component expression can decrease host cell fitness through multiple parameters: o-aaRS-specific perturbations in energy metabolism and o-tRNA-dependent reductions in the fidelity of host protein biosynthesis [68]. These findings highlight the importance of comprehensive characterization of OTS-host interactions when implementing genetic code expansion in GROs.
Table 3: System-Wide Optimization Parameters for GRO Development
| System Component | Optimization Parameter | Impact on GRO Performance |
|---|---|---|
| Genetic Code | Number of reassigned codons | Increased nsAA incorporation sites |
| Orthogonal Systems | tRNA/aaRS/EF specificity | Reduced cross-talk with native translation |
| Host Physiology | Metabolic burden | Improved growth and protein yield |
| Genetic Isolation | Viral resistance | Biocontainment and manufacturing stability |
| Cellular Stress | Stress response activation | Enhanced OTS stability and function |
Experimental Protocol: GRO Construction via Multiplex Genome Editing
Protocol 3: Whole-Genome Recoding Using MAGE and CAGE
Materials:
- E. coli strain C321.ΔA (ΔTAG precursor)
- MAGE oligonucleotide library for TGA→TAA conversion
- Counter-selection markers (e.g., tolC)
- Conjugative assembly strains
- Whole-genome sequencing capabilities
Procedure: Phase 1: Essential Gene Recoding
- Design oligonucleotides for 71 essential genes terminating with TGA
- Divide targets between two genomic subdomains (A' and B') across two clones
- Perform iterative MAGE cycles targeting distinct genomic regions
- Screen for successful conversions using multiplex allele-specific PCR
- Assemble recoded subdomains via CAGE to create intermediate strain rEcΔ2E.ΔA
- Validate essential gene function and cellular viability
Phase 2: Genome-Wide Recoding
- Design oligonucleotides for 1,012 additional ORFs terminating with TGA
- Implement refactoring strategies for 380 overlapping ORFs where single-nucleotide substitutions might affect neighboring gene expression
- Perform concurrent MAGE targeting across eight distinct genomic subdomains (A-H) split among rEcΔ2E.ΔA clones
- Delete 229 non-essential ORFs containing TGA via marker placement
- Assemble fully recoded genome through sequential CAGE steps
- Verify complete TGA removal and correct ORF refactoring via whole-genome sequencing
Phase 3: Translation Factor Engineering
- Engineer release factor 2 (RF2) to attenuate UGA recognition while preserving UAA termination
- Modify tRNATrp to reduce near-cognate suppression at UGA codons
- Introduce orthogonal aminoacyl-tRNA synthetases for UAG and UGA reassignment
- Optimize expression levels of engineered factors to minimize metabolic burden
Troubleshooting:
- If recoding efficiency is low, optimize MAGE conditions and oligonucleotide design
- If assembly fails at CAGE steps, identify and resolve toxic combinations of edits
- If growth impairment occurs after recoding, adapt cultivation conditions and identify compensatory mutations
Application Notes and Implementation Guidelines
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for GRO Development and Genetic Code Expansion
| Reagent/Cell Line | Function/Application | Key Features |
|---|---|---|
| C321.ΔA E. coli | First-generation GRO with all UAG codons replaced | ΔprfA (RF1 deletion); enables UAG reassignment |
| Ochre GRO (rEcΔ2.ΔA) | Second-generation GRO with single stop codon | ΔTAG/ΔTGA; UAA sole stop; UAG/UGA for nsAAs |
| pSerOTS System | Phosphoserine incorporation at stop codons | pSerRS, tRNA^pSer^, EF-pSer; orthogonal system |
| MAGE/CAGE System | Multiplex genome editing | Enables large-scale, precise genomic modifications |
| Nano-tRNAseq Protocol | tRNA abundance/modification analysis | Simultaneous quantification of tRNA features |
Implementation Strategies for Industrial and Therapeutic Applications
The development of GRO platforms enables innovative approaches to biomanufacturing and therapeutic protein production. GRO-based systems offer enhanced capabilities for producing programmable protein biologics with reduced immunogenicity and extended half-life [70]. These properties are particularly valuable for biopharmaceutical applications, where controlling protein stability and immune recognition is critical. The Ochre GRO platform provides a foundation for constructing multi-functional biologics containing multiple distinct non-standard amino acids with site-specific precision [40] [70].
For industrial implementation, GROs provide inherent biocontainment and viral resistance by creating genetic isolation from natural organisms and viruses [69] [71]. This genetic isolation addresses crucial safety concerns in biomanufacturing while reducing susceptibility to viral contamination in production facilities. Additionally, the reassignment of multiple codons enables the synthesis of proteins with novel chemical properties not achievable with the standard 20 amino acids, opening possibilities for advanced biomaterials with enhanced conductivity, stability, or catalytic functions [40] [70].
Visualizing System Relationships and Experimental Workflows
Diagram 1: GRO Development and Optimization Workflow
Diagram 2: Translation System Optimization Network
Application Note
This application note addresses a central challenge in genetic code expansion (GCE) research: the cellular toxicity and fitness costs imposed by introducing orthogonal translation systems. The duplication of tRNAs, while essential for creating new coding capacity, can disrupt native cellular processes. We provide a structured framework, backed by recent quantitative studies, to measure, understand, and mitigate these detrimental effects, enabling more robust and efficient GCE implementations.
Quantitative Framework for Fitness Costs
The fitness cost of genetic alterations, including gene duplications essential for GCE, is not random but follows predictable patterns. Key determinants identified through comparative modeling in yeast models include:
Table 1: Molecular Determinants of Gene Duplication Fitness Costs
| Determinant | Effect on Fitness | Experimental Evidence |
|---|---|---|
| Cumulative Single-Gene Duplication Cost | Primary driver of aneuploidy toxicity; explains 74-94% of growth rate variance [72]. | Measured by profiling growth rates of strains with single-gene duplications from a genomic library [72]. |
| tRNA Gene Duplication | Beneficial; can improve fitness and partially compensate for deficits [72] [73]. | Deletion of specific tRNA genes in mice reduced total tRNA levels and impaired development; increased expression of other tRNAs provided compensatory buffering [73]. |
| snoRNA Gene Duplication | Deleterious; worsens the fitness cost of aneuploidy [72]. | Modeling shows snoRNA duplication contributes negatively to growth rate [72]. |
| Gene Length | Best predictor of deleterious gene duplications; longer genes confer higher cost [72]. | Machine learning analysis of properties affecting duplication toxicity [72]. |
These findings indicate that the fitness impact of introducing orthogonal tRNA systems is multi-factorial. The "copy number" of tRNA genes is particularly critical, as a multi-copy configuration is required to buffer translation and ensure viability in mammals [73].
Core Experimental Protocols for Assessment and Mitigation
Protocol 1: Quantifying Fitness Cost of Orthogonal System Components
Objective: To empirically measure the fitness burden imposed by candidate orthogonal tRNAs/RS pairs before full system integration.
- Step 1: Generate Duplication Panel. Create a library of host strains (e.g., S. cerevisiae) individually harboring duplications of the candidate orthogonal tRNA genes. Include controls with empty vectors and essential native tRNAs.
- Step 2: Growth Rate Assay. Inoculate strains in triplicate in appropriate liquid medium. Use a microplate reader to monitor optical density (OD600) over 24-48 hours.
- Key Parameters: Calculate maximum growth rate (μmax) and doubling time from the exponential phase.
- Step 3: Competitive Fitness Assay. Mix each test strain with a genetically marked wild-type reference strain at a 1:1 ratio. Co-culture for ~20 generations. Use flow cytometry or selective plating to determine the ratio of test to reference cells at the start and end of the competition.
- Calculation: The selection rate coefficient (s) is calculated as
s = ln[(F_test_end / F_ref_end) / (F_test_start / F_ref_start)] / generations, where F is the frequency. A negative (s) indicates a fitness cost.
- Calculation: The selection rate coefficient (s) is calculated as
- Step 4: Data Integration. Relate the measured fitness costs to gene-specific properties (e.g., length, function) to build a predictive model for future component selection [72].
Protocol 2: Assessing tRNA Expression and Function In Situ
Objective: To verify the expression and aminoacylation status of orthogonal tRNAs within the host and ensure they do not deplete native tRNA pools.
- Step 1: Cell Lysis and RNA Extraction. Harvest cells expressing the orthogonal system. Use acidic phenol-chloroform extraction to enrich for small RNAs, including tRNAs.
- Step 2: Northern Blotting.
- Separate total RNA on a denaturing urea-polyacrylamide gel.
- Transfer to a nylon membrane.
- Hybridize with DNA probes specific to the orthogonal tRNA's unique sequence and to essential native tRNAs (e.g., tRNA-Phe).
