Validating Directed Evolution Outcomes: A Comprehensive Guide to NGS Coverage Analysis
Directed evolution is a powerful protein engineering tool, but its success hinges on accurately identifying enriched variants from complex libraries.
Validating Directed Evolution Outcomes: A Comprehensive Guide to NGS Coverage Analysis
Abstract
Directed evolution is a powerful protein engineering tool, but its success hinges on accurately identifying enriched variants from complex libraries. Next-Generation Sequencing (NGS) has become the cornerstone for this analysis; however, the reliability of the data is fundamentally dependent on appropriate sequencing depth and coverage. This article provides researchers, scientists, and drug development professionals with a complete framework for validating directed evolution outcomes through robust NGS coverage analysis. We cover foundational principles, detailed methodological workflows, strategies for troubleshooting and optimizing sequencing parameters, and finally, methods for the statistical validation and comparative analysis of enriched variants. By establishing clear guidelines for NGS coverage, this guide aims to enhance the efficiency and success rate of directed evolution campaigns for therapeutic and biotechnological applications.
The Pillars of Success: Understanding Directed Evolution and NGS Fundamentals
Directed Evolution as an Adaptive Walk on a Fitness Landscape
Directed evolution is a powerful protein engineering method that mimics natural selection in a laboratory setting to steer proteins or nucleic acids toward a user-defined goal [1]. The process consists of iterative rounds of mutagenesis (creating a library of variants), selection (isolating members with the desired function), and amplification [1]. This approach circumvents our profound ignorance of how a protein's sequence encodes its function by using iterative rounds of random mutation and artificial selection to discover new and useful proteins [2].
The conceptual framework of directed evolution is best understood as an adaptive walk on a high-dimensional fitness landscape [2] [3]. In this analogy, first articulated by John Maynard Smith, all possible protein sequences of length L are arranged such that sequences differing by one amino acid mutation are neighbors [2]. Each sequence is assigned a "fitness" value—in artificial selection, this is defined by the experimenter based on desired properties like enzymatic activity, thermostability, or binding affinity [2] [3]. The vastness of this sequence space is incomprehensible; for a small protein of 100 amino acids, there are 20¹⁰⁰ (∼10¹³⁰) possible sequences [2].
Protein evolution can then be envisioned as a walk on this fitness landscape, where regions of higher elevation represent more desirable proteins [2]. The structure of this landscape profoundly influences the effectiveness of evolutionary search strategies [2]. Landscapes range from smooth, single-peaked 'Fujiyama' landscapes to rugged, multi-peaked 'Badlands' landscapes [2]. The rougher the landscape, the harder it is for evolution to climb, as local optima create traps that evolution cannot escape unless temporary decreases in fitness are permitted or multiple simultaneous mutations enable jumps to new peaks [2].
Quantitative Comparison of Directed Evolution Strategies
Directed evolution methodologies have diversified significantly, from traditional iterative approaches to modern machine-learning assisted platforms. The table below summarizes the performance characteristics of these different strategies based on experimental data.
Table 1: Performance Comparison of Directed Evolution Strategies
| Strategy | Typical Library Size | Key Advantages | Limitations | Reported Fitness Gain | Optimal Application Context |
|---|---|---|---|---|---|
| Traditional DE [2] [1] | 10³-10⁶ variants | • Simple implementation• No prior structural knowledge needed• Proven success across many proteins | • Resource-intensive screening• Susceptible to local optima• Multiple rounds required | Varies by protein (e.g., >40°C thermostability increase in lipase A [2]) | Rugged landscapes with fewer local optima; when high-throughput screening is available |
| Machine Learning-Assisted DE (MLDE) [4] | 10⁴-10⁵ training variants | • More efficient exploration of sequence space• Better navigation of epistatic landscapes• Can predict high-fitness variants in silico | • Requires initial training data• Model performance depends on landscape structure | Consistently matches or exceeds traditional DE across 16 diverse protein landscapes [4] | Landscapes with significant epistasis and local optima |
| Focused Training MLDE (ftMLDE) [4] | 10⁴-10⁵ variants | • Enhanced training set quality using zero-shot predictors• Leverages evolutionary, structural, and stability knowledge | • Dependent on quality of zero-shot predictors | Outperforms random sampling for both binding and enzyme activities [4] | Landscapes with challenging attributes (fewer active variants, more local optima) |
| Continuous Evolution (T7-ORACLE) [5] | Effectively unlimited over time | • Extremely rapid (rounds with each cell division)• No manual intervention between rounds• 100,000x higher mutation rate than normal | • Technical complexity of system setup• Currently limited to E. coli host | Evolved antibiotic resistance up to 5,000x higher in less than a week [5] | When extremely rapid evolution is needed; for exploring vast sequence spaces |
Table 2: Effect of Selection Parameters on Directed Evolution Outcomes in Polymerase Engineering [3]
| Selection Parameter | Impact on Recovery Yield | Impact on Variant Enrichment | Impact on Variant Fidelity | Optimization Recommendation |
|---|---|---|---|---|
| Mg²⁺/Mn²⁺ Concentration | Significant impact | Crucial for shaping polymerase activity | Influences polymerase/exonuclease equilibrium | Requires careful titration to balance activity and fidelity |
| Nucleotide Chemistry | Affects background noise | Directly determines selective pressure | Impacts mechanism of incorporation | Should match desired substrate specificity |
| Selection Time | Influences parasite recovery | Affects stringency | Longer times may favor proofreading | Optimize to minimize false positives while maintaining diversity |
| Additives | Can improve or suppress yield | Modifies enzyme kinetics | Can stabilize specific conformations | Screen common PCR additives systematically |
Experimental Protocols for Key Directed Evolution Approaches
Traditional Directed Evolution Workflow
The standard directed evolution protocol involves iterative cycles of diversification, selection, and amplification [1]. The initial step involves generating genetic diversity in a parental sequence through random mutagenesis techniques such as error-prone PCR or DNA shuffling [6]. Error-prone PCR can be performed using standard PCR protocols with modified conditions, including increased magnesium concentration, addition of manganese, unequal dNTP concentrations, and using Taq polymerase which lacks proofreading activity [6]. This generates a library of variant genes with point mutations across the entire sequence.
The library is then subjected to selection or screening based on the desired function [1]. For binding proteins, phage display is commonly employed, where the target molecule is immobilized on a solid support, the library of variant proteins is flowed over it, poor binders are washed away, and the remaining bound variants are recovered [1]. For enzymatic activities, screening systems individually assay each variant using colorimetric or fluorogenic substrates [6]. High-throughput screening via fluorescence-activated cell sorting (FACS) can achieve throughput of up to 10⁸ variants per day when the evolved property can be linked to a change in fluorescence [6].
The selected variants are amplified, either by PCR or through bacterial hosts, and the process is repeated for multiple rounds [1]. The entire process typically requires 1-2 weeks per round, with 3-6 rounds needed to achieve significant improvements [5].
Machine Learning-Assisted Directed Evolution Protocol
MLDE enhances traditional directed evolution by incorporating machine learning models to predict high-fitness variants [4]. The protocol begins with creating an initial training library of 10⁴-10⁵ variants, which should be randomly sampled from the full combinatorial space [4]. Each variant in this library is experimentally characterized to determine its fitness value.
The sequence-fitness data is used to train supervised machine learning models, such as Gaussian process regression or neural networks, which capture non-additive epistatic effects [4]. For focused training (ftMLDE), the training set quality is enriched using zero-shot predictors that leverage evolutionary, structural, or stability knowledge to selectively sample variants that avoid low-fitness regions [4]. The trained model then predicts fitness across the entire sequence space, identifying high-fitness variants for experimental validation [4].
In active learning DE (ALDE), this process becomes iterative—the model's predictions guide the selection of additional variants for experimental testing, which are then incorporated into the training set to refine the model [4]. This approach is particularly advantageous on rugged landscapes rich in epistasis, where it provides greater benefits compared to traditional DE [4].
Emulsion-Based Selection for Polymerase Engineering
Emulsion-based selection platforms enable the directed evolution of DNA polymerases with novel functions [3]. The protocol involves creating water-in-oil emulsions where individual aqueous droplets serve as microreactors, each containing a single cell expressing a unique polymerase variant, along with substrates and products [3]. This compartmentalization minimizes cross-reactivity and cross-catalysis, allowing partitioning of libraries based on the enzyme function of individual variants [3].
Key steps include:
- Library design: Creating focused libraries targeting specific residues, such as metal-coordinating residues and their neighbors in DNA polymerase [3].
- Emulsion formation: Using microfluidics or vigorous mixing to generate monodisperse emulsion droplets with cell, substrate, and product compartments [3].
- Selection: Applying specific selection pressures through factors like nucleotide concentration, nucleotide chemistry, selection time, and divalent metal ion concentration [3].
- Recovery and analysis: Breaking the emulsion, recovering selected variants, and using next-generation sequencing to identify enriched mutants [3].
This method has successfully isolated polymerase variants with improved thermostability, altered substrate specificity, and reverse transcription activity [3].
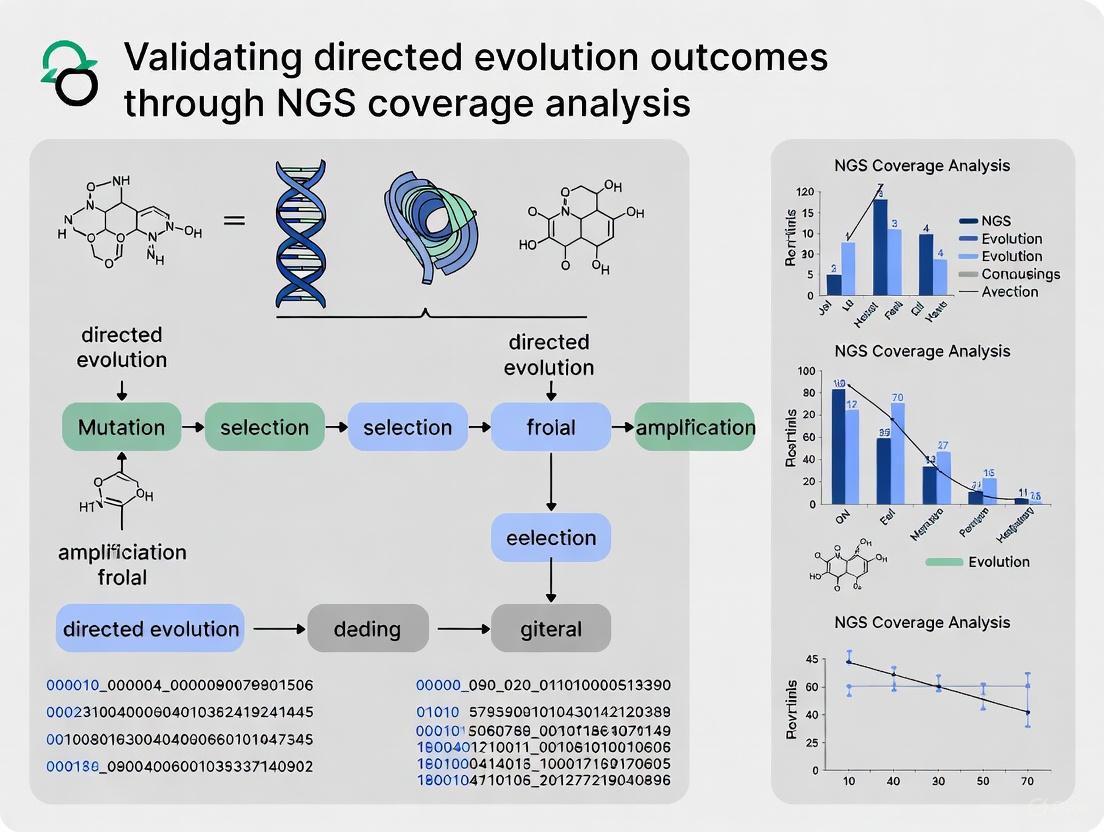
Validating Directed Evolution Outcomes with NGS Coverage Analysis
Next-generation sequencing has become an indispensable tool for analyzing directed evolution outcomes, enabling comprehensive characterization of variant libraries and their enrichment patterns. Adequate sequencing coverage is critical for accurate identification of significantly enriched mutants [3].
The optimal sequencing coverage depends on the specific goals of the analysis. For identifying enriched variants in selection outputs, a threshold of 50-100x coverage per variant provides precise and accurate identification of active mutants [3]. This coverage is significantly lower than required for genome assembly but sufficient for variant identification in directed evolution contexts [3].
For clinical applications or high-stakes validation, more extensive coverage is recommended. One study on gastrointestinal cancer detection achieved >99% sensitivity for single-nucleotide variants with allele frequencies >10% using NGS coverage that provided consistent detection sensitivity down to 10% variant frequency [7]. The same study demonstrated 97.2% sensitivity and 99.2% specificity in formalin-fixed, paraffin-embedded specimens [7].