- Analysis: Quantify band intensity. Reduced levels of native tRNAs suggest competitive interference. This method directly assessed tissue-specific tRNA abundance in tRNA-Phe knockout mice [73].
- Step 3: tRNA Charging Status (Optional).
- Use acid-urea polyacrylamide gel electrophoresis, where charged (aminoacylated) tRNAs migrate slower than uncharged tRNAs, to determine the efficiency of orthogonal tRNA charging.
- Step 4: RT-qPCR for tRNA Quantification. For higher throughput, use a protocol based on regular reverse transcription followed by quantitative PCR (RT-qPCR) to estimate the relative levels of specific tRNA species [74]. Note: This method may not distinguish between charged/uncharged or modified/unmodified tRNAs.
Conceptual Framework for Toxicity Mitigation
The following workflow outlines the logical process for designing, implementing, and optimizing a GCE system with minimal fitness cost, integrating the protocols and data above.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Implementing and Assessing GCE Systems
| Research Reagent | Function / Rationale | Considerations |
|---|---|---|
| Orthogonal tRNA/RS Pair | The core engine for ncAA incorporation; must not cross-react with host aaRSs or tRNAs [38]. | Select for minimal gene length and high orthogonality to reduce intrinsic fitness cost [72]. |
| tRNA Gene Knockout Strains | Models to study how loss of specific endogenous tRNAs affects fitness and if orthogonal tRNAs can compensate [73]. | Useful for validating that your orthogonal system does not exacerbate native tRNA deficits. |
| CRISPR-Cas9 System | Enables precise genomic editing for creating knockout models or for stable genomic integration of orthogonal components [73]. | Genomic integration can offer more stable expression and reduce plasmid-borne fitness costs. |
| RT-qPCR Kit for tRNAs | A convenient method to quantify relative levels of individual tRNA species after orthogonal system introduction [74]. | Cannot distinguish between charged/uncharged tRNAs; Northern blotting is required for this. |
| Non-Canonical Amino Acid (ncAA) | The target novel monomer for incorporation. Must be cell-permeable, non-toxic, and stable within the cellular environment [38]. | Toxicity or poor uptake of the ncAA itself can be a major source of observed fitness costs. |
Proof of Concept: Validating Systems and Comparing Therapeutic Platforms
Genetic code expansion (GCE) technology enables the site-specific incorporation of non-canonical amino acids (ncAAs) into proteins, providing powerful tools for probing biological function and engineering novel protein therapeutics [38]. This approach relies on engineered orthogonal aminoacyl-tRNA synthetase/tRNA pairs (OTSs) that charge ncAAs onto tRNAs that recognize blank codons, most commonly the amber stop codon (UAG) [28] [23]. Central to the successful implementation of any GCE system is the rigorous biochemical validation of two critical parameters: incorporation efficiency, which measures the yield of full-length target protein containing the ncAA relative to wild-type protein, and incorporation fidelity, which quantifies the ratio of ncAA incorporation versus mis-incorporation of canonical amino acids at the target site [38]. This Application Note provides detailed protocols for the quantitative assessment of these essential parameters, framed within emerging research on tRNA gene duplication and evolution [21].
Key Validation Parameters and Their Quantitative Assessment
Defining and accurately measuring efficiency and fidelity is fundamental for comparing and optimizing GCE systems. The table below outlines the core validation metrics, their definitions, and standard assessment methodologies.
Table 1: Key Validation Parameters for ncAA Incorporation Systems
| Parameter | Definition | Common Assessment Methods |
|---|---|---|
| Efficiency | Yield of full-length ncAA-containing protein compared to wild-type protein produced under identical conditions [38]. | - Western blot quantification- Mass spectrometry (MS) of full-length protein- Fluorescent reporter assays (e.g., RFP/GFP ratios) [28] |
| Fidelity | Ratio of ncAA incorporation versus mis-incorporation of canonical amino acids at the target codon [38]. | - Tandem MS (LC-MS/MS) to detect mis-incorporated residues- Negative selection in absence of ncAA [28] |
| Orthogonality | Specificity of the OTS for its cognate tRNA and ncAA without cross-reactivity with endogenous host tRNAs, aaRSs, or canonical amino acids [23]. | - Growth-based assays in auxotrophic strains- Proteomic analysis for global mis-incorporation |
| Permissivity | Ability of a single engineered OTS to incorporate a variety of different ncAAs, which can be advantageous for certain applications [38]. | - Testing incorporation of multiple structurally similar ncAAs using the same reporter system |
Research Reagent Solutions for GCE Validation
A successful validation workflow requires specific genetic constructs, reagents, and analytical tools. The following table details essential components for establishing and assessing ncAA incorporation.
Table 2: Essential Research Reagents and Materials
| Reagent/Material | Function in Validation | Examples & Notes |
|---|---|---|
| Orthogonal aaRS/tRNA Pair | Charges the ncAA onto the orthogonal tRNA that decodes the blank codon [28]. | - PylRS/tRNAPyl from Methanosarcina species [75]- EcTyrRS/tRNA pair from E. coli |
| Reporter Construct | Provides a quantifiable readout for incorporation efficiency and fidelity. | - RFP-GFP with interceding amber codon (e.g., RXG reporter) [28]- Amber-containing therapeutic protein of interest |
| Non-Canonical Amino Acid (ncAA) | The novel chemical moiety to be incorporated into the protein. | - Must be cell-permeable, non-toxic, and stable in culture [38]- e.g., Nε-acetyl-lysine (AcK) [75] |
| Stable Cell Line | Ensures homogeneous and reproducible expression of the OTS and target protein [75]. | - HEK293, CHO, or mouse ES cells with genomically integrated OTS (e.g., via PiggyBac) [75] |
| Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) | The gold-standard method for confirming ncAA incorporation and identifying mis-incorporation events with high sensitivity and specificity. | - Used for analyzing intact proteins and tryptic peptides |
Experimental Protocols
Protocol 1: Assessing Incorporation Efficiency Using a Ratiometric Fluorescent Reporter
This protocol uses a dual-fluorescent reporter system to quantitatively measure ncAA incorporation efficiency in living cells, providing a rapid and reliable screening method [28].
Workflow Overview:
Materials:
- Plasmid encoding ratiometric RXG reporter (RFP-TAG-GFP) [28]
- Plasmid(s) encoding orthogonal aaRS/tRNA pair
- Appropriate host cells (e.g., HEK293, S. cerevisiae)
- ncAA stock solution
- Cell culture media and transfection reagents
- Flow cytometer or fluorescence plate reader
Procedure:
- Construct the Ratiometric Reporter: Use a plasmid where RFP and GFP are separated by a linker containing a single amber stop codon. A control plasmid with a sense codon (e.g., RYG) is essential for normalization [28].
- Cell Transfection: Co-transfect the host cells with the RXG reporter plasmid and the plasmids for the orthogonal aaRS/tRNA pair.
- Cell Culture and Induction: Divide the transfected cells into two populations. Culture one population in media supplemented with the target ncAA, and the other in media without the ncAA. Allow sufficient time for protein expression (e.g., 24-48 hours).
- Fluorescence Measurement: Harvest the cells and measure the RFP and GFP fluorescence intensities for each sample using a flow cytometer or fluorescence plate reader.
- Data Analysis and Calculation: Calculate the Relative Readthrough Efficiency (RRE) as follows [28]:
- For each sample, compute the GFP/RFP fluorescence ratio.
- RRE = (GFP/RFP for RXG with ncAA) / (GFP/RFP for RYG control)
- Incorporation efficiency is directly proportional to the RRE value. A high RRE in the presence of ncAA indicates high incorporation efficiency.
Protocol 2: Confirming Fidelity and Identity via Mass Spectrometry
This protocol provides definitive confirmation of site-specific ncAA incorporation and detects potential mis-incorporation of canonical amino acids, serving as the cornerstone for validating fidelity [38].
Workflow Overview:
Materials:
- Purified target protein containing the ncAA
- Trypsin or other proteolytic enzyme
- LC-MS/MS system
- C18 reverse-phase chromatography column
- Standard solvents for LC-MS (water, acetonitrile, formic acid)
Procedure:
- Protein Expression and Purification: Express the target protein containing an amber codon at the desired site in the presence of the ncAA and the orthogonal OTS. Purify the protein using standard chromatography methods (e.g., affinity, SEC).
- Intact Protein Mass Analysis: Analyze the purified protein using LC-MS under soft ionization conditions. The observed mass of the intact protein should match the theoretical mass of the protein containing the desired ncAA, providing the first confirmation of successful incorporation.
- Proteolytic Digestion: Denature the purified protein and digest it with a sequence-specific protease like trypsin. This should generate a peptide fragment containing the ncAA incorporation site.
- LC-MS/MS Analysis: Separate the resulting peptides using reverse-phase liquid chromatography and analyze them with tandem mass spectrometry. The MS1 chromatogram should show a peak for the target peptide with a mass shift corresponding to the ncAA.
- Data Analysis: Isolate the target peptide ion for fragmentation (MS/MS). The resulting fragmentation spectrum should provide sequence coverage confirming the identity of the peptide and the site of incorporation. The absence of a peptide with the same retention time and mass corresponding to a canonical amino acid at that position is a key indicator of high fidelity.
Troubleshooting and Data Interpretation
Addressing Low Efficiency:
- Optimize OTS Expression: Ensure adequate expression levels of the orthogonal tRNA and aaRS. Multi-copy tRNA expression cassettes can significantly enhance incorporation in mammalian cells [75].
- Check ncAA Delivery: Verify the ncAA is cell-permeable, stable in the media, and used at a non-toxic, effective concentration.
- Use Evolved OTSs: Consider using directed evolution platforms like OrthoRep in yeast to generate aaRS variants with enhanced activity and specificity for the target ncAA [28].
Addressing Low Fidelity (Mis-incorporation):
- Stringent Negative Selection: Employ selection strategies that kill or inhibit cells that exhibit readthrough of the amber codon in the absence of the ncAA. This enriches for OTSs that are strictly dependent on the ncAA [28].
- Engineer aaRS Specificity: The active site of the aaRS can be engineered to more strictly exclude canonical amino acids, particularly the one whose mis-incorporation is observed (often tyrosine or lysine for amber suppression) [23].
- Utilize Genomically Recoded Organisms (GROs): For prokaryotic systems, using GROs where all amber stop codons have been replaced with ochre codons eliminates competition with release factor 1, reducing background and improving fidelity [23].
Genetic code expansion (GCE) technology enables the site-specific incorporation of unnatural amino acids (UAAs) into proteins, creating opportunities to engineer proteins with novel properties for therapeutic and basic research [14]. The core of this technology relies on orthogonal aminoacyl-tRNA synthetase/tRNA (aaRS/tRNA) pairs—components that must function without cross-reacting with the host's endogenous translation machinery [14] [6]. Maintaining high fidelity in UAA incorporation is paramount, as even low levels of misincorporation can compromise experimental results and therapeutic protein quality [76]. This application note examines the crystallographic and structural evidence underlying orthogonal system function, detailing the molecular mechanisms that enable and sometimes compromise orthogonality, and provides validated protocols for enhancing translational fidelity in GCE experiments.
Structural Principles of tRNA Orthogonality
The Conserved Architecture of tRNA
Transfer RNA (tRNA) molecules adopt a highly conserved L-shaped three-dimensional structure that is critical for their function in translation. This structure consists of two primary domains:
- Acceptor Domain: Formed by the stacking of the acceptor stem (AAS) and the T-stem loop (T-arm). This region includes the 3′ CCA sequence where the amino acid is covalently attached [77] [14].
- Anticodon Domain: Comprising the D-stem loop (D-arm) and the anticodon stem loop (ASL). This domain is responsible for mRNA codon recognition on the ribosome [77] [14].
These two domains converge at the tRNA elbow region, a critical structural feature stabilized by conserved nucleotide interactions between the D- and T-arms [77]. The characteristic L-shape was first revealed through X-ray crystallography of yeast tRNAPhe in 1974 and has since been confirmed as a universal feature across nearly all organisms [14].
Identity Elements and Molecular Recognition
The specificity of tRNA aminoacylation is governed by identity elements—specific nucleotides and structural features that are recognized by cognate aminoacyl-tRNA synthetases (aaRS) [14]. These identity elements vary between different tRNA species and across evolutionary domains, providing the structural basis for orthogonality:
Table 1: Key Identity Elements in tRNA Recognition
| Identity Element | Location | Function in Recognition |
|---|---|---|
| Acceptor Stem Base Pairs | Positions 1-72 | Major determinant for many aaRS; differs between archaeal and bacterial systems |
| Discriminator Base | Position 73 | Critical for specific recognition by many aaRS |
| Anticodon Nucleotides | Anticodon Loop | Recognized by most aaRS (except SerRS, AlaRS, LeuRS, PylRS) |
| Variable Loop | Between Anticodon & T arms | Length and structure provide distinguishing features for specific tRNAs |
The strategic exploitation of these divergent identity elements enables the creation of orthogonal pairs. For instance, the Methanocaldococcus jannaschii tyrosyl-tRNA synthetase (MjTyrRS) recognizes a C1:G72 base pair in its cognate tRNA, while most bacterial aaRS recognize a G1:C72 base pair [76]. This fundamental difference in recognition patterns forms the structural basis for developing orthogonal systems that can be imported into non-native hosts.
Crystallographic Evidence of Orthogonal System Function
Structural Basis of Orthogonality and Its Limitations
Crystallographic studies have revealed how orthogonal systems maintain their specificity in non-native hosts. The M. jannaschii tyrosyl-tRNA synthetase/tRNA pair exhibits orthogonality in E. coli because its identity elements (C1:G72 and A73) are distinct from those recognized by most endogenous E. coli aaRS [76]. However, these structural investigations have also uncovered a significant limitation: the E. coli prolyl-tRNA family shares similar acceptor stem identity elements (C1:G72) with the archaeal Mj tRNATyr [76].
This structural similarity creates a molecular vulnerability where the evolved MjTyrRS (specific for p-acetylphenylalanine or pAcF) can misrecognize and misaminoacylate E. coli prolyl-tRNAs, leading to pAcF misincorporation at proline sites with a frequency of approximately 0.5% per proline codon [76]. In a protein with 22 proline residues, this misincorporation rate results in readily detectable impurities, as demonstrated by a +92 Da mass shift corresponding to pAcF-for-Pro substitution [76].
Structural Insights from Misincorporation Studies
The investigation of this misincorporation phenomenon provides compelling evidence for the importance of structural compatibility in orthogonal systems:
- Elimination Experiments: Removal of the Mj aaRS gene eliminated misincorporation, while deletion of the suppressor tRNACUA had no detectable effect, confirming the misaminoacylation originates from the aaRS [76].
- Conserved Structural Features: Beyond the acceptor stem identity elements, conserved nucleotides in the region between A14 and U18 and the short variable loop in both E. coli tRNAPro and Mj tRNACUA contribute to the misrecognition [76].
- Competition Rescue: Overexpression of the endogenous E. coli prolyl-tRNA synthetase (ProS) gene reduced misincorporation by competing with the archaeal aaRS for tRNAPro binding, demonstrating that orthogonality can be enhanced through manipulation of cellular components [76].
The following diagram illustrates the structural basis of this misincorporation and its resolution:
Experimental Protocols for Enhancing Orthogonal System Fidelity
Protocol: Assessing and Mitigating Misincorporation in Recombinant Proteins
Purpose: To identify and eliminate misincorporation of unnatural amino acids at canonical amino acid sites in recombinant proteins expressed via genetic code expansion.
Materials:
- E. coli expression strain with orthogonal aaRS/tRNA system
- Plasmid encoding target protein with amber stop codon at desired position
- Unnatural amino acid (e.g., p-acetylphenylalanine)
- Proline-enriched media or NaCl for osmotic stress control
- Plasmid for ProS overexpression (e.g., pProS)
Procedure:
- Protein Expression and Purification:
- Transform expression strain with target plasmid and orthogonal pair plasmid.
- Induce expression in media supplemented with UAA.
- Purify recombinant protein using affinity chromatography.
Misincorporation Detection:
- Perform intact protein mass analysis to detect mass shifts (+92 Da for pAcF at Pro).
- Conduct tryptic digest and peptide mapping by RP-HPLC to identify modification sites.
- Compare mass spectra across multiple peptides to confirm widespread vs. site-specific modifications.
Confirmation Experiments:
- Express wild-type protein (without amber codon) in presence of orthogonal pair.
- Assess if +92 Da species persists, indicating misincorporation independent of amber suppression.
- Test different growth conditions (media composition, temperature) to evaluate effects on misincorporation frequency.
Mitigation Strategies:
- Co-express endogenous ProS gene on high-copy plasmid.
- Evaluate misincorporation reduction via intact mass analysis.
- For persistent issues, consider genomic manipulations to increase intracellular ProS:tRNAPro ratio.
Troubleshooting:
- If misincorporation increases under osmotic stress (0.8 M NaCl), focus on ProS overexpression.
- If misincorporation is media-dependent, optimize growth conditions to reduce effect.
- Multiple +92 Da modifications across peptides indicate widespread proline replacement rather than specific site issue.
Protocol: Structural Engineering of tRNA Orthogonality
Purpose: To engineer tRNA sequences with improved orthogonality and folding properties using computational and structure-guided approaches.