NGS analysis enables not only identification of enriched variants but also assessment of selection quality through metrics like:
- Variant enrichment: Measuring the proportional increase of specific variants after selection [3]
- Recovery yield: The percentage of input variants recovered after selection [3]
- Variant fidelity: For polymerases, the balance between synthesis efficiency and accuracy [3]
- Parasite identification: Detection of variants enriched through non-desired functions [3]
Diagram 1: Directed Evolution Workflow with NGS Validation. This flowchart illustrates the iterative process of directed evolution, highlighting the integration of machine learning and NGS coverage analysis for validation.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential Research Reagents and Platforms for Directed Evolution
| Tool Category | Specific Products/Platforms | Primary Function | Key Applications |
|---|---|---|---|
| Directed Evolution Platforms | T7-ORACLE [5], OrthoRep [5], EcORep [5] | Continuous evolution systems enabling rapid protein optimization | Evolving therapeutic proteins, antibiotic resistance, enzyme engineering |
| Specialized DNA Polymerases | KAPA HiFi DNA Polymerase [8], KOD DNA Polymerase variants [3] | High-fidelity amplification, XNA synthesis, reverse transcription | NGS library preparation, xenobiotic nucleic acid processing |
| Library Preparation Kits | KAPA HyperPrep Kit [8], KAPA RNA HyperPrep Kit [8] | Efficient construction of sequencing libraries from limited input | RNA-seq, whole transcriptome analysis, NGS workflow optimization |
| Screening Technologies | Phage Display [1] [6], FACS-based methods [6], Emulsion platforms [3] [8] | High-throughput identification of variants with desired properties | Antibody engineering, enzyme evolution, binding protein optimization |
| NGS Validation Solutions | Custom NGS panels [7], Targeted sequencing assays [3] | Validation of directed evolution outcomes, variant enrichment analysis | Gastrointestinal cancer profiling, polymerase variant characterization |
Diagram 2: Landscape Ruggedness Determines Optimal Evolution Strategy. This diagram illustrates how fitness landscape structure influences the choice between traditional and machine learning-assisted directed evolution.
Directed evolution represents a powerful experimental framework for protein engineering, conceptualized as an adaptive walk on a fitness landscape. The efficacy of different evolution strategies—from traditional iterative approaches to modern machine-learning assisted platforms—varies significantly based on landscape characteristics, with MLDE providing particular advantages on rugged landscapes rich in epistasis. The integration of NGS coverage analysis has become indispensable for validating directed evolution outcomes, with optimal coverage thresholds enabling accurate identification of enriched variants. As the field advances, continuous evolution platforms like T7-ORACLE and sophisticated MLDE approaches are dramatically accelerating our ability to engineer proteins with novel functions, opening new frontiers in therapeutic development, industrial biocatalysis, and fundamental evolutionary science.
The Indispensable Role of NGS in Decoding Evolutionary Outcomes
Next-Generation Sequencing (NGS) has revolutionized our ability to decode evolutionary outcomes by providing unprecedented resolution for analyzing genetic changes over time. This transformative technology enables researchers to move beyond theoretical models to empirical validation of evolutionary processes, from directed evolution experiments in laboratory settings to natural population studies in diverse ecosystems. The capacity of NGS to simultaneously sequence millions of DNA fragments in a high-throughput, cost-effective manner has established it as an indispensable tool for modern evolutionary biology [9]. By capturing comprehensive genetic information across entire genomes, NGS provides the quantitative data necessary to validate evolutionary hypotheses, track adaptive trajectories, and understand the complex interplay between selection, genetic drift, and other evolutionary forces.
In directed evolution specifically, NGS serves as a critical validation tool that connects experimental design with functional outcomes. Where traditional methods might only identify a handful of optimized variants, NGS reveals the complete spectrum of mutations underlying improved function, providing insights into the sequence-function relationships that govern protein evolution [10]. This detailed perspective enables researchers to move beyond simply observing that evolution occurred to understanding how it occurred at a molecular level – what mutations arose, how they interacted, and which evolutionary pathways were navigated to reach functional optima.
NGS Technology Platforms for Evolutionary Studies
Comparative Analysis of Sequencing Platforms
The selection of an appropriate NGS platform is fundamental to designing effective evolutionary studies, as each technology offers distinct advantages for specific applications. The table below summarizes the key characteristics of major sequencing platforms relevant to evolutionary research:
Table 1: Comparison of NGS Platforms for Evolutionary Studies
| Platform | Technology | Read Length | Key Strengths | Limitations | Best Applications in Evolutionary Studies |
|---|---|---|---|---|---|
| Illumina | Sequencing-by-synthesis | 36-300 bp | High accuracy (∼99.9%), low cost per base | Short reads limit structural variant detection | Variant calling in populations, tracking mutation trajectories [9] |
| PacBio SMRT | Single-molecule real-time sequencing | 10,000-25,000 bp | Excellent for structural variants, haplotype resolution | Higher cost, lower throughput | Resolving complex genomic regions, detecting recombination events [9] |
| Oxford Nanopore | Nanopore sensing | 10,000-30,000 bp | Ultra-long reads, real-time analysis, portable | Higher error rate (∼5-15%) | Field applications, complete genome assembly [9] |
| Ion Torrent | Semiconductor sequencing | 200-400 bp | Fast run times, simple workflow | Homopolymer errors | Rapid screening of mutant libraries [9] |
Platform Selection Considerations
Choosing the optimal NGS platform requires balancing multiple factors specific to evolutionary research questions. For directed evolution experiments where tracking specific mutations across rounds of selection is paramount, Illumina platforms provide the cost-effective, high-accuracy sequencing needed to identify enriched variants [10]. For studies of population history and divergence dating in natural populations, long-read technologies like PacBio SMRT sequencing enable more complete assembly of genomic regions and better resolution of structural variants that often underlie adaptive evolution [11]. Each platform's characteristics directly influence the evolutionary inferences that can be drawn from the resulting data, making platform selection a critical first step in experimental design.
NGS in Directed Evolution: Experimental Framework
Workflow for Validating Directed Evolution Outcomes
Directed evolution mimics natural selection in laboratory settings to engineer biomolecules with improved or novel functions. NGS integrates throughout this pipeline, both informing selection strategies and validating outcomes. The following diagram illustrates the comprehensive workflow:
Experimental Protocol for NGS-Based Validation
The following detailed methodology enables comprehensive analysis of directed evolution outcomes:
Library Preparation for NGS: For protein engineering studies, extract plasmid DNA from pre-selection and post-selection populations. Amplify target genes using barcoded primers to enable multiplexing. For Illumina platforms, use tagmentation-based library preparation (Nextera) or amplification-based methods (TruSeq). For studies requiring maximum accuracy, consider hybrid capture-based approaches that minimize allele dropout [12].
Sequencing Depth Optimization: Determine appropriate sequencing coverage based on library diversity. For typical directed evolution libraries containing 10⁶-10⁹ variants, ensure sufficient depth to detect pre-selection variants at ≥10x coverage. As demonstrated in polymerase engineering studies, cost-effective identification of enriched variants is achievable even at moderate coverages (50-100x), though higher coverage (200x) improves accuracy for low-frequency variants [10].
Variant Calling and Filtering: Process raw sequencing data through standardized bioinformatics pipelines. After demultiplexing, align reads to reference sequences using BWA-MEM or similar aligners [11]. Call variants using GATK Best Practices or specialized tools for engineered libraries. Filter based on quality scores, strand bias, and mapping quality. For critical clinical applications, confirm NGS-identified variants using Sanger sequencing, which remains a best practice for validation [13].
Enrichment Calculation and Statistical Analysis: Calculate enrichment scores for variants by comparing frequencies between pre-selection and post-selection populations. Apply statistical frameworks (such as Fisher's exact test with multiple testing correction) to identify significantly enriched mutations. For polymerase engineering studies, this approach has successfully identified mutations that confer improved activity toward xenobiotic nucleic acids (XNAs) [10].
NGS in Natural Population Evolution Studies
Tracking Evolutionary Forces in Wild Populations
Beyond laboratory evolution, NGS enables detailed investigation of evolutionary processes in natural populations. By applying various sequencing strategies to population samples, researchers can reconstruct demographic history, identify signatures of selection, and quantify gene flow:
Table 2: NGS Approaches for Studying Natural Population Evolution
| Method | Key Features | Data Output | Evolutionary Insights | Example Application |
|---|---|---|---|---|
| Whole Genome Sequencing | Comprehensive genomic coverage | High-density SNPs, structural variants | Demographic history, selective sweeps, local adaptation | Rhodomyrtus tomentosa population history [11] |
| RAD-Seq | Reduced representation, cost-effective | Thousands of SNPs across many individuals | Population structure, gene flow, outlier loci for selection | Genetic diversity assessment across multiple populations [11] |
| Hybrid Capture | Targeted sequencing of specific regions | Sequence data for loci of interest | Evolution of gene families, phylogenetic relationships | Comparative genomics of adaptive traits [14] |
Experimental Protocol for Population Evolutionary Studies
Implementing NGS in population genetics requires specialized methodological considerations:
Sample Collection and Preservation: For population studies like the Rhodomyrtus tomentosa investigation, collect tissue samples (leaves for plants, blood/tissue for animals) from multiple individuals across geographical ranges, ensuring adequate spatial sampling to resolve population structure. Immediately preserve samples using silica gel, liquid nitrogen, or appropriate preservatives to prevent DNA degradation [11].
Library Preparation for Population Sequencing: Extract high-quality DNA using modified CTAB or commercial kits. For RAD-seq, digest genomic DNA with appropriate restriction enzymes (e.g., MseI and EcoRI), ligate with barcoded adapters, size-select fragments (300-500bp), and amplify with indexing primers. Sequence on Illumina platforms (2×150bp recommended) [11]. For WGS, use mechanical shearing or transposase-based fragmentation followed by library preparation with platform-specific adapters.
Variant Calling and Filtering for Population Data: Process raw sequencing data through quality control (FastQC), demultiplex using process_radtags in Stacks, align to reference genome using BWA-MEM, and call variants using Stacks populations program or similar pipelines. Apply rigorous filtering: remove samples with >20% missing data, exclude SNPs with minor allele count <10, filter based on Hardy-Weinberg equilibrium (p<10⁻⁷), and prune for linkage disequilibrium if needed for specific analyses [11].
Population Genetic Analyses: Calculate standard diversity statistics (π, FIS), population differentiation (FST), and structure (PCA, ADMIXTURE). Test for isolation-by-distance using Mantel tests. For demographic inference, apply PSMC methods to whole-genome data or coalescent-based approaches to SNP data. Identify regions under selection using outlier approaches (e.g., BayPass) or environmental association analyses (RDA) [11].
Essential Research Reagent Solutions
Successful implementation of NGS in evolutionary studies requires specific reagents and tools optimized for particular applications:
Table 3: Key Research Reagents for NGS-Based Evolutionary Studies
| Reagent/Tool Category | Specific Examples | Function in Evolutionary Studies | Performance Considerations |
|---|---|---|---|
| High-Fidelity Polymerases | KAPA HiFi DNA Polymerase, Q5 High-Fidelity DNA Polymerase | Library amplification with minimal errors, inverse PCR for library construction | Industry-leading fidelity for accurate representation of variant frequencies [8] |
| Library Preparation Kits | KAPA HyperPrep Kits, KAPA RNA HyperPrep Kits | Efficient library construction from diverse input materials | Higher library yields, reduced duplicates, improved coverage uniformity [8] |
| Directed Evolution Enzymes | Evolved polymerases for XNA synthesis | Enable novel substrate incorporation for expanded functional selection | Engineered through directed evolution for specialized activities not found in nature [8] [10] |
| Variant Calling Pipelines | Stacks, GATK, BWA-MEM, SAMtools | Identify genetic variants from raw sequencing data | Critical for accurate mutation tracking in directed evolution and population studies [11] |
| Selection Reagents | Custom nucleotide analogs (2′F-rNTPs), specialized cofactors | Create selective environments for specific enzyme functions | Concentration optimization crucial for success of directed evolution campaigns [10] |
Data Analysis and Coverage Considerations
Determining Optimal Sequencing Coverage
A critical aspect of NGS experimental design for evolutionary studies is determining appropriate sequencing coverage, which varies significantly based on research goals. The following diagram illustrates the coverage decision process:
Addressing Technical Challenges in NGS-Based Evolutionary Studies
Despite its transformative potential, NGS implementation in evolutionary studies presents specific technical challenges that require strategic solutions:
Error Rate Management: All NGS platforms exhibit characteristic error profiles that can be misconstrued as evolutionary mutations. For Illumina platforms, error rates typically range from 0.1-1% [9]. Effective error mitigation includes using unique molecular identifiers (UMIs) to distinguish true biological variants from sequencing artifacts, implementing duplicate removal, and applying Bayesian statistical approaches that model error probabilities during variant calling.
Tumor Purity Considerations in Somatic Evolution: For cancer evolution studies, accurate variant detection requires careful assessment of tumor purity. Pathologist review of hematoxylin and eosin-stained slides enables estimation of tumor cell fraction, which is critical for interpreting mutant allele frequencies and copy number alterations [12]. Conservative estimation is recommended, as inflammatory infiltrates can lead to underestimation of tumor proportion.
Bioinformatics Challenges: The enormous data volumes generated by NGS necessitate sophisticated computational infrastructure and analytical approaches [15]. Beyond standard variant calling, specialized algorithms are required for detecting copy number alterations (CNAs) and structural variants (SVs) in cancer evolution studies, or for identifying introgression and selective sweeps in population genomic datasets [12].
Next-Generation Sequencing has fundamentally transformed our ability to decode evolutionary outcomes across biological scales, from single proteins to entire ecosystems. By providing comprehensive, high-resolution genetic data, NGS enables researchers to move beyond inference to direct observation of evolutionary processes. In directed evolution, NGS reveals the complex mutational patterns underlying functional optimization, guiding protein engineering efforts. In natural populations, NGS illuminates the historical demographic events and selective pressures that shape contemporary biodiversity. As sequencing technologies continue to advance, becoming more accessible and cost-effective, their integration with evolutionary studies will undoubtedly yield deeper insights into the fundamental mechanisms driving biological change. The continued development of specialized analytical frameworks and experimental approaches will further enhance our ability to extract meaningful evolutionary understanding from the vast datasets generated by these powerful technologies.