Materials:
- tRNA sequence analysis software (RNAfold, Chi-T)
- Plasmid library for tRNA expression
- Reporter system for orthogonality assessment (e.g., GFP with amber codon)
- Cognate aaRS for orthogonal pair
Procedure:
- Computational Analysis:
- Input candidate tRNA sequences into RNAfold to predict minimum free energy (MFE) structures.
- Calculate frequency of cloverleaf structure in predicted ensemble.
- Identify sequences with >80% predicted cloverleaf structure, as these are strongly correlated with orthogonal activity.
Rational Design:
- Identify and modify non-orthogonal identity elements in acceptor stem, D-arm, and T-arm.
- Introduce 2-3 point mutations to stabilize cloverleaf folding while maintaining cognate aaRS recognition.
- Use Chi-T method to generate chimeric tRNAs from segmented parts of millions of isoacceptor sequences.
Experimental Validation:
- Clone engineered tRNAs into expression vectors.
- Co-express with cognate aaRS and GFP reporter containing amber codon at position 3.
- Measure fluorescence to assess suppression efficiency and orthogonality.
- Test engineered tRNAs in absence of cognate aaRS to confirm no mischarging by endogenous synthetases.
Orthogonality Optimization:
- For tRNAs with poor orthogonality, use directed evolution to introduce negative selection elements against endogenous aaRS.
- Employ RS-ID computational tool to identify synthetases that may acylate engineered tRNAs.
- Iterate between computational design and experimental validation until optimal orthogonality is achieved.
Table 2: Quantitative Assessment of Engineered tRNA Orthogonality
| tRNA Construct | Predicted Cloverleaf Frequency | Orthogonality Status | Suppression Efficiency | Misacylation by Endogenous aaRS |
|---|---|---|---|---|
| CsProtRNACUA (parent) | Low | Orthogonal Inactive | Minimal | None |
| CsProtRNACUAfix (engineered) | High | Orthogonal Active | 6x improvement | None |
| ApHistRNACUA (parent) | No unambiguous cloverleaf | Non-orthogonal Active | High | Lysine, Glutamine |
| ApHistRNACUAfix (engineered) | High | Non-orthogonal Active | Further activated | Lysine, Glutamine |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Orthogonal System Development
| Reagent / Tool | Function / Application | Example / Source |
|---|---|---|
| Orthogonal aaRS/tRNA Pairs | Base system for UAA incorporation | MjTyrRS/tRNA, PylRS/tRNA pairs |
| Chi-T Computational Tool | Automated generation of orthogonal tRNAs | Segments and reassembles tRNA parts from millions of sequences |
| RNAfold Software | Predicts tRNA secondary structure and folding stability | ViennaRNA Package |
| RS-ID Computational Tool | Identifies potential synthetases for engineered tRNAs | Complementary to Chi-T for orthogonal pair development |
| PtNTT2 Nucleotide Transporter | Enables import of unnatural triphosphates in SSOs | From Phaeodactylum tricornutum |
| Unnatural Base Pairs (UBPs) | Creates novel codons for genetic code expansion | dNaM-dTPT3 pair |
| Genomically Recoded Organisms (GROs) | Host organisms with reassigned codons for reduced cross-talk | E. coli with amber codon removed |
Implications for tRNA Duplication Research
The structural insights into orthogonal system function have significant implications for understanding tRNA gene evolution through duplication events. Research in plant species has revealed that tandem duplication of tRNA genes is a fundamental evolutionary force, with conserved sequence and structural features maintained across diverse species [21]. Notably, tandemly located tRNA gene pairs with anticodons to proline are widely distributed across 33 plant species, suggesting evolutionary conservation of this arrangement [21].
These findings connect to experimental observations in orthogonal systems, where the unique identity elements of prokaryotic tRNAPro molecules (C1:G72) make them vulnerable to misaminoacylation by archae-derived aaRS [76]. The conservation of these identity elements across species, maintained through duplication events, creates predictable patterns of molecular recognition that can be exploited or must be engineered around in synthetic biological systems.
The development of automated orthogonal tRNA generation tools like Chi-T, which leverages natural tRNA diversity created through evolutionary processes including duplication, demonstrates how understanding these fundamental evolutionary mechanisms directly enables advances in genetic code expansion technology [78].
The expansion of the genetic code through tRNA research has unveiled novel therapeutic strategies, among which the simultaneous targeting of multiple functional sites on essential enzymes represents a frontier in anti-infective drug development. Aminoacyl-tRNA synthetases (aaRSs), crucial for protein synthesis, have emerged as validated drug targets due to their conservation across pathogens and the presence of structurally distinct substrate-binding pockets. This case study explores the innovative "double drugging" approach applied to prolyl-tRNA synthetase (PRS), demonstrating how concurrent inhibition of neighboring substrate subsites can achieve potent anti-parasitic effects while potentially circumventing resistance mechanisms. The PRS enzyme features three adjacent subsites that bind its natural substrates: ATP, L-proline (L-pro), and the 3'-end of tRNAPro [79]. Traditional single-target inhibition faces limitations regarding efficacy and resistance emergence. However, recent advances demonstrate that simultaneous occupation of these subsites with multiple inhibitors creates a synergistic blocking mechanism, establishing a new paradigm for drug development against infectious diseases like toxoplasmosis and avian coccidiosis [79] [80].
Background and Significance
Prolyl-tRNA Synthetase as a Therapeutic Target
PRS catalyzes the covalent attachment of proline to its corresponding tRNA molecule, a critical step in protein synthesis. As a member of the aminoacyl-tRNA synthetase family, PRS is essential for translation fidelity and parasite survival [80]. The enzyme's active site comprises three distinct pockets that accommodate ATP, proline, and the 3'-terminal adenosine of tRNAPro (A76), providing multiple strategic points for therapeutic intervention [80]. The high proline content in structural proteins like collagen makes collagen synthesis particularly vulnerable to PRS inhibition, extending its therapeutic relevance to fibrotic diseases [81].
Structural analyses reveal PRS consists of several domains: a catalytic domain (CD) directly involved in the aminoacylation reaction, an insertion (INS) domain crucial for substrate binding and activation, an anticodon binding domain (ABD), and a C-terminal zinc-binding-like domain (Z-domain) [80]. Significant conservation of these domains across species underscores PRS's fundamental role while highlighting opportunities for selective pathogen targeting [80].
tRNA Biology and Genetic Code Expansion Context
Transfer RNA (tRNA) serves as the molecular bridge between genetic information and functional proteins, with a highly conserved L-shaped structure comprising approximately 76-90 nucleotides [6]. Key structural elements include the acceptor stem (where aminoacylation occurs), the D arm, the anticodon arm (which recognizes mRNA codons), the variable arm, and the T arm [6].
Research on genetic code expansion (GCE) has been instrumental in advancing tRNA-targeted therapeutic strategies. GCE technology relies on orthogonal aminoacyl-tRNA synthetase/tRNA pairs that incorporate unnatural amino acids into proteins, bypassing the constraints of the standard genetic code [6] [43]. These approaches have revealed critical insights into tRNA engineering principles, including:
- Identity elements: Specific nucleotides and structural features that ensure correct tRNA recognition by its cognate synthetase [6]
- Orthogonality: Engineered tRNAs that avoid recognition by endogenous synthetases while maintaining functionality with translational machinery [43]
- Binding optimization: Strategic modifications to enhance interactions with elongation factors and ribosomes [6]
These fundamental discoveries in tRNA biology directly inform drug development strategies targeting synthetase-tRNA interactions, providing the conceptual framework for multi-site inhibition approaches.
Experimental Data and Findings
Dual Inhibitor Strategy AgainstToxoplasma gondiiPRS
A groundbreaking study demonstrated simultaneous targeting of Toxoplasma gondii PRS (TgPRS) with two inhibitors—halofuginone (HFG) and a novel ATP mimetic (L95)—that bind neighboring sites to collectively block all three substrate subsites [79]. This ternary complex formation represents a novel mechanism wherein HFG occupies the L-pro and tRNA binding sites while L95 occupies the ATP pocket [79].
Table 1: Quantitative Profiling of TgPRS Dual Inhibitors
| Parameter | Halofuginone (HFG) | L95 | Combined (HFG + L95) |
|---|---|---|---|
| IC₅₀ Values | Nanomolar range | Nanomolar range | Not specified |
| EC₅₀ Values | Nanomolar range | Nanomolar range | Not specified |
| Binding Sites | L-pro and tRNA subsites | ATP pocket | All three substrate subsites |
| Mode of Action | Dual-site inhibition | Single-site inhibition | Additive effect |
This "double drugging" approach resulted in additive parasite inhibition without apparent antagonism, suggesting independent binding and complementary mechanisms [79]. The structural basis for this compatibility was elucidated through high-resolution crystallography, confirming simultaneous occupancy without steric clash [79].
PRS Inhibition in Avian Coccidiosis
Computational screening against Eimeria tenella PRS (EtPRS) identified several natural compounds with strong binding affinity, including Chelidonine, Bicuculline, and Guggulsterone [80]. These compounds demonstrated stable interactions within the active site, favorable ADMET profiles, and binding stability in molecular dynamics simulations [80].
Table 2: Computational Screening Results for Novel EtPRS Inhibitors
| Compound | Binding Affinity | Key Interactions | ADMET Profile |
|---|---|---|---|
| Chelidonine | Strong | Stable interactions within active site | Favorable |
| Bicuculline | Strong | Stable interactions within active site | Favorable |
| Guggulsterone | Strong | Stable interactions within active site | Favorable |
Sequence alignment of EtPRS with human (HsPRS) and chicken (GgPRS) homologs revealed significant conservation within catalytic domains while identifying unique EtPRS variations that enable species-specific targeting [80].
Structural Insights and Safety Optimization
Research on human PARS1 inhibition for fibrotic diseases yielded critical safety insights relevant to anti-infective strategies. The development of DWN12088, a novel PARS1 catalytic inhibitor, revealed an asymmetric binding mode to PARS1 homodimers, wherein the compound binds with different affinity to each protomer [81]. This unique mechanism creates a decreased responsiveness at higher doses, effectively expanding the safety window—a crucial consideration for translational applications [81].
Furthermore, bacterial PRS inhibition efforts have employed fluorine scanning (F-scanning) strategies to optimize selectivity and potency. The dual-fluorinated derivative PAA-38 achieved exceptional binding affinity (Kd = 0.399 ± 0.074 nM) and inhibitory activity (IC₅₀ = 4.97 ± 0.98 nM) against Pseudomonas aeruginosa ProRS (PaProRS), demonstrating the power of strategic chemical modification for enhancing drug properties [82].
Experimental Protocols
Protocol 1: In Vitro Assessment of PRS Inhibition
Objective: Evaluate compound efficacy against target PRS enzyme through biochemical inhibition assays.
Materials:
- Purified recombinant PRS enzyme (pathogen-specific)
- Test compounds (e.g., HFG, L95, or novel inhibitors)
- Reaction buffer (50 mM HEPES-KOH pH 7.5, 20 mM KCl, 10 mM MgCl₂, 2 mM DTT, 0.1 mg/mL BSA)
- ATP, L-proline, and tRNAPro substrates
- Radioactive [³H]-proline or ATP for detection
- Filtration equipment or scintillation counter
Procedure:
- Prepare reaction mixtures containing buffer, PRS enzyme (10-50 nM), and varying concentrations of test compounds
- Pre-incubate for 15 minutes at room temperature to allow inhibitor binding
- Initiate reactions by adding substrates (typically 1 mM ATP, 10-50 μM L-proline, and 1-10 μM tRNAPro)
- Incubate at parasite physiological temperature (typically 37°C) for 30-60 minutes
- Terminate reactions by adding trichloroacetic acid or EDTA
- Quantify reaction products using radioactive measurement or alternative detection methods
- Calculate IC₅₀ values using non-linear regression of inhibition curves [79] [80]
Protocol 2: Structural Characterization of Inhibitor Complexes
Objective: Determine atomic-level binding modes of single and combined inhibitors.
Materials:
- Purified PRS at high concentration (>5 mg/mL)
- Inhibitor compounds (HFG, L95, or novel inhibitors)
- Crystallization screening kits
- X-ray diffraction facility
- Structural analysis software (e.g., PyMOL, Coot, Phenix)
Procedure:
- Incubate PRS with individual or combined inhibitors (1:2-1:5 molar ratio) for 1 hour on ice
- Screen crystallization conditions using vapor diffusion methods
- Optimize crystal growth for optimal diffraction quality
- Collect X-ray diffraction data at synchrotron facilities
- Solve structures by molecular replacement using existing PRS coordinates
- Model inhibitors into electron density maps, refining positions and interactions
- Analyze binding interfaces, conformational changes, and simultaneous binding compatibility [79]
Protocol 3: Cellular Efficacy Assessment in Parasite Cultures
Objective: Evaluate inhibitor efficacy in whole-cell systems against relevant pathogens.
Materials:
- Mammalian cell lines (host environment)
- Toxoplasma gondii tachyzoites or other relevant parasites
- Cell culture media and supplements
- Compound dilution series
- Cell viability assays (e.g., MTT, plaque assays, flow cytometry)
Procedure:
- Culture host cells in appropriate media to 70-80% confluency
- Infect cultures with freshly harvested parasites at optimized multiplicity of infection
- Add test compounds at various concentrations immediately post-infection
- Include untreated infected controls and uninfected controls
- Incubate for appropriate parasite replication cycles (typically 48-72 hours)
- Quantify parasite proliferation through plaque counting, intracellular replication assays, or reporter systems
- Calculate EC₅₀ values from dose-response curves [79]
Visualization of Mechanisms and Workflows
PRS Dual Inhibition Mechanism
Diagram Title: PRS Dual Inhibition Mechanism
Experimental Workflow for Dual Inhibitor Evaluation
Diagram Title: Dual Inhibitor Evaluation Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for PRS Inhibition Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Validated PRS Inhibitors | Halofuginone (HFG), L95, DWN12088, PAA-38 | Reference compounds for assay validation; mechanistic studies [79] [81] [82] |
| Recombinant PRS Enzymes | TgPRS, EtPRS, PaProRS, HsPRS | Biochemical characterization; inhibitor screening; structural studies [79] [80] [82] |
| Structural Biology Tools | Crystallization screens; X-ray diffraction facilities; Cryo-EM equipment | Determination of inhibitor binding modes; ternary complex analysis [79] |
| Computational Screening Platforms | Molecular docking software; virtual compound libraries; MD simulation packages | Identification of novel inhibitor scaffolds; binding affinity predictions [80] |
| Cell-Based Assay Systems | Parasite cultures (T. gondii, Eimeria spp.); infection models; viability assays | Evaluation of cellular efficacy and selectivity [79] [80] |
The dual inhibition of prolyl-tRNA synthetase represents a transformative approach in anti-infective development, moving beyond single-target paradigms to multi-site intervention strategies. By simultaneously targeting the ATP, L-proline, and tRNA binding subsites with complementary inhibitors, this approach achieves comprehensive enzyme blockade with additive effects and potentially superior resistance profiles. The structural and mechanistic insights from PRS "double drugging" provide a template for targeting other aminoacyl-tRNA synthetases and multi-domain enzymes. As genetic code expansion research continues to reveal intricate details of tRNA-synthetase interactions and identity elements, new opportunities will emerge for precision targeting of pathogen translation machinery. This case study establishes a framework for future therapeutic development that leverages fundamental tRNA biology to overcome limitations of conventional single-drug approaches.
Genetic code expansion (GCE) technology enables the incorporation of unnatural amino acids (UAAs) into proteins, thereby surpassing the constraints of the natural genetic code and creating new opportunities for engineering proteins with novel properties [14]. The core of this technology lies in the use of orthogonal aminoacyl-tRNA synthetase/tRNA (AARS/tRNA) pairs, which must function efficiently within host systems such as E. coli, yeast, and mammalian cells without being cross-reactive with the host's native translation machinery [14]. The selection of an appropriate production platform is critical, as no single system is optimal for all recombinant proteins [83]. This analysis evaluates the performance of these three major GCE platforms—prokaryotic (E. coli), lower eukaryotic (yeast), and higher eukaryotic (mammalian) systems—within the specific context of research involving tRNA duplication and engineering. We provide a comparative quantitative summary, detailed experimental protocols for assessing platform performance, essential reagent information, and visual workflows to guide researchers and drug development professionals in selecting and implementing the most suitable GCE platform for their specific applications in therapeutic protein development [83] [14] [84].
GCE platforms are built on orthogonal AARS/tRNA pairs that are imported from different domains of life to ensure they do not cross-react with the host's native translation machinery [14]. The efficiency of UAA incorporation hinges on the orthogonality of the pair and its ability to interface effectively with the host's transcriptional, translational, and metabolic systems [14]. Table 1 summarizes the key characteristics of E. coli, yeast, and mammalian cell platforms, highlighting their respective advantages and challenges.