In the field of next-generation sequencing (NGS), particularly when validating outcomes from directed evolution experiments, a precise understanding of sequencing depth and coverage is non-negotiable. These two metrics form the bedrock of data quality and reliability, directly influencing the confidence with which scientists can call genetic variants, assemble genomes, and interpret functional selections. Despite being often used interchangeably, depth and coverage describe distinct, complementary aspects of sequencing data. Sequencing depth (or read depth) refers to the number of times a specific nucleotide is read during the sequencing process, providing a measure of confidence at individual base positions [16]. Sequencing coverage, however, describes the proportion of the target genome or region that has been sequenced at least once, indicating the completeness of the data [16] [17].
The confusion between these terms is pervasive, yet overcoming it is critical for rigorous NGS experimental design, especially in applied fields like directed evolution where identifying enriched mutants amidst a diverse library depends entirely on data completeness and accuracy [10]. This guide objectively compares these core metrics, outlines their practical implications, and provides a framework for their application in validating directed evolution outcomes, supported by current experimental data and protocols.
Defining the Metrics: A Comparative Analysis
Sequencing Depth: The Measure of Confidence
- Definition: Sequencing depth is a localized metric, defined as the number of sequencing reads that align to a specific genomic coordinate [16]. For example, if a single nucleotide is sequenced 30 times, the depth at that position is 30x.
- Purpose and Importance: Depth underpins the statistical confidence of variant calls. A higher depth means more independent observations of a base, making it easier to distinguish a true genetic variant from a random sequencing error. This is paramount in directed evolution for detecting low-frequency variants within a heterogeneous pool or for confirming a mutation with high certainty [16] [18].
- Measurement: It is typically expressed as an average across the entire target region (e.g., "The library was sequenced to 100x depth") [16].
Sequencing Coverage: The Measure of Completeness
- Definition: Sequencing coverage is a global metric, referring to the percentage of the target region (e.g., a genome, exome, or gene panel) that is represented by at least one sequencing read [16] [17].
- Purpose and Importance: Coverage ensures the experiment captures the entire region of interest. Low coverage results in "gaps"—genomic segments with no data—which can lead to missed variants and an incomplete picture of the genetic landscape. In directed evolution, low coverage could mean entire mutant sequences are absent from the data, severely biasing the interpretation of selection outcomes [16].
- Measurement: It is usually expressed as a percentage (e.g., "95% of the exome was covered at 1x") [16].
Table 1: Core Differences Between Sequencing Depth and Coverage
| Feature | Sequencing Depth | Sequencing Coverage |
|---|---|---|
| Definition | Number of times a nucleotide is sequenced [16] | Proportion of the target region sequenced [16] |
| Answers the Question | "How confident can I be in this base call?" | "How much of my target has been sequenced?" |
| Primary Role | Confidence & accuracy for variant calling [16] | Completeness & comprehensiveness of data [16] |
| Impact of Low Values | Inability to call variants reliably; false positives/negatives | Gaps in data; entire variants missed |
| Typical Unit | Multiplier (e.g., 30x, 100x) | Percentage (e.g., 95%) |
The Interrelationship and Trade-offs
Depth and coverage are intrinsically linked but are not the same. It is possible to have high depth but low coverage if a small subset of the genome is sequenced an enormous number of times while other regions are missed entirely. Conversely, one can have high coverage but low depth if every region is sequenced, but only once or twice, providing little confidence in the base calls [16].
The relationship is often governed by the Lander/Waterman equation: C = LN / G, where C is coverage, L is read length, N is the number of reads, and G is the haploid genome length [18] [17]. This equation highlights that for a fixed amount of sequencing capacity (LN), larger genomes (G) will result in lower coverage. In practice, achieving both high depth and high breadth of coverage requires a careful balance and is often constrained by cost and sequencing resources [16] [18].
Figure 1: The Interdependent Relationship Between Sequencing Goals and Resource Allocation. Achieving high depth and coverage are both key objectives, but they compete for finite sequencing resources, requiring researchers to prioritize based on their primary experimental goal.
Quantitative Guidelines and Technology Comparisons
Established Coverage Recommendations
Required depth and coverage are not one-size-fits-all; they are dictated by the specific application and the biological question. The following table summarizes standard recommendations for common NGS methods.
Table 2: Recommended Sequencing Coverage for Common NGS Applications [17]
| Sequencing Method | Recommended Coverage | Rationale |
|---|---|---|
| Whole Genome Sequencing (WGS) | 30x - 50x for human | Balances cost with high-confidence variant calling across the genome. |
| Whole-Exome Sequencing (WES) | 100x | Higher depth needed as exomes capture only 1-2% of the genome, focusing on protein-coding regions of high interest. |
| RNA Sequencing | Often calculated in millions of reads | Detecting lowly expressed genes requires greater sampling (depth). |
| ChIP-Seq | 100x | Needed to confidently identify transcription factor binding sites. |
Recent technological advances are reshaping these guidelines. Pacific Biosciences highlights that with their high-fidelity long-read (HiFi) technology, a 20x human genome can achieve over 99% of the variant detection performance (F1 score) of a 30x genome for single nucleotide variants (SNVs) and structural variants (SVs), and over 98% for indels [19]. This demonstrates that read accuracy is as important as raw depth.
The Critical Role of Coverage Uniformity
A key factor in this efficiency is coverage uniformity—how evenly reads are distributed across the target [19]. Two datasets with the same average depth (e.g., 30x) can have vastly different scientific value. One may have poor uniformity, with depths ranging from 0x in some areas to 60x in others, creating gaps and over-sampled regions. The other, with high uniformity (e.g., most bases covered between 25x-35x), provides reliable information genome-wide [19]. Hybridization-capture methods generally offer better uniformity than amplicon-based approaches, which can suffer from dropout due to primer mismatches [12].
Application in Directed Evolution: A Case Study
Directed evolution mimics natural selection to engineer proteins with improved properties. Validating these experiments requires NGS to identify which mutants are enriched after selection, placing unique demands on depth and coverage.
Experimental Protocol for Variant Enrichment Analysis
A 2024 study on polymerase engineering provides a robust methodological framework [10]:
- Library Construction & Selection: Create a focused saturation mutagenesis library targeting specific active-site residues (e.g., a 5-point library targeting residues L403, D404, F405, L408, Y409). Subject the library to emulsion-based compartmentalization for activity-based selection (e.g., Compartmentalized Self-Replication - CSR) under different selective pressures (varying nucleotide chemistry, metal cofactors, time).
- NGS Library Prep and Sequencing: Isolve DNA from pre- and post-selection populations. Prepare sequencing libraries (e.g., using Illumina kits) from both to track enrichment. Sequence to a depth that ensures comprehensive sampling of the variant library.
- Variant Identification and Analysis: Map reads to a reference sequence. Count the frequency of each unique variant in the pre- and post-selection libraries. Calculate fold-enrichment for variants that survive selection. The study confirmed that cost-effective, precise, and accurate identification of active variants is possible even at low coverages, though the specific threshold must be determined empirically [10].
The Scientist's Toolkit: Key Reagents for NGS in Directed Evolution
Table 3: Essential Research Reagents for Directed Evolution NGS Workflows
| Reagent / Material | Function in the Workflow |
|---|---|
| Saturation Mutagenesis Library | Provides the genetic diversity for selection; the starting point of the experiment [10]. |
| Emulsion Reagents (Oil, Surfactants) | Creates water-in-oil droplets for compartmentalizing individual variants and linking genotype to phenotype [10]. |
| NGS Library Prep Kit (e.g., Illumina) | Prepares the genetic material from selected variants for sequencing by fragmenting and adding platform-specific adapters [12]. |
| Selection Substrates (e.g., 2'F-rNTPs) | The challenging substrate or condition that defines the selective pressure for enriching functional mutants [10]. |
| High-Fidelity DNA Polymerase | Used for accurate amplification steps during library construction and PCR validation [10]. |
Optimizing Depth and Coverage for Your Experiment
Selecting the right metrics is a strategic decision. The following flowchart provides a logical framework for determining optimal sequencing depth, tailored to different research goals.
Figure 2: A Strategic Framework for Determining Optimal Sequencing Depth and Technology Selection. This decision tree guides researchers in prioritizing sequencing parameters based on their primary experimental objective.
A Step-by-Step Selection Guide
- Define Study Objectives: The primary goal is the most important factor [16]. Are you searching for rare variants in a mixed population (requiring high depth) or ensuring you miss no variants in a clinical gene panel (requiring high coverage)?
- Consider Sample Characteristics: The quality and quantity of input DNA/RNA can limit achievable coverage. Formalin-fixed, paraffin-embedded (FFPE) samples, for instance, yield degraded nucleic acids that often require higher depth to achieve confident base calling [20].
- Understand Target Variation: Different variant types have different detection requirements. Single Nucleotide Variants (SNVs) can be called at lower depths than insertions/deletions (indels) or complex structural variants [12] [19].
- Account for Technology: As comparative studies show, highly accurate long-read technologies (e.g., PacBio HiFi) can achieve high confidence at lower nominal depths than earlier short-read technologies, due to their longer read lengths and high per-read accuracy [19].
- Balance with Resources: Finally, depth and coverage must be traded off against cost and data storage capabilities [16] [18]. Deeper sequencing generates more data, increasing expenses and computational burdens.
In next-generation sequencing, "depth" and "coverage" are not synonymous; they are distinct, critical metrics for data quality and completeness. Sequencing depth dictates confidence in base calls, while sequencing coverage ensures no part of the genomic target is missed. For researchers validating directed evolution outcomes, a deliberate strategy that prioritizes both sufficient depth to identify low-frequency, enriched mutants and sufficient coverage to ensure the entire mutant library is sampled is essential for drawing meaningful, accurate conclusions. By applying the frameworks, guidelines, and experimental precedents outlined here, scientists can design more efficient, reliable, and cost-effective NGS experiments, fully leveraging the power of sequencing to decode complex biological selections.
In the rigorous validation of directed evolution experiments, next-generation sequencing (NGS) coverage is not merely a technical metric; it is the foundational determinant of statistical confidence. The precise identification of enriched protein variants—the very outcome of a successful directed evolution campaign—hinges on the depth and breadth of sequencing data. Coverage threshold refers to the minimum number of times a specific nucleotide base must be sequenced to ensure a variant call is accurate and reproducible. Within the context of directed evolution, where distinguishing true beneficial mutations from background noise is paramount, applying appropriate coverage thresholds transforms NGS from a simple sequencing tool into a powerful engine for functional discovery. This guide examines the critical link between coverage thresholds and variant discovery confidence, providing a framework for researchers to validate directed evolution outcomes with statistical rigor.
Understanding Coverage and Depth in NGS
Core Definitions and Distinctions
Although often used interchangeably, "coverage" and "depth" describe distinct but related concepts in NGS data analysis. Precise understanding of these terms is essential for experimental design and data interpretation.
- Sequencing Depth (Read Depth): This metric describes the number of times a specific nucleotide is read during sequencing. It is expressed as an average across the genome or target region (e.g., 100x) and directly impacts base-calling accuracy. Higher depth provides redundancy, enabling error correction and confident variant calling [16].
- Sequencing Coverage: This refers to the percentage of the target genome or region that has been sequenced at least once. It is typically expressed as a percentage (e.g., 95% coverage) and indicates the comprehensiveness of the sequencing effort. Gaps in coverage mean certain regions are completely missing from the data [16].
The relationship between these metrics is symbiotic. While increasing sequencing depth generally improves the likelihood of achieving comprehensive coverage, biases in library preparation or genomic complexities can still leave some regions under-represented or entirely missing despite high overall depth [16].
The Lander/Waterman Equation: Predicting Coverage
The theoretical foundation for understanding sequencing coverage was established by the Lander/Waterman equation, which predicts genome coverage based on known parameters [17]:
C = LN / G
Where:
- C = Coverage
- L = Read length
- N = Number of reads
- G = Haploid genome length
This equation provides a statistical framework for experimental planning, allowing researchers to calculate the sequencing effort required to achieve a desired coverage level for their specific target, whether it's a full genome, exome, or a custom directed evolution library.
Coverage Requirements Across NGS Applications
Coverage requirements vary significantly across different NGS applications, reflecting their distinct biological questions and technical considerations. The following table summarizes recommended coverage thresholds for common applications:
| Sequencing Method | Recommended Coverage | Primary Rationale | Key Considerations |
|---|---|---|---|
| Whole Genome Sequencing (Human) | 30× to 50× [17] | Balance of comprehensive mapping & cost | Dependent on application and statistical model; sufficient for variant calling in diploid genomes |
| Whole-Exome Sequencing | 100× [17] | Focus on protein-coding regions | Enables reliable detection of heterozygous variants in critical regions |
| RNA Sequencing | Varies (often 20-50 million reads) [17] | Capture dynamic expression range | Depth requirements increase for detection of rare transcripts and splice variants |
| ChIP-Seq | 100× [17] | Identify protein-DNA binding sites | Must account for antibody efficiency and background signal |
| Directed Evolution Libraries | Varies by design [10] | Distinguish enriched variants from background | Must cover full library diversity; higher depth for rare variants |
Special Considerations for Directed Evolution
In directed evolution experiments, coverage requirements extend beyond standard genomic applications. The primary goal is to confidently identify enriched variants resulting from functional selection pressures. A recent study demonstrated that cost-effective, precise identification of active variants is possible even at relatively low coverages with appropriate statistical support [10]. However, the optimal coverage depends on multiple factors:
- Library Diversity: Larger libraries require greater sequencing depth to ensure all variants are adequately represented.
- Selection Stringency: Highly stringent selections with few surviving variants may require deeper sequencing to reliably detect enriched sequences.
- Variant Frequency: Detection of rare variants (e.g., present at <0.1% frequency) demands significantly higher coverage than common variants.