Table 1: Comparative Analysis of Major GCE Host Platforms
| Feature | E. coli (Prokaryotic) | Yeast (Lower Eukaryotic) | Mammalian Cells (Higher Eukaryotic) |
|---|---|---|---|
| General Cost & Scalability | Low cost, highly scalable fermentation [83] | Moderate cost and scalability [83] | High cost, complex scalability [83] |
| Expression Speed | Rapid protein production (hours) [84] | Moderate speed (days) [84] | Slower production (days to weeks) [84] |
| Post-Translational Modifications | Limited, lacks eukaryotic-specific modifications [83] | Capable of many eukaryotic-like modifications [83] | Native human-like PTMs (e.g., complex glycosylation) [83] |
| tRNA Engineering Context | Well-established orthogonal pairs (e.g., M. jannaschii tyrosyl) [14] | Suitable for tRNA duplication studies in a eukaryotic context [14] | Directly relevant for human therapeutic protein production [14] |
| Key Challenge | Inability to produce complex human glycoproteins [83] | May require further engineering to humanize glycosylation patterns [83] | Lower yields and higher costs compared to simpler systems [83] |
| Ideal Application in GCE | High-throughput screening of UAAs, production of simple proteins and enzymes [14] [84] | Production of secreted eukaryotic proteins requiring disulfide bonds or simple glycosylation [83] [84] | Production of complex therapeutic glycoproteins requiring precise human PTMs [83] |
The performance of a GCE platform is quantitatively assessed by measuring the yield and fidelity of the target protein incorporating the UAA. Table 2 provides a comparative summary of performance metrics for the production of a model protein, hGAD65, across different systems, illustrating the typical yield ranges and methodologies used in each platform.
Table 2: Quantitative Performance Metrics for Recombinant hGAD65 Production across Platforms [83]
| Host Platform | Reported Yield | Key Methodological Notes |
|---|---|---|
| E. coli | Up to 12.5 g/L | Achieved as soluble, immunogenic product only when expressed as an N-terminal fusion with thioredoxin or glutathione S-transferase [83]. |
| Yeast (S. cerevisiae) | Up to 3.52 mg/L | Production of an active protein; yield increased to 12.16 mg/L using a soluble form generated by substituting the N-terminal domain [83]. |
| Insect Cells (Baculovirus) | Up to 50 mg/L | Highest yields ever reported for hGAD65; yield dropped to 3–5 mg/L when expressed with a C-terminal His6 tag [83]. |
| Mammalian (CHO cells) | ~1.7 mg/L | Recombinant protein was soluble and retained its native structure without a fusion partner [83]. |
| Plant-based Systems | Up to 143.6 μg/g FW (tobacco leaves) | Yield achieved for a catalytically inactive mutant (hGAD65mut), which accumulates to higher levels [83]. |
Experimental Protocols for GCE Platform Evaluation
Protocol 1: Assessing UAA Incorporation Efficiency and Protein Yield
This protocol describes a standardized method to compare the performance of different GCE platforms in incorporating a UAA and producing the target protein.
Step 1: Plasmid Construction
- Clone the gene of interest, containing an amber stop codon (TAG) at the desired site for UAA incorporation, into an expression vector suitable for the host system (e.g., pET series for E. coli, pPICZ for yeast, or pcDNA for mammalian cells).
- Co-transform/transfect the host cells with a second plasmid expressing the orthogonal AARS/tRNA pair specific to the desired UAA (e.g., the M. jannaschii tyrosyl pair).
Step 2: Cell Culture and Induction
- E. coli: Grow transformed cells in LB medium at 37°C to an OD600 of ~0.6. Induce protein expression with IPTG (e.g., 0.5 mM) in the presence of the UAA (1-5 mM).
- Yeast: Grow transformed cells in appropriate selective medium (e.g., YPD) and induce expression with methanol (for P. pastoris) according to standard protocols, adding UAA at the time of induction.
- Mammalian Cells: Transfect cells (e.g., HEK293 or CHO) using a standard method (e.g., PEI). Add the UAA to the culture medium 1-2 hours post-transfection.
Step 3: Protein Analysis and Yield Quantification
- Harvesting: Pellet cells by centrifugation 12-24 hours post-induction/transfection. Lyse cells using sonication (E. coli) or lysis buffer (yeast, mammalian).
- SDS-PAGE and Western Blot: Analyze the total protein and specifically detect the full-length target protein using an antibody. Compare the intensity of the full-length band to a standard curve of the wild-type protein to quantify yield.
- Purification: If the protein is tagged, purify it using affinity chromatography (e.g., Ni-NTA for a His-tag). Measure the concentration of the purified protein using a Bradford or BCA assay.
- Mass Spectrometry: Confirm the precise incorporation of the UAA by liquid chromatography-mass spectrometry (LC-MS) analysis of the purified protein.
Protocol 2: Evaluating Orthogonality and Fidelity
This protocol assesses the orthogonality of the AARS/tRNA pair and the fidelity of UAA incorporation, which are critical for minimizing mis-incorporation of natural amino acids.
Step 1: Testing for Natural Amino Acid Mis-incorporation
- Express the target protein with the amber stop codon in the presence of the orthogonal AARS/tRNA pair but in the absence of the UAA.
- Analyze the cell lysate via SDS-PAGE and Western Blot. The absence of a full-length protein band indicates no mis-incorporation of natural amino acids and high orthogonality.
- For a more sensitive assay, use a fluorescent reporter gene (e.g., GFP) with an amber stop codon at a permissive site. Measure fluorescence in the presence and absence of the UAA. High fluorescence only in the presence of UAA indicates high fidelity.
Step 2: Assessing tRNA Expression and Processing
- Isolate total RNA from cells expressing the orthogonal tRNA.
- Perform Northern Blot analysis using a probe complementary to the orthogonal tRNA sequence to confirm the tRNA is expressed and processed to its correct length without degradation.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful implementation of GCE relies on a suite of specialized reagents and genetic tools. The following table details essential components for designing and executing GCE experiments.
Table 3: Essential Research Reagents for Genetic Code Expansion
| Reagent / Genetic Element | Function in GCE Experiment |
|---|---|
| Orthogonal AARS/tRNA Pair | The core engine of GCE; charges the specific unnatural amino acid (UAA) onto the orthogonal tRNA without interacting with endogenous host pairs [14]. |
| Unnatural Amino Acid (UAA) | The novel chemical moiety to be incorporated into the protein; often contains bio-orthogonal functional groups like azides, alkynes, or photo-crosslinkers [14]. |
| Suppressor tRNA | A tRNA engineered to recognize a stop codon (e.g., TAG) on the mRNA; delivers the UAA to the ribosome during translation, allowing protein synthesis to continue [14]. |
| Expression Vector with Stop Codon | The plasmid carrying the gene of interest, which has been modified to include a premature stop codon (e.g., amber/TAG) at the site where the UAA is to be inserted [14]. |
| CRISPR-Cas9 System | A genome editing tool used for advanced host engineering, such as knocking out competing endogenous tRNAs or integrating orthogonal pairs into the host genome for stable expression [84]. |
| High-Throughput Screening Assay | A method (e.g., fluorescence-activated cell sorting, FACS; or phage display) coupled with a selection marker to rapidly evolve more efficient AARS/tRNA pairs [14] [84]. |
Workflow and Pathway Visualizations
The following diagrams, generated using DOT language, illustrate the core concept of GCE and the experimental workflow for platform evaluation.
GCE Orthogonal Pair Mechanism
GCE Platform Evaluation Workflow
The fundamental role of transfer RNA (tRNA) in protein translation has expanded beyond its canonical adapter function to become a pivotal platform for therapeutic intervention. With nonsense mutations—which introduce premature termination codons (PTCs)—accounting for approximately 11-24% of pathogenic alleles in genetic disease databases, suppressor tRNAs (sup-tRNAs) represent a promising therapeutic strategy to restore full-length protein production [49] [85]. This Application Note explores recent advances in tRNA-based therapies, focusing on two primary approaches: prime editing-installed endogenous sup-tRNAs and engineered sup-tRNA delivery via lipid nanoparticles (LNPs). These modalities enable targeted readthrough of PTCs and offer potential treatments for hundreds of genetic disorders through a mutation-driven rather than disease-specific approach. The content is framed within the broader context of genetic code expansion research, highlighting how tRNA gene duplication and remolding principles inform therapeutic development [86].