Research indicates that establishing a systematic pipeline for optimizing selection parameters, including coverage requirements, can significantly enhance the efficiency of directed evolution strategies for polymerase engineering and other enzyme optimization projects [10].
Experimental Protocols for Coverage Analysis
Establishing Minimum Coverage Thresholds
Determining appropriate coverage thresholds requires a methodical approach that considers the specific goals of the directed evolution experiment:
- Define Variant Detection Goals: Establish the minimum variant frequency that must be detected with confidence (e.g., variants present at 0.1% frequency post-selection).
- Calculate Theoretical Requirements: Use statistical models (e.g., Poisson distribution) to calculate the depth needed to detect variants at the desired frequency with 95% confidence.
- Account for Technical Variation: Include buffer for technical variation, sequencing errors, and amplification biases that may require additional coverage.
- Validate Empirically: Use control samples with known variants at various frequencies to empirically verify detection sensitivity at different coverage levels.
For clinical NGS applications, the Association of Molecular Pathology and College of American Pathologists have established best practice guidelines that emphasize an error-based approach, identifying potential sources of errors throughout the analytical process and addressing these through test design and validation [12]. While directed evolution experiments may not require clinical-grade validation, these principles provide a robust framework for establishing confidence in variant calls.
Coverage-Based Variant Calling Methodology
The following workflow illustrates the process of establishing and applying coverage thresholds in directed evolution experiments:
Experimental Workflow for Coverage-Based Variant Calling
This workflow emphasizes that applying coverage thresholds (Step 5) is a critical gatekeeping step that occurs after initial data processing but before final variant calling. This ensures that only positions with sufficient data quality contribute to the identification of putative enriched variants.
Validation Using Orthogonal Methods
Even with appropriate coverage thresholds, orthogonal validation of key variants remains essential for high-stakes applications. Sanger sequencing has traditionally served as the gold standard for variant confirmation, though studies now show >99% concordance between NGS and Sanger sequencing for single nucleotide variants (SNVs) in high-complexity regions [21]. For directed evolution outcomes, functional validation of enriched variants through individual expression and characterization provides the most biologically relevant confirmation.
Emerging approaches include machine learning models that can classify variants into high or low-confidence categories based on multiple quality metrics including read depth, allele frequency, sequencing quality, and mapping quality [21]. These models can significantly reduce the burden of confirmatory testing while maintaining high precision and specificity.
Quantitative Data on Coverage and Variant Detection
Coverage Requirements for Different Variant Types
The relationship between sequencing coverage and variant detection confidence varies significantly by variant type and context. The following table summarizes key findings from empirical studies:
| Variant Type | Minimum Recommended Coverage | Detection Confidence | Application Context |
|---|---|---|---|
| Single Nucleotide Variants (SNVs) | 20-30× [12] | >99% [21] | Germline variants in clinical testing |
| Heterozygous SNVs | 30-50× [22] | High with balanced allele ratio | Diploid genomes |
| Rare Somatic Variants | 100-1000× [12] | Varies with variant allele frequency | Cancer genomics (5-10% VAF) |
| Insertions/Deletions (Indels) | 50-100× [12] | Lower than SNVs due to alignment issues | Complex regions require higher depth |
| Gene Amplifications | 50-100× [23] | Strong correlation with FISH (ρ=0.847) [23] | Copy number variation in cancer |
| Directed Evolution Variants | Varies by library size | Enables identification of significantly enriched mutants [10] | Functional screening outputs |
Coverage Impact on False Positive and Negative Rates
The relationship between coverage and variant calling accuracy follows a predictable statistical pattern. In a study evaluating germline genetic variants, researchers found that integrating machine learning models with quality metrics achieved 99.9% precision and 98% specificity in identifying true positive heterozygous SNVs [21]. This highlights how appropriate coverage thresholds combined with quality filters can dramatically improve variant calling accuracy.
For copy number variation, a 2025 study demonstrated that NGS fold changes correlated strongly with FISH metrics (Spearman's ρ = 0.847 for gene copy number) when detecting MET and HER2 amplifications in non-small cell lung cancer [23]. The researchers established a fold change cutoff of 2.0 to effectively distinguish amplified from non-amplified cases, demonstrating how coverage-based metrics can reliably predict molecular events previously requiring orthogonal confirmation.
Research Reagent Solutions for Coverage Optimization
Successful NGS library preparation and coverage analysis depend on specialized reagents and systems. The following table details key solutions referenced in the literature:
| Reagent/Solution | Manufacturer | Primary Function | Application in Directed Evolution |
|---|---|---|---|
| KAPA HiFi DNA Polymerase | Roche [8] | High-fidelity library amplification | Maintains sequence integrity during library prep |
| KAPA HyperPrep Kits | Roche [8] | Library preparation efficiency | Higher library yields, reduced duplicates |
| KAPA HyperPlus Reagents | Roche [21] | Enzymatic fragmentation & library prep | Automated workflow compatibility |
| Twist Target Enrichment | Twist Bioscience [21] [23] | Hybridization-based capture | Custom panel design for specific targets |
| NovaSeq Sequencing System | Illumina [21] [23] | High-throughput sequencing | Enables deep coverage for large libraries |
| BL21 (DE3) Expression Strain | NEB [10] | Protein expression for functional testing | Validation of enriched variant function |
The Relationship Between Coverage and Statistical Confidence
Statistical Models for Coverage and Variant Calling
The confidence in variant calling increases logarithmically with coverage depth according to statistical principles. At low coverage (10-15x), base calling uncertainties lead to higher false negative rates, particularly for heterozygous variants. As coverage increases to 30x, the probability of missing a heterozygous variant drops significantly, approaching the gold standard for germline variant detection [22].
For directed evolution applications, the optimal coverage represents a balance between statistical confidence and practical constraints. A recent study noted that establishing a sequencing coverage threshold for accurate identification of significantly enriched mutants allowed researchers to streamline selection processes using smaller libraries and more cost-effective NGS sequencing [10]. This approach demonstrates how understanding coverage thresholds can improve the efficiency of directed evolution pipelines.
Impact of Coverage Uniformity
While average coverage provides a useful summary metric, coverage uniformity across the target region significantly impacts variant discovery confidence. The Inter-Quartile Range (IQR) metric quantifies statistical variability, reflecting the non-uniformity of coverage across the entire data set [17]. A high IQR indicates high variation in coverage, meaning some regions are significantly under-covered while others are over-covered, potentially leading to gaps in variant detection.
In a directed evolution context, uneven coverage could lead to preferential detection of variants in high-coverage regions while missing potentially beneficial variants in low-coverage regions. Methods to improve uniformity include optimized probe design for hybrid capture-based approaches, PCR optimization to minimize amplification bias, and utilizing molecular barcodes to accurately quantify unique molecules [12].
In directed evolution and other applications requiring high-confidence variant discovery, coverage thresholds serve as the critical link between raw sequencing data and biologically meaningful conclusions. The evidence consistently demonstrates that appropriate coverage thresholds—tailored to the specific variant type, application, and required confidence level—directly dictate the reliability of variant discovery. As NGS technologies continue to evolve and directed evolution libraries increase in complexity, the principles of coverage optimization remain foundational. By applying the systematic approaches outlined here—from experimental design using the Lander/Waterman equation to implementing coverage thresholds in variant calling pipelines—researchers can significantly enhance the statistical rigor and biological relevance of their variant discovery efforts, ultimately accelerating the development of novel enzymes and therapeutics through directed evolution.
Directed evolution serves as a powerful protein engineering tool, mimicking natural selection to optimize enzymes and receptors for industrial and therapeutic applications. For researchers in drug development, validating the outcomes of these experiments is paramount. This guide examines the critical success metrics and compares different analytical approaches, with a specific focus on how Next-Generation Sequencing (NGS) coverage analysis provides the foundation for rigorous, data-driven validation of your directed evolution campaigns.
Core Success Metrics in Directed Evolution
The success of a directed evolution experiment is multi-faceted, quantified through a combination of performance, stability, and functional output measurements. The table below summarizes the key metrics used for a comprehensive assessment.
Table 1: Key Success Metrics for Directed Evolution Experiments
| Metric Category | Specific Metric | Description | Measurement Methods |
|---|---|---|---|
| Functional Production | Functional Protein Yield | Quantity of properly folded, active protein produced [24]. | Spectrophotometry (e.g., Bradford assay), functional activity assays |
| Stability | Thermostability (Tm) | Melting temperature; indicator of protein's resistance to heat denaturation. | Differential Scanning Fluorimetry (DSF), Thermofluor assays |
| Soluble Expression | Level of protein expressed in soluble fraction versus insoluble aggregates [24]. | SDS-PAGE, Western Blot of soluble vs. insoluble fractions | |
| Binding & Kinetics | Binding Affinity (Kd) | Dissociation constant; measures strength of ligand binding. | Surface Plasmon Resonance (SPR), Isothermal Titration Calorimetry (ITC) |
| Catalytic Efficiency (kcat/Km) | Specificity constant for enzyme activity. | Kinetic assays with varying substrate concentrations | |
| Sequencing Outcomes | Variant Enrichment | Significant increase in frequency of beneficial mutants over selection rounds [3]. | Next-Generation Sequencing (NGS) |
| Mutation Load | Average number of mutations per variant in the final enriched pool [3]. | NGS data analysis |
Experimental Protocols for Key Assays
Protocol for Assessing Functional Production and Stability
This protocol is adapted from methods used to engineer stable GPCRs and polymerases [24] [3].
Materials:
- Expression Vector: Contains gene library of protein variants.
- Host Cells: E. coli (e.g., BL21(DE3)) or eukaryotic cells (e.g., S. cerevisiae).
- Lysis Buffer: Suitable for the target protein and host cell.
- Purification Resin: e.g., Ni-NTA resin for His-tagged proteins.
- Analytical Equipment: Spectrophotometer, SDS-PAGE gel system, real-time PCR machine for DSF.
Method:
- Library Transformation: Transform the mutagenic library into the appropriate expression host cell line.
- Protein Expression: Induce expression in small-scale cultures. For stability screening, cultures can be grown at different temperatures.
- Cell Lysis and Fractionation: Lyse cells and separate soluble and insoluble fractions via centrifugation.
- Analysis:
- Functional Yield: Purify the protein from the soluble fraction and quantify the yield.
- Thermostability: Use Differential Scanning Fluorimetry (DSF). Mix purified protein with a fluorescent dye (e.g., SYPRO Orange) that binds hydrophobic patches exposed upon unfolding. Run a thermal ramp in a real-time PCR machine and calculate the melting temperature (Tm) from the resulting fluorescence curve.
Protocol for NGS-Based Variant Enrichment Analysis
This protocol is critical for validating directed evolution outcomes as described in modern pipelines [3] [25].
Materials:
- DNA Library: Plasmid DNA from the initial library and from populations after each selection round.
- PCR Reagents: High-fidelity DNA polymerase (e.g., KAPA HiFi Polymerase, developed via directed evolution for superior performance [8]), NGS library preparation kit.
- Sequencing Platform: e.g., Illumina, PacBio.
- Bioinformatics Tools: Software for sequence alignment (e.g., BWA) and variant calling (e.g., GATK).
Method:
- Library Preparation: Amplify the target gene region from the population DNA using barcoded primers for multiplexing.
- Sequencing: Sequence the prepared libraries on an NGS platform. Long-read technologies (e.g., PacBio) can be valuable for haplotyping, but short-read Illumina is common [24].
- Bioinformatic Analysis:
- Demultiplexing and Quality Control: Separate sequences by sample and remove low-quality reads.
- Variant Calling: Align reads to a reference sequence and identify mutations.
- Variant Enrichment Analysis: Calculate the frequency of each mutation or haplotype in each selection round. Identify variants that show a statistically significant increase in frequency over successive rounds. Studies show that a sequencing coverage of 50-100x per variant in the library can be sufficient for accurate identification of enriched mutants [3].
Visualizing the Directed Evolution Workflow and Success Metrics
The following diagram illustrates the integrated stages of a directed evolution campaign and where key success metrics are applied.
Directed Evolution Workflow and Metrics
The Scientist's Toolkit: Essential Research Reagents
The quality of reagents is critical for reproducibility. The table below details essential solutions used in modern directed evolution experiments, including commercially engineered options.
Table 2: Key Research Reagent Solutions for Directed Evolution
| Reagent / Solution | Critical Function | Example & Key Feature |
|---|---|---|
| High-Fidelity DNA Polymerase | Amplifies mutant libraries for sequencing and cloning with minimal errors. | KAPA HiFi DNA Polymerase: Engineered via directed evolution for ultra-high fidelity and robust amplification, ideal for NGS library prep [8]. |
| NGS Library Preparation Kit | Prepares the genetic library for high-throughput sequencing. | KAPA HyperPrep Kit: Designed for high efficiency and reduced duplicates, improving sequencing data quality [8]. |
| Emulsion Reagents | Creates microreactors for high-throughput screening, linking genotype to phenotype. | Water-in-oil emulsion systems enable compartmentalization for functional screening of millions of variants in parallel [3] [8]. |
| Stable Cell Line | Expresses challenging proteins like GPCRs for functional assays. | Engineered eukaryotic (e.g., HEK293) or prokaryotic (e.g., E. coli BL21) cells optimized for membrane protein expression [24]. |
Defining success in directed evolution requires a multi-parametric approach. While traditional metrics like binding affinity and thermostability remain fundamental, the integration of NGS-based analysis provides an unparalleled, quantitative view of the evolutionary process. By combining these metrics—functional production, biophysical stability, and NGS-driven variant enrichment—researchers can move beyond simple functional screens to a comprehensive understanding of their evolved proteins. This rigorous validation, powered by high-quality engineered reagents, is essential for advancing robust candidates in the drug development pipeline.
From Library to Data: A Practical NGS Workflow for Directed Evolution
In directed evolution, the goal is to mimic natural selection in the laboratory to develop proteins with enhanced functions, such as improved thermostability, specific activity, or resistance to inhibitors [8]. The success of these campaigns hinges on accurately identifying beneficial protein variants through next-generation sequencing (NGS). Library preparation serves as the foundational bridge, transforming the protein variants of interest—via their coding nucleic acids—into sequencable DNA libraries. This process converts a diverse pool of DNA sequences into a format compatible with NGS platforms, ensuring that the resulting data truly represents the underlying genetic diversity created during directed evolution. The fidelity of this step is therefore paramount; any introduction of bias or error can obscure the identification of genuinely improved variants, compromising the entire validation process [26].
Core Technologies and Methodologies for DNA Library Construction
The process of NGS library preparation involves a series of molecular steps designed to fragment DNA, repair the ends, attach universal adapters, and often amplify the final construct. The following workflow diagram outlines the two primary methodological pathways for constructing sequencing libraries.
Detailed Breakdown of Library Preparation Steps
- Fragmentation: DNA is fragmented into manageable sizes (e.g., 200–600 bp) through mechanical or enzymatic methods. Mechanical shearing (e.g., acoustic shearing) offers minimal sequence bias, while enzymatic methods (e.g., tagmentation) are automation-friendly and suitable for lower input amounts [26].
- End Repair & A-Tailing: The fragmented DNA ends are blunted and phosphorylated. A single adenine (A) nucleotide is then added to the 3' ends to facilitate ligation to thymine (T)-overhang adapters [26].
- Adapter Ligation: Sequencing adapters containing flow-cell binding sites, barcodes (indexes), and primer binding sequences are ligated to the fragments. These adapters are essential for cluster generation and sequencing on platforms like Illumina [26].
- Cleanup & Size Selection: Libraries are purified to remove adapter dimers, enzymes, and buffer components. Size selection ensures fragments are within the optimal size range for sequencing, improving data quality [26].
- Library Amplification: A limited-cycle PCR amplifies the adapter-ligated DNA to generate sufficient material for sequencing. Using high-fidelity polymerases is critical to minimize amplification bias and errors, especially critical for detecting true positive variants in directed evolution outcomes [8] [26].
Comparison of Major Library Preparation Approaches
Two principal methods are employed for targeted sequencing, each with distinct advantages for specific applications in validating directed evolution experiments.
Table 1: Comparison of Targeted Library Preparation Methods
| Feature | Hybrid Capture-Based | Amplification-Based (Amplicon) |
|---|---|---|
| Principle | Solution-based hybridization of biotinylated probes to genomic regions of interest, followed by pull-down [12]. | PCR amplification using primers designed for specific genomic targets [12]. |
| Primary Input | DNA [12]. | DNA or RNA (cDNA) [12]. |
| Variant Detection | SNVs, Indels, Copy Number Alterations (CNAs), Structural Variants (SVs) [12]. | Excellent for SNVs and small Indels [12]. |
| Key Advantage | High flexibility in panel design; less prone to allele dropout; suitable for detecting a wide range of variant types [12]. | High sensitivity and specificity for targeted regions; fast and cost-effective for smaller gene sets [12]. |
| Consideration | Requires more input DNA and longer workflow [12]. | Prone to allele-specific dropout if primers overlap with variants [12]. |
Quantitative Performance Comparison of Library Preparation Systems
Selecting an appropriate library preparation system requires a careful evaluation of its performance characteristics. The following data, compiled from independent studies, provides a quantitative basis for this decision-making process.
Table 2: Performance Metrics of Commercial Library Preparation Systems
| System / Kit | Evaluation Context | Key Quantitative Findings | Implication for Directed Evolution |
|---|---|---|---|
| Tecan MagicPrep NGS [27] | Clinical microbial WGS vs. Illumina Nextera DNA Flex | - Hands-on time: 5 hours less per run- Concordance: 100% with reference method- Library Output: Higher concentration and molarity | Improves workflow efficiency for high-throughput screening of microbial enzyme variants. |
| Collibri PS DNA Library Prep Kit [28] | General NGS workflows on Illumina systems | - Workflow Time: ~1.5 hours for PCR-free protocol- Feature: Visual feedback for reagent mixing | Rapid protocol with built-in QC checks reduces preparation errors. |
| Four Exome Capture Platforms [29] | WES on DNBSEQ-T7 sequencer (BOKE, IDT, Nanodigmbio, Twist) | - Reproducibility: Comparable across platforms- Accuracy: Superior technical stability and detection accuracy on DNBSEQ-T7- Uniformity: Achieved with a standardized hybridization workflow | A robust, platform-agnostic capture workflow ensures consistent performance for exome-level variant detection. |
Experimental Protocols for Key Applications
Protocol 1: Automated Library Preparation for High-Throughput Screening
This protocol is adapted from the evaluation of the Tecan MagicPrep NGS system [27] and is ideal for processing large numbers of samples from directed evolution campaigns, such as screening microbial libraries for enzyme variants.
- Sample Input: Use 50-100 ng of genomic DNA from bacterial or fungal clones in a 96-well plate format.
- Automated Processing: Transfer the plate to the automated liquid handling system. The system executes:
- Tagmentation: Fragments DNA and adds adapter sequences in a single-step reaction.
- PCR Amplification: Performs a limited-cycle PCR (e.g., 12-15 cycles) with indexed primers to amplify the library and add unique sample barcodes.
- Normalization and Pooling: Normalizes the concentrations of the individual libraries and pools them into a single tube for sequencing.
- QC Check: Quantify the final library pool using fluorometry (e.g., Qubit) and assess the size distribution and integrity via a fragment analyzer (e.g., Bioanalyzer, TapeStation) [27] [26].
- Sequencing: Dilute the pool to the appropriate molarity and load it onto the sequencer.
Supporting Data: A study implementing this automated approach demonstrated a 5-hour reduction in hands-on time per run while maintaining 100% concordance with manual methods, proving its reliability for high-throughput variant validation [27].
Protocol 2: A Standardized Hybridization-Capture Workflow for Exome Sequencing
This protocol, derived from a comparative study of exome capture platforms, ensures uniform performance across different probe sets, which is critical for comprehensive variant discovery in all protein-coding regions [29].
- Library Construction: Prepare pre-capture libraries from 50 ng of sheared genomic DNA using a universal library prep kit. Include unique dual indexes (UDIs) during the PCR amplification step (e.g., 8 cycles).
- Pre-Capture Pooling: Quantify the pre-capture libraries and pool 8 libraries equimolarly for a multiplexed capture reaction, with a total input of 2000 ng.
- Target Enrichment: Perform hybridization capture using a consistent set of reagents and a 1-hour hybridization time, regardless of the commercial exome probe panel used (e.g., from Twist, IDT, etc.).
- Post-Capture Amplification: Amplify the captured libraries with 12 cycles of PCR.
- Quality Control: Quantify the final yield and validate enrichment efficiency before sequencing on the desired platform (e.g., DNBSEQ-T7 or Illumina systems) [29].
Supporting Data: This standardized workflow was successfully applied to four commercial exome panels, demonstrating uniform and outstanding performance across all of them, which enhances the reliability of variant calling for directed evolution studies [29].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for NGS Library Preparation
| Item | Function in Workflow | Key Considerations |
|---|---|---|
| High-Fidelity DNA Polymerase [8] | Amplifies adapter-ligated fragments with minimal errors. | Essential for accurate representation of variant libraries; enzymes engineered via directed evolution (e.g., KAPA HiFi) offer superior fidelity and robustness [8]. |
| Sequencing Adapters & Barcodes [26] | Attaches fragments to the flow cell and allows sample multiplexing. | Proper design and ligation efficiency are critical for library complexity and avoiding index hopping. |
| Magnetic Beads (e.g., AMPure XP) [26] | Purifies nucleic acids between steps and performs size selection. | The ratio of beads to sample determines the size cutoff, crucial for selecting the ideal insert size. |
| Targeted Enrichment Probes [12] [29] | Hybridizes to and enriches specific genomic regions of interest. | Probe design (e.g., for hybrid capture) impacts coverage uniformity and the ability to detect all variant types without bias. |
| Universal Library Prep Kits [29] | Provides all core reagents for end repair, A-tailing, and ligation in an optimized buffer system. | Using a single kit for all samples, as in the standardized exome workflow, reduces batch effects and improves reproducibility [29]. |
The transition from protein variants to sequencable DNA through meticulous library preparation is a critical determinant for the success of NGS-based validation in directed evolution. As demonstrated, the choice between automated and manual systems, or hybrid-capture versus amplicon-based approaches, carries significant implications for data accuracy, throughput, and operational efficiency. The quantitative data and standardized protocols provided here offer a framework for researchers to build robust, reproducible NGS workflows. By selecting the appropriate library preparation strategy and implementing rigorous quality control, scientists can ensure that the valuable variant information encoded in their directed evolution libraries is accurately captured and translated into reliable sequencing data, thereby accelerating the development of novel enzymes and therapeutics.
Next-Generation Sequencing (NGS) has become an indispensable tool for validating the outcomes of directed evolution experiments. The choice between short-read and long-read sequencing technologies profoundly impacts the accuracy, depth, and scope of the analysis researchers can perform. Within the context of a broader thesis on validating directed evolution outcomes with NGS coverage analysis, this platform selection is a critical methodological step. This guide provides an objective comparison of these technologies, focusing on their performance in analyzing complex variant libraries, supported by experimental data and detailed protocols to inform researchers and drug development professionals.
Technology Comparison: Core Methodologies and Specifications
The two dominant NGS approaches differ fundamentally in their chemistry and output, leading to distinct advantages and limitations.
Short-Read Sequencing Technologies
Short-read technologies, often termed "next-generation sequencing," generate reads typically ranging from 50 to 300 base pairs (bp). They operate on a massive parallel scale.
- Illumina: This dominant technology uses sequencing-by-synthesis. Single-stranded DNA-binding proteins facilitate bridge amplification on a flow cell, followed by the cyclic addition of fluorescently labelled nucleotides. The emitted light determines the base identity [30].
- Ion Torrent: This semiconductor-based method also uses emulsion PCR for clonal amplification. However, it detects incorporated nucleotides through the release of a hydrogen ion, which causes a measurable change in pH, rather than via optics [30].
- Element Biosciences AVITI System: This platform employs a unique "sequencing by binding" (SBB) chemistry. Fluorescently labelled nucleotides bind transiently to the DNA synthesis complex for imaging but are not permanently incorporated, leading to a more natural synthesis process and reported high accuracy (Q40+) [30].
- MGI DNBSEQ Platforms: Utilizing DNA nanoball (DNB) technology, this method is a significant player, particularly in Asia. It involves rolling circle amplification to create DNBs which are then sequenced combinatorially [30].
A key challenge for ensemble-based short-read sequencing is the multi-step library preparation, which includes DNA fragmentation, end repair, adapter ligation, and amplification. This process can introduce biases and is a noted burden [30].
Long-Read Sequencing Technologies
Long-read, or third-generation, sequencing platforms directly sequence single DNA molecules, producing reads that can span thousands to hundreds of thousands of base pairs.
- Pacific Biosciences (PacBio): This technology uses Single Molecule Real-Time (SMRT) sequencing. DNA polymerase is immobilized at the bottom of a zero-mode waveguide with a single DNA molecule template. The fluorescence from nucleotide incorporation is detected in real-time. The revolutionary HiFi method sequences a circularized template multiple times, generating highly accurate long reads (Q30-Q40+) [30].
- Oxford Nanopore Technologies (ONT): ONT threads a single DNA strand through a biological nanopore. As nucleotides pass through the pore, they cause characteristic disruptions to an ionic current, which are decoded to determine the sequence. This technology is capable of producing extremely long reads, theoretically up to millions of base pairs [30].
- Roche Sequencing by Expansion (SBX): An emerging technology currently in early access, SBX uses a combination of nanopores and expandable nucleotides. It is designed to provide high-throughput, accurate mid-length single-molecule sequencing and is expected to be commercially available in 2026 [30].
The following table provides a direct, quantitative comparison of the key specifications of these platforms.
Table 1: Technical Specifications of Major Sequencing Platforms
| Platform (Technology) | Read Length | Accuracy | Key Strengths | Common Applications in Directed Evolution |
|---|---|---|---|---|
| Illumina (Short-read) | 50-300 bp | Very High (Q30+) [30] | High throughput, low per-base cost | Deep variant calling, high-coverage population analysis |
| PacBio HiFi (Long-read) | 5,000 - 20,000 bp | Very High (Q30-Q40+) [30] | Long, accurate reads; detects structural variants | Phasing mutations, resolving complex haplotypes |
| Oxford Nanopore (Long-read) | Up to ~1 Mb | Moderate (improving with depth) [30] | Ultra-long reads, real-time analysis, portable | Sequencing entire plasmids/gene clusters, rapid feedback |
Performance Analysis in Directed Evolution and Metagenomics
The structural differences between the technologies lead to divergent performance in assembling and interpreting the complex genomic data typical of directed evolution libraries.
Assembly Contiguity and Variant Discovery
A critical 2025 metagenomic study directly compared long-read (PacBio HiFi) and short-read (Illumina) assemblies from a complex soil microbiome, offering insights relevant to the heterogeneous populations in directed evolution [31].
- Factor 1: Coverage and Diversity: The study identified that low coverage and high sequence diversity are the two main factors leading to misassemblies in short-read data. Many genomic regions "missed" by short-read assemblies were variable parts of the genome, such as integrated viruses or defense system islands [31]. This directly parallels the challenge of capturing the full diversity of a directed evolution library, where high sequence variation around beneficial mutations can lead to assembly failures.