Quantitative Analysis of Therapeutic tRNA Efficacy
Protein Restoration Levels Across Disease Models
Table 1: Efficacy Metrics of sup-tRNA Interventions in Preclinical Models
| Disease Model | Mutation | Platform | Protein Restoration | Functional Outcome |
|---|---|---|---|---|
| Hurler syndrome (in vivo) | IDUA p.W392X | Prime editing-installed sup-tRNA | ~6% enzyme activity | Near-complete pathology rescue |
| Methylmalonic acidemia (in vivo) | Arg-TGA PTC | LNP-AP003 (Alltrna) | Up to 25% functional protein | Above clinical benefit threshold |
| Phenylketonuria (in vivo) | Arg-TGA PTC | LNP-AP003 (Alltrna) | ~7% functional protein | 76% reduction in phenylalanine |
| Batten disease (in cellulo) | TPP1 p.L211X/p.L527X | Prime editing-installed sup-tRNA | 20-70% normal enzyme activity | Protein function restoration |
| Reporter system (in vivo) | GFP nonsense mutation | Prime editing-installed sup-tRNA | ~25% full-length GFP | Successful PTC readthrough |
sup-tRNA Engineering Optimization Parameters
Table 2: Engineering Strategies for Enhanced sup-tRNA Efficacy
| Engineering Target | Structural Impact | Functional Optimization | Representative Outcome |
|---|---|---|---|
| Anticodon stem (Ai variants) | Modulates decoding accuracy | Improved ribosomal A-site geometry | tSA1 variant showed enhanced readthrough |
| TΨC stem (Ti variants) | Alters eEF1A binding affinity | Fine-tunes thermodynamic stability | tST5 variant increased suppression efficiency |
| Combined stem modifications | Optimizes both decoding & factor binding | Synergistic enhancement of PTC readthrough | tSA1T5 most effective for UGA/UAG PTCs |
| Anticodon loop replacement | Enables stop codon recognition | Converts endogenous tRNA to sup-tRNA | 29% average conversion rate of endogenous tRNAs |
| Leader and terminator sequences | Regulates transcription & processing | Enhances expression from single genomic locus | Improved potency without overexpression |
Experimental Protocols for sup-tRNA Development
Protocol 1: Prime Editing-Mediated Installation of Endogenous sup-tRNAs
Principle: Utilize prime editing to permanently convert a dispensable endogenous tRNA gene into an optimized sup-tRNA at its native genomic locus, enabling endogenous-level expression without overexpression-associated toxicity [49].
Workflow:
- Endogenous tRNA Selection: Identify redundant human tRNA genes with minimal essential functions using genomic databases (e.g., 418 high-confidence human tRNA genes). Select targets with high expression but low functional constraint.
- sup-tRNA Design: Engineer sup-tRNA sequences through iterative screening of thousands of tRNA variants. Optimize the 40-bp leader sequence, tRNA coding sequence via saturation mutagenesis, and terminator sequence.
- Prime Editor Construction: Design prime editing guide RNAs (pegRNAs) encoding the desired sup-tRNA sequence and nicking gRNAs to facilitate the conversion of the endogenous tRNA anticodon to complement the targeted PTC.
- Delivery & Editing: Transfect cells with prime editor components (nickase and reverse transcriptase fused to engineered reverse transcriptase) using appropriate delivery systems (e.g., lipid nanoparticles, viral vectors).
- Validation: Isolve genomic DNA and sequence the edited tRNA locus to assess conversion efficiency (typically 19-37%). Evaluate PTC readthrough using mCherry-STOP-GFP reporters or disease-relevant protein functional assays.
Troubleshooting Tips:
- If conversion efficiency is low, optimize pegRNA design with improved 3' homology arm extensions.
- To minimize off-target effects, perform whole-genome sequencing to verify editing specificity.
- For in vivo applications, optimize LNP formulations for tissue-specific delivery.
Protocol 2: Engineered sup-tRNA Delivery via Lipid Nanoparticles
Principle: Design and chemically synthesize optimized sup-tRNAs in vitro, then encapsulate them in LNPs for in vivo delivery to enable PTC readthrough without permanent genomic changes [85] [87].
Workflow:
- sup-tRNA Optimization: Select appropriate tRNA scaffold (e.g., tRNASer, tRNAArg, tRNAGly) based on the amino acid to be incorporated. Systematically engineer anticodon stem and TΨC stem to fine-tune decoding accuracy and eEF1A affinity.
- In Vitro Transcription: Synthesize sup-tRNAs using T7 RNA polymerase with optimized reaction conditions to ensure proper folding and homogeneity.
- Quality Control: Analyze sup-tRNA integrity and homogeneity using denaturing urea-PAGE (12%) and confirm identity by MALDI-MS.
- LNP Formulation: Encapsulate sup-tRNAs in ionizable lipid-based nanoparticles using microfluidic mixing technology. Characterize particle size (70-100 nm), encapsulation efficiency (>90%), and stability.
- In Vivo Administration: Administer LNP–sup-tRNA formulations via intravenous or intratracheal routes. For liver-targeted delivery, utilize standard intravenous injection.
- Efficacy Assessment: Measure functional protein restoration by disease-relevant biochemical assays (e.g., enzyme activity, metabolite reduction) and monitor phenotypic rescue.
Troubleshooting Tips:
- If TLR activation is observed, optimize LNP composition to reduce immune stimulation.
- For enhanced tissue specificity, incorporate targeting ligands into LNP formulations.
- If readthrough efficiency is suboptimal, adjust sup-tRNA dosing regimen and administration frequency.
Visualizing sup-tRNA Mechanisms and Workflows
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Therapeutic tRNA Development
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| Prime editing system | Permanent genomic installation of sup-tRNA sequences | Converting endogenous tRNA-Gln-CTG-6-1 to sup-tRNA [49] |
| LNP formulation reagents | In vivo delivery of engineered sup-tRNAs | AP003 candidate for liver stop codon diseases [87] |
| PTC reporter constructs | Quantification of readthrough efficiency | mCherry-STOP-GFP or Fluc-STOP reporters [49] [85] |
| Nano-tRNAseq | Simultaneous quantification of tRNA abundance and modifications | Assessing sup-tRNA expression and modification status [17] |
| tRNA demethylase cocktails | Improving reverse transcription for NGS-based tRNA profiling | Overcoming modification-induced biases in sequencing [17] |
| ADAT2/3 enzymes | A-to-I tRNA editing at wobble position 34 | Exploring codon-biased mRNA translation in disease [88] |
| Fluorous affinity chromatography | Isolation of fully modified wild-type tRNAs | Obtaining functional tRNAs for structural studies [89] |
Therapeutic tRNA applications represent a paradigm shift in genetic medicine, moving from disease-specific to mutation-targeted approaches. The two primary modalities—prime editing-installed endogenous sup-tRNAs and LNP-delivered engineered sup-tRNAs—offer complementary advantages: the former provides permanent correction with endogenous regulation, while the latter enables transient, titratable protein restoration. Both approaches have demonstrated compelling preclinical efficacy across multiple disease models, with protein restoration levels often exceeding established therapeutic thresholds.
Future development will focus on expanding the scope of treatable conditions beyond liver diseases to include muscle and central nervous system disorders, requiring advanced delivery solutions. Additionally, basket trial designs—grouping patients by mutation rather than disease—will accelerate clinical validation and regulatory approval [87]. As tRNA engineering principles continue to evolve and delivery technologies advance, sup-tRNA therapies are poised to transform treatment for thousands of patients with diverse genetic disorders caused by nonsense mutations.
Conclusion
The strategic exploitation of tRNA duplication provides a powerful and biologically inspired framework for genetic code expansion, effectively bridging evolutionary history and synthetic biology innovation. The key takeaways reveal that natural duplication events have conserved essential tRNA features, offering a blueprint for engineering highly efficient and orthogonal translation systems. Methodological advances in directed evolution and high-throughput screening are critical for refining these systems, while comprehensive optimization of the entire cellular translation apparatus is necessary to achieve high-yield ncAA incorporation. Validated through rigorous biochemical and structural studies, GCE technologies are now poised to revolutionize biomedical research and therapeutic development. Future directions will involve the creation of more sophisticated multi-drugging strategies to combat resistance, the application of tRNA-based medicines for treating genetic diseases through nonsense mutation readthrough, and the continuous expansion of the synthetic amino acid repertoire to engineer proteins with unprecedented functions. This convergence of evolutionary insight and engineering precision is set to unlock the next frontier of biomedicine.