- Factor 2: Gene Enrichment Analysis: The research performed gene enrichment analysis on consistently assembled versus missed regions. It found that short-read assemblies underestimated the diversity of variable genome regions, suggesting a bias towards conserved sequences [31]. In directed evolution, this could translate to a systematic under-sampling of clones with more significant, structurally complex genomic alterations.
Coverage Requirements for Variant Identification
The coverage depth required for reliable variant detection differs between the technologies and is a crucial economic and design consideration. Research into directed evolution pipelines has specifically explored this.
- Sequencing Coverage Threshold: A 2024 study established that cost-effective, precise, and accurate identification of active variants in directed evolution is possible even at low coverages [10]. This analysis identified a sequencing coverage threshold for the accurate identification of significantly enriched mutants, which is distinct from the requirements for de novo genome assembly. This allows for more efficient sequencing strategies when screening selection outputs.
Error Profiles and Validation
The nature of sequencing errors differs and must be accounted for in bioinformatic pipelines.
- Short-read errors are rare but can be systematic, related to specific sequence contexts or amplification.
- Long-read errors were historically a major limitation. However, PacBio HiFi reads now achieve high accuracy by using multiple passes on a single molecule to generate a consensus read, effectively eliminating random errors [30]. While raw Oxford Nanopore reads can have a higher error rate, consensus accuracy from deep coverage data is now sufficient for many applications [30].
Table 2: Experimental Performance in Complex Sequence Analysis
| Performance Metric | Short-Read Sequencing | Long-Read Sequencing |
|---|---|---|
| Assembly of Repetitive Regions | Struggles; leads to fragmentation [31] | Excels; long reads span repeats [30] [31] |
| Variant (Haplotype) Phasing | Limited to short distances | Presents a key strength; links distant mutations |
| Detection of Structural Variants | Limited | Highly effective [30] |
| Recovery of Variable Genomic Regions | Can underestimate diversity [31] | Improves recovery of variable regions [31] |
Experimental Protocols for Technology Comparison
To objectively compare platforms for a specific directed evolution project, the following experimental protocols can be implemented.
Protocol: Hybrid Assembly for Directed Evolution Validation
Objective: To leverage the high accuracy of short reads and the contiguity of long reads to generate a high-quality reference for validating directed evolution outcomes.
Methodology:
- Sample Preparation: Extract genomic DNA from a pooled population of evolved clones.
- Parallel Library Preparation & Sequencing:
- Prepare and sequence a library on a short-read platform (e.g., Illumina) to achieve high depth (>100x).
- Prepare and sequence a library on a long-read platform (e.g., PacBio HiFi) to achieve a shallower depth but long contiguity.
- Hybrid Assembly: Use a hybrid assembler (e.g., metaSPAdes [31]) to combine the short and long reads. The short reads correct base-level errors in the long reads, while the long reads scaffold the assembly.
- Variant Calling and Haplotype Phasing: Map all sequencing reads (both short and long) back to the hybrid assembly. Use the long reads to phase mutations and determine which combinations of mutations exist on the same DNA molecule.
Protocol: Analysis of Selection Enrichment and Parasites
Objective: To identify truly enriched variants and discriminate them from "parasite" sequences that are recovered due to non-specific processes.
Methodology:
- Pre- and Post-Selection Sequencing: Perform deep sequencing on the plasmid library both before and after a round of selection.
- Variant Frequency Calculation: Calculate the frequency of every unique variant in the pre- and post-selection libraries.
- Enrichment Score: Compute an enrichment score (e.g., log2(Post-Frequency / Pre-Frequency)) for each variant.
- Statistical Analysis: As described in directed evolution research, establish a sequencing coverage threshold to accurately identify variants with statistically significant enrichment, distinguishing them from background noise and parasites [10].
Visualizing the Experimental and Decision Workflow
The following diagrams map the logical workflows for experimental comparison and platform selection.
Directed Evolution NGS Validation Workflow
Sequencing Platform Selection Logic
The Scientist's Toolkit: Key Research Reagent Solutions
The following reagents and materials are essential for executing the NGS workflows described in this comparison.
Table 3: Essential Research Reagents for NGS in Directed Evolution
| Reagent / Material | Function in Workflow | Technology Example |
|---|---|---|
| High-Fidelity DNA Polymerase | Amplifies library for sequencing with ultra-low error rate to avoid introducing false mutations. | KAPA HiFi DNA Polymerase, engineered via directed evolution for high fidelity and robustness [8]. |
| Hybrid Capture Probes | Enriches sequencing libraries for specific target genes from a complex genomic background. | Biotinylated oligonucleotide probes that hybridize to regions of interest, used in hybrid capture-based NGS [12]. |
| NGS Library Preparation Kit | Prepares DNA fragments for sequencing by adding required adapters and barcodes. | KAPA HyperPrep Kits for increased library preparation efficiency and improved coverage [8]. |
| Emulsion PCR Reagents | Used in some short-read platforms (Ion Torrent) and directed evolution screens to create clonally amplified DNA beads. | Water-in-oil emulsion reagents that physically separate individual amplification reactions [30] [10]. |
| SMRTbell Adapters | Creates a circular DNA template essential for the PacBio HiFi consensus sequencing process. | Hairpin adapters ligated to double-stranded DNA to enable multiple passes of the same insert [30]. |
In the context of validating directed evolution outcomes, determining the optimal sequencing depth is a fundamental prerequisite for reliable variant identification. Next-Generation Sequencing (NGS) does not read each base in a genome just once; instead, it generates millions of short fragments ("reads"), and the number of times a specific nucleotide is sequenced is known as its sequencing depth or read depth [16]. This parameter is distinct from coverage, which refers to the percentage of the target genome region that has been sequenced at least once [16].
The profound importance of depth lies in its direct relationship to data accuracy and sensitivity. In a directed evolution experiment, where identifying novel mutations is the goal, sufficient depth is required to distinguish true, low-frequency variants from background sequencing errors with high statistical confidence [32]. Achieving this balance is not trivial, as insufficient depth risks missing true variants (false negatives), while excessive depth makes the experiment inefficient and costly [16] [32]. This guide synthesizes current experimental data and methodologies to help researchers make evidence-based decisions on sequencing depth for their specific projects.
Core Concepts and Quantitative Guidelines
Defining Depth and Its Relationship to Variant Detection
Sequencing Depth is quantitatively defined as the number of times a particular nucleotide is read during the sequencing process [16]. It is often expressed as an average across the target region (e.g., 100x depth). Variant Allele Frequency (VAF) describes the proportion of sequencing reads that contain a specific variant at a given genomic position [32].
The power to detect a variant is fundamentally governed by its VAF and the available sequencing depth. The lower limit of reliable variant detection is directly tied to depth; detecting a variant present in only 1% of a population (VAF=1%) with high confidence requires a much higher depth than detecting a common variant (VAF=50%) [32]. The following diagram illustrates this core relationship and the key influencing factors.
Data-Driven Depth Recommendations
The optimal sequencing depth is not a single universal value but is determined by the specific application and the type of genetic variation under investigation. The following table consolidates recommended depth ranges based on current practices and research, which should serve as a starting point for experimental design.
Table 1: Recommended Sequencing Depth for Variant Identification Applications
| Application / Variant Type | Recommended Depth | Key Rationale and Context |
|---|---|---|
| Rare Somatic Variants / MRD (Measurable Residual Disease) | ≥ 1000x [32] | Essential for confidently detecting variants with very low Variant Allele Frequencies (VAF < 1%) [32]. |
| Germline Variant Discovery (e.g., Rare Disease Diagnosis) | ~50x (for WGS) [33] [34] | A widely accepted threshold for large-scale genomic studies. One optimized study reported >93% of bases with Q>30 and a mean coverage of >50x [34]. |
| Tumor Profiling | High Depth (Specifics vary) [35] | Required to account for tumor heterogeneity and identify subclonal populations. Often uses targeted panels to enable very high depth. |
| Low-Frequency Variants | Varies based on VAF [32] | Depth must be increased inversely to the VAF you aim to detect. The lower the VAF, the higher the required depth. |
| General Variant Calling | Balanced based on goal [16] | Must balance confidence in variant calling with cost and resource constraints [16]. |
Experimental Protocols and Optimization Strategies
A Protocol for Determining Depth and Batching
A critical practical consideration in achieving optimal depth is sequencing batching—pooling multiple samples in a single sequencing run to maximize throughput and cost-efficiency [32]. The following protocol provides a framework for designing a sequencing run that balances depth, sensitivity, and cost.
Table 2: Key Reagents for NGS Library Preparation and Optimization
| Research Reagent / Solution | Function in Workflow |
|---|---|
| Tn5 Transposase | Enzyme that simultaneously fragments DNA and attaches adapter sequences in a single "tagmentation" step, significantly streamlining library preparation [36]. |
| Unique Molecular Identifiers (UMIs) | Short nucleotide sequences that tag individual DNA molecules before amplification. They help distinguish true biological variants from errors introduced during PCR or sequencing, which is crucial for low-VAF detection [32]. |
| High-Fidelity DNA Polymerase | An accurate PCR enzyme with low error rates, used during library amplification to minimize the introduction of mutations during the preparation process [37]. |
| Size Selection Beads | Magnetic beads used to isolate and purify DNA fragments within a specific size range, ensuring a uniform library and removing unwanted adapter dimers or too-short/long fragments [37]. |
| Dual-Indexed Barcoded Primers | Primers containing unique molecular barcodes that allow multiple samples to be pooled (multiplexed) in a single sequencing run and later bioinformatically separated [37]. |
Workflow:
- Define the Limit of Detection (LOD): Determine the lowest VAF that is biologically or clinically significant for your directed evolution experiment (e.g., 0.1%, 1%, 5%).
- Calculate Total Data Needed: Based on your LOD and the size of your target region (e.g., whole genome, exome, or a specific amplicon), use statistical models (e.g., based on Poisson distribution) to estimate the total number of reads required to detect a variant at that VAF with a desired confidence level.
- Account for Sample Quality: If working with degraded or low-input DNA, consider increasing the calculated depth by 10-20% to compensate for potential technical noise and lower data quality [32].
- Determine Batch Size: Divide the total output capacity of your sequencing flow cell (e.g., in Gb) by the total data needed per sample. The result is the maximum number of samples you can batch together while maintaining the required depth.
- Example: For a flow cell with a 120 GB capacity and a requirement of 5 GB per sample, the maximum batch size is 24 samples.
- Validate with a Pilot Run: Before running all precious samples, perform a pilot sequencing run using a control sample with known variants at different frequencies. This validates that your chosen depth and bioinformatic pipeline can reliably detect variants at your desired LOD [32].
This workflow is summarized in the following diagram, which outlines the key decision points and their consequences for a sequencing project.
Case Study: Optimized Viral Genome Sequencing
A 2025 study on Influenza A virus (IAV) genomics provides an excellent example of wet-lab protocol optimization to improve sequencing efficiency and data quality, even from challenging samples [37].
Objective: To enhance the recovery of complete IAV genomes from clinical samples with low viral loads, ensuring all eight genomic segments are well-represented [37].
Optimized Methodology:
- Reverse Transcription (RT): Used the LunaScript RT Master Mix Kit with a modified primer ratio (MBTuni-12 and MBTuni-12.4 at a 1:4 ratio) and optimized cycling conditions (2 min at 25°C, 30 min at 55°C) to improve cDNA yield of large segments [37].
- PCR Amplification: Employed 0.02 U/μL of Q5 Hot Start High-Fidelity DNA Polymerase with 35 PCR cycles to amplify all segments from minimal cDNA input [37].
- Library Preparation: Introduced a dual-barcoding approach for the Oxford Nanopore platform, enabling high-throughput multiplexing of at least eight samples without significant loss of sensitivity [37].
Outcome: This optimized mRT-PCR protocol demonstrated improved amplification of all eight IAV segments, particularly the large polymerase genes (PB1, PB2, PA), which are often challenging to recover from low-concentration samples. The workflow proved robust for avian, swine, and human IAV samples, strengthening genomic surveillance capabilities [37].
The Scientist's Toolkit for Variant Prioritization
Once sequencing data is generated, the subsequent challenge is the bioinformatic prioritization of variants. For rare disease diagnosis—a process analogous to identifying meaningful mutations in a directed evolution pool—Exomiser is a widely adopted open-source tool.
A 2025 study on Undiagnosed Diseases Network data established an optimized protocol for this software [33]:
- Key Parameters for Optimization: Performance is highly dependent on the configuration of gene-phenotype association data, variant pathogenicity predictors, the quality and quantity of phenotype terms (using Human Phenotype Ontology/HPO), and the accuracy of family variant data [33].
- Performance Gains: By systematically optimizing these parameters, researchers significantly improved Exomiser's performance. The percentage of coding diagnostic variants ranked within the top 10 candidates increased from 49.7% to 85.5% for genome sequencing (GS) data, and from 67.3% to 88.2% for exome sequencing (ES) data [33].
- Practical Recommendation: This case highlights that beyond wet-lab depth, the selection and configuration of bioinformatic tools are critical for efficient and accurate variant identification. Researchers should leverage such evidence-based optimizations for their own analyses.
In directed evolution experiments, the precise bioinformatic processing of next-generation sequencing (NGS) data is crucial for validating enrichment outcomes and identifying beneficial mutations. This analysis transforms raw sequencing data into actionable biological insights, enabling researchers to quantify mutation frequencies, track variant enrichment across selection rounds, and confirm the success of evolutionary experiments. The bioinformatic workflow for enrichment analysis follows a structured pipeline of primary, secondary, and tertiary analysis stages [38], each employing specialized tools and algorithms to ensure accurate interpretation of complex NGS datasets. As the field advances, the integration of long-read sequencing technologies and machine learning-enhanced variant callers is pushing the boundaries of analytical precision for directed evolution studies [39].
The NGS Bioinformatics Workflow: From Raw Data to Enrichment Metrics
The standard bioinformatic processing of NGS data follows a structured, three-tiered analytical framework that systematically converts raw sequencing output into validated enrichment profiles.
The following diagram illustrates the complete bioinformatic processing pipeline for NGS enrichment analysis:
Primary Analysis: Quality Assessment and Demultiplexing
Primary analysis begins with converting raw sequencing data from the instrument-specific format (such as Illumina's BCL files) into standardized FASTQ files containing sequence reads and quality scores [38]. This critical first step includes demultiplexing to separate pooled samples using their unique barcodes, generating individual FASTQ files for each library in the experiment [40]. Quality metrics assessed at this stage include:
- Phred Quality Scores (Q-scores): Probability of incorrect base calling, where Q30 represents 99.9% base call accuracy [38]
- Cluster Density: Measure of optimal flow cell loading, with >80% passed filter (%PF) clusters considered optimal [38]
- Error Rate: Percentage of incorrect base calls based on internal controls like PhiX genome [38]
Tools like FastQC provide comprehensive quality assessment through per-base sequence quality plots, sequence duplication levels, and adapter contamination analysis [38]. For enrichment studies, rigorous primary analysis is essential as low-quality data can significantly skew variant frequency calculations.
Secondary Analysis: Read Mapping and Variant Calling
Secondary analysis converts quality-filtered sequencing reads into identified genetic variants through a multi-step process:
Read Cleanup: Adapter sequences and low-quality base calls are trimmed from reads. For enrichment analysis, the use of Unique Molecular Identifiers (UMIs) is particularly valuable as they enable correction for PCR amplification biases and sequencing errors by tracking individual molecules through the library preparation process [38].
Sequence Alignment: Cleaned reads are mapped to a reference sequence using aligners such as BWA (Burrows-Wheeler Aligner) or Bowtie2 [38]. The alignment output is stored in BAM (Binary Alignment Map) format, which can be visually inspected in genome browsers like the Integrative Genomic Viewer (IGV) to verify mapping quality [38].
Variant Calling: Specialized algorithms identify mutations relative to the reference sequence. For directed evolution studies, sensitive detection of low-frequency variants is critical for capturing early enrichment signals. The Genome Analysis Toolkit (GATK) provides a comprehensive suite for variant discovery, offering high accuracy in SNP and indel detection [41]. Emerging tools like DeepVariant use deep learning approaches to improve calling accuracy, particularly for challenging genomic regions [39].
Tertiary Analysis: Enrichment Quantification and Functional Interpretation
Tertiary analysis represents the specialized phase where biological meaning is extracted from variant calls specifically for directed evolution applications:
Variant Annotation: Tools like SnpEff annotate variants with functional consequences (missense, nonsense, synonymous), predicting their impact on protein function [41]. This step categorizes mutations based on their potential biological effects.
Frequency Calculation: Variant allele frequencies are calculated for each selection round, establishing baseline measurements and tracking changes over time. The formula for variant frequency calculation is:
[ \text{Variant Frequency} = \frac{\text{Number of reads containing variant}}{\text{Total reads at position}} \times 100\% ]
Enrichment Scoring: Statistical measures quantify the significant changes in variant frequencies between selection rounds. Enrichment p-values can be calculated using Fisher's exact test or binomial tests to identify variants under positive selection.
Pathway Analysis: For complex phenotypes, enriched variants are mapped to biological pathways using databases like KEGG or Reactome to identify selected functional modules [39].
Comparative Analysis of Bioinformatics Tools for Enrichment Studies
The selection of appropriate bioinformatics tools significantly impacts the sensitivity, accuracy, and interpretability of enrichment analysis in directed evolution experiments.
Key Tool Categories and Performance Metrics
Table 1: Comparison of Bioinformatics Tools for NGS Enrichment Analysis
| Tool Category | Tool Name | Key Features | Accuracy Metrics | Best For | Limitations |
|---|---|---|---|---|---|
| Sequence Alignment | BWA [34] | Memory-efficient, supports paired-end reads | High mapping accuracy for short reads | General purpose alignment | Not optimized for long reads |
| Bowtie2 [38] | Ultra-fast alignment, FM-index based | Excellent speed with good accuracy | Large-scale studies requiring speed | Slightly lower accuracy than BWA | |
| Variant Calling | GATK [41] | Comprehensive variant discovery pipeline | >99% for SNP detection | Population-level variant calling | Computationally intensive |
| DeepVariant [39] | Deep learning-based variant caller | Superior accuracy in repetitive regions | Challenging genomic regions | Requires significant computational resources | |
| Strelka2 [39] | Optimized for somatic and low-frequency variants | High sensitivity for indels | Detection of rare variants | Complex installation and configuration | |
| Visualization | IGV [38] | Interactive exploration of alignment data | N/A | Debugging alignment issues | Not for batch processing |
| Cytoscape [41] | Network visualization of enriched pathways | N/A | Pathway analysis and integration | Steep learning curve | |
| Workflow Management | Nextflow [39] | Reproducible pipeline development | N/A | Complex, multi-step analyses | Requires programming knowledge |
Tool Selection Guidelines for Directed Evolution Studies
For directed evolution applications, tool selection should prioritize sensitivity for low-frequency variants and accurate indel detection, as these often represent key functional mutations. The GATK toolkit remains the gold standard for variant calling due to its rigorous validation and high accuracy, though DeepVariant shows promise for detecting complex variants [41] [39]. For alignment, BWA-MEM provides excellent balance between accuracy and computational efficiency for short-read data [34].
When working with long-read sequencing technologies (increasingly used for complex structural variants), specialized aligners like minimap2 and variant callers optimized for PacBio HiFi or Oxford Nanopore data are essential [42] [39]. The integration of UMI-based error correction significantly improves accuracy for low-frequency variant detection, crucial for identifying early enrichment signals in initial selection rounds [38].
Experimental Protocols for Enrichment Validation
Robust experimental design and validation are essential for confirming directed evolution outcomes through NGS enrichment analysis.
Protocol 1: Variant Enrichment Tracking Across Selection Rounds
This protocol enables quantitative monitoring of mutation frequency changes throughout directed evolution campaigns.
Methodology:
- Sample Collection: Collect biomass from each selection round for genomic DNA extraction
- Library Preparation: Prepare NGS libraries using fragmentation and adapter ligation, incorporating UMIs to control for amplification biases [40]
- Sequencing: Sequence libraries to sufficient depth (typically 100-200x coverage) to ensure statistical power for detecting frequency changes
- Variant Calling: Process data through the bioinformatic pipeline outlined above, using consistent parameters across all samples
- Frequency Normalization: Normalize variant frequencies to internal control sequences to account for technical variations
- Enrichment Calculation: Compute enrichment ratios between consecutive rounds using the formula:
[ \text{Enrichment Ratio} = \frac{\text{Variant Frequency}{round\,n}}{\text{Variant Frequency}{round\,n-1}} ]
Validation Metrics:
- Statistical Significance: Apply multiple testing correction (e.g., Benjamini-Hochberg) to enrichment p-values
- Replicate Concordance: Compare biological replicates to identify consistently enriched variants
- Dose-Response Correlation: Correlate variant enrichment with selection pressure intensity
Protocol 2: Functional Validation of Enriched Variants
This protocol confirms the functional contribution of enriched mutations to the selected phenotype.
Methodology:
- Variant Prioritization: Select top enriched variants based on statistical significance and magnitude of enrichment
- Site-Directed Mutagenesis: Introduce individual mutations into the parental background using CRISPR-Cas9 or other genome editing tools [42]
- Phenotypic Screening: Assay individual variants for the selected phenotype using medium-throughput functional assays
- Additivity Testing: Combine multiple enriched mutations to test for synergistic effects
- Structural Analysis: Map enriched mutations to protein structures to rationalize functional mechanisms
Interpretation Framework:
- Beneficial Variants: Show significant improvement in selected phenotype over parental sequence
- Neutral Variants: No significant functional effect despite enrichment (may represent hitchhiker mutations)
- Synergistic Combinations: Variant pairs showing greater-than-additive improvement
Research Reagent Solutions for NGS Enrichment Analysis
Table 2: Essential Research Reagents and Platforms for NGS Enrichment Studies
| Category | Product/Platform | Key Features | Application in Enrichment Studies |
|---|---|---|---|
| Library Prep | Illumina DNA Prep | Efficient fragmentation and adapter ligation | Standardized library construction for variant detection |
| MGIEasy FS DNA Library Prep Kit [34] | Enzymatic fragmentation, exome capture compatible | Target enrichment for focused evolution studies | |
| Target Enrichment | SureSelect (Agilent) [43] | Hybrid capture-based enrichment | Focusing sequencing on specific genomic regions |
| AmpliSeq (Ion Torrent) [43] | Amplicon-based targeted sequencing | High-sensitivity variant detection in defined regions | |
| Sequencing Platforms | Illumina NovaSeq Series | Ultra-high throughput, short reads | Comprehensive variant discovery in complex populations |
| PacBio Revio [42] | HiFi long reads with >99.9% accuracy | Resolving complex structural variants | |
| Oxford Nanopore PromethION | Ultra-long read capability | Haplotype phasing in evolved populations | |
| Automation & Analysis | Nextflow [39] | Reproducible workflow management | Standardized processing across multiple experiments |
| Unique Molecular Identifiers (UMIs) [38] | Molecular barcoding for error correction | Accurate frequency measurement for rare variants |
Emerging Trends and Future Directions
The field of NGS bioinformatics for enrichment analysis is rapidly evolving, with several key trends shaping future methodologies:
AI-Enhanced Variant Calling: Machine learning approaches, particularly deep learning models, are increasingly being integrated into variant calling pipelines to improve accuracy, especially for challenging variant types like complex indels and structural variants [39]. Tools like DeepVariant demonstrate how convolutional neural networks can outperform traditional statistical methods.
Multi-Omics Integration: Researchers are combining NGS data with transcriptomic, proteomic, and metabolomic datasets to gain comprehensive understanding of how enriched genetic variants influence cellular phenotypes [39]. This systems biology approach is particularly valuable for complex traits influenced by multiple genetic factors.
Single-Cell Sequencing Applications: The integration of single-cell RNA sequencing with enrichment analysis enables tracking of variant effects at cellular resolution, revealing how mutations influence transcriptional heterogeneity in evolved populations [39].
Real-Time Analysis Platforms: Cloud-based platforms and serverless computing architectures are making large-scale NGS analysis more accessible, with tools like the Illumina Connected Software portfolio offering streamlined analysis solutions for researchers without extensive bioinformatics expertise [44].
As these technologies mature, they will further enhance our ability to extract meaningful biological insights from directed evolution experiments, accelerating the engineering of improved enzymes, biosynthetic pathways, and microbial chassis for biotechnology applications.
Xenobiotic nucleic acids (XNAs) are synthetic genetic polymers with immense potential in biotechnology and molecular medicine, offering properties like nuclease resistance that natural DNA lacks [45] [46]. A significant barrier to their application is that natural DNA polymerases cannot efficiently synthesize or reverse-transcribe these unnatural genetic polymers. Directed evolution has emerged as a powerful strategy to engineer novel polymerase enzymes capable of processing XNAs, bypassing the need for complete structural knowledge of these proteins [10].
This case study examines the practical application of a directed evolution workflow to engineer XNA polymerases, with a specific focus on validating outcomes through Next-Generation Sequencing (NGS) coverage analysis. We will objectively compare the performance of several engineered polymerase variants and detail the experimental protocols required to replicate this research, providing a framework for scientists engaged in enzyme engineering and synthetic biology.
The Directed Evolution and Validation Workflow
The process of engineering and validating novel XNA polymerases follows a systematic workflow that integrates directed evolution, functional screening, and deep sequencing analysis. The diagram below illustrates this multi-stage pipeline.
Diagram 1: The directed evolution workflow for engineering XNA polymerases. The process begins with library design targeting key polymerase residues, proceeds through iterative selection rounds, and culminates in NGS-based validation of enriched variants.
The successful execution of this workflow relies on several critical considerations. Library design often targets specific polymerase domains, such as the palm and thumb subdomains, which are crucial for substrate specificity [10]. During the directed evolution phase, a bacterial selection system is frequently employed. This system links the survival of E. coli expressing a polymerase variant to its ability to synthesize XNA, thereby cleaving a lethal gene (ccdB) placed under arabinose-inducible control [47]. Following selection, NGS analysis is paramount. It moves beyond simple variant identification to analyze enrichment levels and ensure sufficient sequencing coverage, which is critical for distinguishing genuinely improved mutants from background noise or parasitic false positives that survive the selection without the desired activity [10].
Comparative Performance of Engineered XNA Polymerases
Directed evolution campaigns have yielded several notable XNA polymerase variants. The table below summarizes the synthesis fidelity and key characteristics of leading engineered mutants, primarily from the Taq DNA polymerase family, for the synthesis of 2'-fluoroarabino nucleic acid (2′F XNA).