References
- 1. Conservation and Tandem Duplication of tRNA Genes in Plant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12652284/]
- 2. The Origin of tRNA Is in Replication, not Translation [https://www.nationalacademies.org/read/4910/chapter/4]
- 3. The diverse structural modes of tRNA binding and ... [https://www.sciencedirect.com/science/article/pii/S0021925823019944]
- 4. Sequencing and Comparing Whole Mitochondrial ... [https://www.sciencedirect.com/science/article/abs/pii/S0076687905950192]
- 5. Different sequence signatures in the upstream regions of plant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3082873/]
- 6. tRNA engineering strategies for genetic code expansion [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2024.1373250/full]
- 7. tRNA-derived small RNAs in plant response to biotic and ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2023.1131977/full]
- 8. Researchers Use Living Fossils to Uncover a Wealth of ... [https://www.nyu.edu/about/news-publications/news/2025/november/living-fossils-genes-seed-improvement.html]
- 9. Recurrent evolution of selfishness from an essential tRNA ... [https://www.nature.com/articles/s41559-025-02894-2]
- 10. Frontiers | Genome-wide analysis of tandem duplicated genes and their expression under salt stress in seashore paspalum [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.971999/full]
- 11. an integrated database to study tandem duplicated genes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4369376/]
- 12. Importance of Lineage-Specific Expansion of Plant Tandem Duplicates in the Adaptive Response to Environmental Stimuli - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2556807/]
- 13. Genome-wide analysis of tandem duplicated genes and ... [https://www.sciencedirect.com/science/article/pii/S0888754320319625]
- 14. tRNA engineering strategies for genetic code expansion - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10954879/]
- 15. Unveiling the functional fate of duplicated genes through ... [https://pubmed.ncbi.nlm.nih.gov/40813247/]
- 16. High-resolution quantitative profiling of tRNA abundance and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8062790/]
- 17. Quantitative analysis of tRNA abundance and ... [https://www.nature.com/articles/s41587-023-01743-6]
- 18. Rooted tRNAomes and evolution of the genetic code - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5927645/]
- 19. On origin of genetic code and tRNA before translation [https://biologydirect.biomedcentral.com/articles/10.1186/1745-6150-6-14]
- 20. Evolution of tRNA into rRNA secondary structures [https://www.sciencedirect.com/science/article/abs/pii/S2452014419301256]
- 21. Conservation and Tandem Duplication of tRNA Genes in ... [https://www.mdpi.com/2073-4425/16/11/1307]
- 22. tRNA genes rapidly change in evolution to meet novel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3868979/]
- 23. Reaching new heights in genetic code manipulation with high ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11879460/]
- 24. tRNA acceptor stem and anticodon bases form ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4475997/]
- 25. The role of tRNA identity elements in aminoacyl-tRNA editing [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1437528/full]
- 26. New codes on RNA | Nature Chemical Biology [https://www.nature.com/articles/s41589-025-01989-y]
- 27. Quantitative analysis of tRNA modifications by HPLC- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4313537/]
- 28. Directed evolution of aminoacyl-tRNA synthetases through ... [https://www.nature.com/articles/s41467-025-60120-w]
- 29. Engineering aminoacyl-tRNA synthetases for use in ... [https://www.sciencedirect.com/science/article/abs/pii/S1874604720300202]
- 30. Development of orthogonal aminoacyl-tRNA synthetase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11116331/]
- 31. Rapid discovery and evolution of orthogonal aminoacyl-tRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7116527/]
- 32. Engineering tRNA abundances for synthetic cellular systems [https://www.nature.com/articles/s41467-023-40199-9]
- 33. Continuous in situ synthesis of a complete set of tRNAs ... [https://www.nature.com/articles/s41467-025-61671-8]
- 34. Simultaneous synthesis of all 21 types of tRNA in vitro [https://www.eurekalert.org/news-releases/1099147]
- 35. RNA-modification by Base Exchange: Structure, Function ... [https://www.sciencedirect.com/science/article/pii/S0022283625000464]
- 36. Genetic Code Engineering by Natural and Unnatural Base ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9171071/]
- 37. Therapeutic applications of genetic code expansion - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6190509/]
- 38. Genetic Code Expansion - GCE4All - Oregon State University [https://gce4all.oregonstate.edu/research/genetic-code-expansion]
- 39. Sense Codon Reassignment Enables Viral Resistance and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7611380/]
- 40. Engineering a genomically recoded organism with one ... [https://www.nature.com/articles/s41586-024-08501-x]
- 41. An Expanded Genetic Code In Mammalian Cells With A ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4501474/]
- 42. Encoding the unnatural | Nature Methods [https://www.nature.com/articles/nmeth0510-343]
- 43. Improving the Efficiency and Orthogonality of Genetic Code ... [https://www.sciencedirect.com/science/article/pii/S2693125724000785]
- 44. Residue-specific incorporation of non canonical amino ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3008400/]
- 45. Site-specific incorporation of an unnatural amino acid into ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC135798/]
- 46. Using Genetic Code Expansion for Protein Biochemical ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2020.598577/full]
- 47. Screening macrocyclic peptide libraries by yeast display ... [https://www.nature.com/articles/s41467-025-60907-x]
- 48. Optimization of ACE-tRNAs function in translation ... - PMC - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC11662937/]
- 49. Prime editing-installed suppressor tRNAs for disease ... [https://www.nature.com/articles/s41586-025-09732-2]
- 50. Potential vs Challenges of Expanding the Protein Universe ... [https://www.sciencedirect.com/science/article/pii/S0022283624004297]
- 51. Extensive breaking of genetic code degeneracy with non- ... [https://www.nature.com/articles/s41467-023-40529-x]
- 52. The birth of a bacterial tRNA gene by large-scale, tandem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7661048/]
- 53. GCE4ALL leads global advancements in genetic code ... [https://science.oregonstate.edu/impact/2024/07/gce4all-leads-global-advancements-in-genetic-code-expansion-for-advanced-therapies]
- 54. Tuning tRNAs for improved translation - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11231383/]
- 55. EF-Tu [https://en.wikipedia.org/wiki/EF-Tu]
- 56. Human protein synthesis requires aminoacyl-tRNA pivoting ... [https://www.nature.com/articles/s41467-025-63617-6]
- 57. Review Article Studying the Function of tRNA Modifications [https://www.sciencedirect.com/science/article/pii/S0022283624005643]
- 58. Simultaneous in vitro expression of minimal 21 transfer ... [https://www.nature.com/articles/s41467-025-62588-y]
- 59. Exploiting nanopore sequencing advances for tRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12582020/]
- 60. How EF-Tu can contribute to efficient proofreading of aa- ... [https://www.nature.com/articles/ncomms13314]
- 61. Transfer RNA Modulates the Editing Mechanism Used by ... [https://www.sciencedirect.com/science/article/pii/S0021925820571524]
- 62. Determining the fidelity of tRNA aminoacylation via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5253240/]
- 63. Innate Immune and Chemically Triggered Oxidative Stress ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2785853/]
- 64. Naturally occurring aminoacyl-tRNA synthetases editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3111296/]
- 65. Translational Fidelity during Bacterial Stresses and Host ... [https://www.mdpi.com/2076-0817/12/3/383]
- 66. Elongation in translation as a dynamic interaction among the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2832932/]
- 67. Elongation factors on the ribosome [https://www.sciencedirect.com/science/article/abs/pii/S0959440X05000886]
- 68. System‐wide optimization of an orthogonal translation system ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10407733/]
- 69. Genomically Recoded Organisms Expand Biological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4924538/]
- 70. Yale scientists recode the genome for programmable synthetic ... [https://news.yale.edu/2025/02/06/yale-scientists-recode-genome-programmable-synthetic-proteins]
- 71. Spotlight Genomically recoded organisms: redefining and ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925001210]
- 72. Comparative modeling reveals the molecular determinants ... [https://www.sciencedirect.com/science/article/pii/S2666979X24002672]
- 73. Copy number variation in tRNA isodecoder genes impairs ... [https://www.nature.com/articles/s41467-023-37843-9]
- 74. Optimized protocol for tRNA identification in the ribosomal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8220392/]
- 75. Genetic code expansion in stable cell lines enables ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4888942/]
- 76. Role of tRNA Orthogonality in an Expanded Genetic Code [https://pmc.ncbi.nlm.nih.gov/articles/PMC4167058/]
- 77. The diverse structural modes of tRNA binding and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10424219/]
- 78. Automated orthogonal tRNA generation [https://www.nature.com/articles/s41589-024-01782-3]
- 79. Double drugging of prolyl-tRNA synthetase provides a new ... [https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1010363]
- 80. Structure-Based Virtual Screening of Potential Inhibitors ... [https://www.mdpi.com/1420-3049/30/4/790]
- 81. Control of fibrosis with enhanced safety via asymmetric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10331583/]
- 82. Development of potent inhibitors targeting bacterial prolyl- ... [https://pubmed.ncbi.nlm.nih.gov/40253792/]
- 83. Comparative Evaluation of Recombinant Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3972949/]
- 84. Engineering Genetic Elements for Microbial Protein ... [https://www.sciencedirect.com/science/article/pii/S2405805X2500170X]
- 85. Engineered tRNAs suppress nonsense mutations in cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10284701/]
- 86. Duplication and Remolding of tRNA Genes in the ... [https://www.mdpi.com/1422-0067/17/6/951]
- 87. New tRNA tech aims to rewrite rare disease treatment [https://www.drugtargetreview.com/article/160841/new-trna-tech-aims-to-rewrite-rare-disease-treatment/]
- 88. The tRNA editing complex ADAT2/3 promotes cancer cell ... [https://www.sciencedirect.com/science/article/pii/S0022283625004802]
- 89. Expansion of the genetic code through reassignment of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9638912/]