Table 1: Performance Comparison of Engineered Taq XNA Polymerase Variants
| Polymerase Variant | Key Mutations | Error Rate (errors per 10³ bp) | Template Length | Key Findings / Application |
|---|---|---|---|---|
| SFM4-3 | Not specified | 6.9 [46] | Various | Lower error rate among first-gen mutants; used in aptamer generation [46]. |
| SFM4-6 | Not specified | 19.1 [46] | Various | Higher error rate limits applications requiring high fidelity [46]. |
| SFP1 | Not specified | 5.6 [46] | Various | One of the most accurate 2'F XNA synthetases identified [46]. |
| SFP1 with fidelity mutations | Adds E708, A737V | ~2.4 (estimated from template-corrected data) [46] | 100 bp | Demonstrated that mutations improving natural DNA fidelity can also enhance XNA synthesis accuracy [46]. |
| SFM4-3 with fidelity mutations | Adds E708, A737V | ~3.7 (estimated from template-corrected data) [46] | 100 bp | Confirmed the generalizability of rational fidelity enhancement in XNA polymerases [46]. |
| Tgo:TGK (B-family) | Not specified | 0.2 - 8.0 (across various XNAs) [46] | 74 bp (supF tRNA) | Capable of synthesizing various XNAs, but accuracy is highly substrate-dependent [46]. |
The data reveal that while significant progress has been made, engineered XNA polymerases still function with significantly lower efficiency and fidelity than their natural counterparts, which can have error rates below 1.5 x 10⁻⁵ errors per base pair [46]. A study systematically adding fidelity-enhancing mutations (e.g., E708, A737V) to leading XNA polymerase variants demonstrated a rational path toward closing this performance gap, resulting in mutants with significantly improved accuracy for 2'F XNA synthesis [46].
Experimental Protocols
Protocol 1: Library Construction and Directed Evolution for XNA Polymerases
This protocol is adapted from methodologies used to evolve polymerases and other DNA-editing enzymes [47] [10].
Library Generation by Error-Prone PCR:
- Target: Amplify DNA fragments encoding specific polymerase domains known to interact with the substrate, such as the palm or thumb domains.
- Reaction Setup: In a 100 µL reaction, use 10 µL of 10x ThermoPol reaction buffer, 2 µL of each primer (10 mM), 30 ng of template plasmid, 1 µL of ThermoTaq DNA Polymerase, and 2.4 µL of 10 mM MnCl₂ to introduce a controlled mutation rate (e.g., 6–9 mutations per kilobase) [47].
- Cycling Conditions: 28 cycles of standard PCR amplification.
- Cloning: Use Gibson assembly to replace the corresponding wild-type fragment in a bacterial expression plasmid with the error-prone PCR product.
Bacterial Selection for XNA Synthesis Activity:
- Preparation: Electroporate 2 ng of the variant plasmid library into 50 µL of competent E. coli BW25141(DE3) cells harboring a selection plasmid. This selection plasmid contains an arabinose-inducible ccdB lethal gene and an ampicillin resistance marker [47].
- Selection Plate: After a recovery period, plate the bacterial culture on Petri dishes containing chloramphenicol (to select for the variant plasmid) and arabinose (to induce the lethal gene).
- Principle: Only bacterial colonies expressing polymerase variants that can successfully synthesize XNA to cleave and inactivate the ccdB gene will survive [47].
- Variant Recovery: Collect surviving colonies, isolate their plasmids, and sequence the polymerase genes to identify enriched mutations.
Protocol 2: Fidelity Assay for XNA Synthesis
This protocol describes a method to quantitatively measure the error rate of an XNA polymerase, as employed in recent studies [46].
XNA Synthesis:
- Use the mutant polymerase to synthesize a 2′F XNA strand from a defined natural DNA template.
- Reaction Conditions: Perform synthesis in the absence of manganese and with enzyme concentrations close to stoichiometric to resemble cellular conditions and improve native fidelity [46].
Template Removal and Purification:
- Digest the original DNA template and primer using DNase I or a similar nuclease.
- Purify the synthesized 2′F XNA product using column-based purification.
Reverse Transcription and Amplification:
- Reverse transcribe the purified XNA back into DNA using an appropriate reverse transcriptase.
- Amplify the resulting DNA in a one-pot reaction using a high-fidelity DNA polymerase like Q5.
Sequencing and Error Rate Calculation:
- Submit the final DNA product for NGS.
- Analyze the sequencing data by comparing the sequences of the output reads to the original DNA template sequence.
- Quantify the error rate in errors per thousand base pairs synthesized (eptbp), subtracting the background error rate from a template-only control [46].
Protocol 3: NGS Coverage Analysis for Directed Evolution Validation
Optimizing NGS for directed evolution output analysis is crucial for cost-effective and accurate variant identification [10].
Sample Preparation:
- Pool plasmids from enriched colonies after one or more rounds of selection.
- Prepare the NGS library according to the platform's standard protocol (e.g., Illumina, Ion Torrent).
Sequencing and Data Analysis:
- Sequence the library to an appropriate depth. Studies suggest that cost-effective and precise identification of active variants is achievable even at relatively low coverages, which differ from the requirements of genome assembly projects [10].
- Variant Calling: Map the sequencing reads to the wild-type polymerase reference sequence to identify mutations.
- Enrichment Analysis: Calculate the frequency of each mutation or variant in the post-selection pool and compare it to its frequency in the initial library. Significantly enriched mutations are strong candidates for contributing to the desired function.
- Threshold Determination: Establish a sequencing coverage threshold that allows for the confident identification of significantly enriched mutants, minimizing false positives and negatives [10].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Engineering and Evaluating XNA Polymerases
| Reagent / Solution | Function in the Workflow |
|---|---|
| ThermoTaq DNA Polymerase & MnCl₂ | Used in error-prone PCR for random mutagenesis during library generation [47]. |
| Bacterial Selection System (e.g., ccdB lethal gene) | Provides the selection pressure to isolate functional XNA polymerase variants from large libraries [47]. |
| 2′F-rNTPs (dNTP analogs) | The modified nucleoside triphosphate substrates for XNA synthesis, used in both selection and fidelity assays [46] [10]. |
| N-cyanoimidazole (CNIm) & Mn²⁺ | A chemical ligation system used in non-enzymatic template-directed synthesis of some XNAs, relevant for prebiotic chemistry studies [48]. |
| High-Fidelity DNA Polymerase (e.g., Q5) | Used to amplify DNA after reverse transcription in the fidelity assay to avoid introducing additional errors during PCR [46]. |
| NGS Platform (e.g., Illumina, PGM) | Enables deep sequencing of variant libraries before and after selection to identify enriched mutations and validate outcomes [10] [49]. |
The directed evolution workflow, when coupled with rigorous NGS-based validation, provides a robust framework for engineering XNA polymerases. Current research has successfully produced a range of variants with demonstrable activity in synthesizing artificial genetic polymers. However, performance gaps in fidelity and efficiency compared to natural polymerases remain. The ongoing integration of rational design—such as adding known fidelity mutations—into the directed evolution pipeline represents a powerful synergistic approach. This strategy is rapidly advancing the field toward the goal of creating highly efficient and accurate XNA polymerases, which will unlock the full potential of XNAs in synthetic biology, diagnostics, and therapeutic applications.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas12a has emerged as a powerful genome-editing tool with distinct advantages over Cas9, including its ability to process multiple CRISPR RNAs from a single transcript and generate staggered double-strand breaks [47] [50]. However, its widespread application has been constrained by a fundamental limitation: a stringent requirement for a 5'-TTTV-3' protospacer adjacent motif (PAM) sequence adjacent to its DNA target sites. This requirement restricts Cas12a's targeting capability to approximately 1% of a typical genome [47] [51], significantly limiting its utility for both basic research and therapeutic development.
To overcome this constraint, researchers have turned to directed evolution—a powerful protein engineering approach that mimics natural selection in laboratory settings. This process involves introducing random mutations into a target gene and applying selective pressure to isolate variants with enhanced properties [8]. Recent advances have combined this methodology with next-generation sequencing (NGS) analysis, enabling high-throughput screening of Cas12a variants with expanded PAM compatibility [50]. This case study examines how NGS-driven directed evolution has been utilized to engineer Cas12a variants with relaxed PAM requirements, focusing on experimental protocols, data validation, and comparative performance analysis.
Directed Evolution & NGS Workflow
Directed Evolution Strategy for PAM Relaxation
The directed evolution campaign for Cas12a PAM relaxation employed a bacterial-based selection system with stringent positive and negative selection components [47]. The methodology leveraged a dual-plasmid approach in E. coli, where:
- A chloramphenicol-resistant (CAM+) expression plasmid harbored the mutagenized Lachnospiraceae bacterium Cas12a (LbCas12a) gene library under a TetR promoter, along with a specific crRNA under a strong constitutive proD promoter.
- An ampicillin-resistant (Amp+) selection plasmid contained an arabinose-inducible ccdB lethal gene with integrated target sequences adjacent to non-canonical PAMs.
The selection process applied dual pressure: bacterial survival depended on functional Cas12a variants successfully cleaving the lethal ccdB gene only when recognizing non-canonical PAM sequences. This sophisticated selection system enabled efficient enrichment of PAM-relaxed variants through multiple evolution rounds [47].
Library Generation and Mutagenesis
The mutagenesis strategy specifically targeted the PAM-interacting (PI) and wedge (WED) domains of LbCas12a, as structural studies identified these regions as critical for PAM recognition [47]. Researchers employed error-prone polymerase chain reaction with optimized MnCl₂ concentrations to achieve an mutation rate of 6-9 nucleotide mutations per kilobase. This generated four independent libraries, each containing approximately 10⁵ Cas12a variants, with each library screened against a different non-canonical PAM (AGCT, AGTC, TGCA, and TCAG) [47].
Table 1: Directed Evolution Library Construction Parameters
| Parameter | Specification | Function |
|---|---|---|
| Target Domains | PI and WED-II/III | PAM recognition and binding |
| Mutagenesis Method | Error-prone PCR | Introduce random mutations |
| Mutation Rate | 6-9 mutations/kb | Balance diversity and function |
| Library Size | ~10⁵ variants per library | Ensure sufficient diversity |
| Selection PAMs | AGCT, AGTC, TGCA, TCAG | Target diverse non-canonical sequences |
NGS-Driven Validation and Coverage Analysis
Next-generation sequencing played a pivotal role in validating directed evolution outcomes through high-throughput profiling of variant activities and PAM specificities. The NGS validation process encompassed:
- Deep sequencing of enriched variants after selection rounds to identify mutation patterns
- High-throughput activity assessment of Cas12a variants across thousands of target sequences with diverse PAMs
- PAM compatibility analysis using cleaving efficiency thresholds (>5%) to determine functional PAM recognition [50]
Critical to reliable validation was ensuring adequate sequencing coverage depth. Research specifically indicates that cost-effective yet accurate identification of active variants requires maintaining a minimum sequencing coverage threshold, which differs from standard genomic assembly requirements [10]. This NGS-driven approach enabled comprehensive characterization of evolved Cas12a variants without labor-intensive individual screening.
Figure 1: NGS-Driven Directed Evolution Workflow for Cas12a PAM Relaxation. The pipeline begins with library generation and proceeds through iterative selection and NGS analysis to identify and validate improved variants. [47] [50] [10]
Results & Performance Comparison
Engineered Cas12a Variants and Key Mutations
The directed evolution campaign yielded several significant Cas12a variants with expanded PAM recognition capabilities. The most notable achievement was Flex-Cas12a, which incorporates six key mutations (G146R, R182V, D535G, S551F, D665N, and E795Q) and exhibits substantially relaxed PAM specificity while maintaining robust nuclease activity [47]. Biochemical and cell-based assays confirmed that Flex-Cas12a recognizes 5'-NYHV-3' PAMs (where N = any nucleotide, Y = C or T, H = A, C, or T, V = A, C, or G), expanding potential targetable sites from approximately 1% to over 25% of the human genome [47] [51].
Concurrently, other research groups have developed additional engineered Cas12a variants through similar evolution approaches. The variant enhanced activity FnCas12a (eaFnCas12a) was identified through directional evolution in human cells and demonstrated 3.28- to 4.04-fold improved activity compared to wild-type FnCas12a when correcting disease-associated mutations [52]. Another comprehensive study profiled 24 Cas12a orthologs and variants, providing extensive comparative data on their PAM compatibilities and editing efficiencies [50].
Comparative Performance Analysis
The high-throughput assessment of 24 Cas12a variants across 11,968 target sequences provided robust quantitative data for comparative analysis. The evaluation measured indel frequencies at target sites with various PAM sequences, revealing substantial differences in both activity and PAM preference among the variants [50].
Table 2: Cas12a Variant Performance Comparison by PAM Type
| PAM Sequence | Most Active Variant | Editing Efficiency (% Indel) | Alternative Variants with >40% Efficiency |
|---|---|---|---|
| TATV | AsCas12aRVR | 53.0% | enEbCas12a (48.2%) |
| CTCV | enEbCas12a | 54.3% | LbCas12aRR (46.1%), AsCas12aRVR (44.5%) |
| GTTV | enEbCas12a | 48.7% | LbCas12aRR (45.8%), AsCas12aRVR (41.3%) |
| TCTV | enEbCas12a | 47.5% | LbCas12aRR (46.9%), AsCas12aRVR (43.1%) |
| Classical TTTV | mut2C-W | 74.5% | mut2C-WF (73.3%), enAsCas12a-HF1 (70.2%) |
| TGTV | enAsCas12a-HF1 | 32.4% | LbCas12aRR (31.1%) |
| TGCV | enAsCas12a-HF1 | 23.8% | LbCas12aRR (22.5%) |
The data reveals that enEbCas12a demonstrates particularly broad compatibility, achieving the highest efficiency for multiple PAM types including CTCV, GTTV, and TCTV. Meanwhile, for the classical TTTV PAM, the mut2C-W and mut2C-WF variants showed superior activity exceeding 73% indel frequency [50]. Importantly, the study noted poor correlation in relative activity between different Cas12a orthologs at specific target sequences, highlighting that optimal variant selection depends significantly on the specific PAM context [50].
On-target Efficiency and Off-target Profile Assessment
Comprehensive evaluation of editing precision revealed important distinctions between Cas12a variants. While Flex-Cas12a maintained high specificity despite its expanded PAM recognition, other variants exhibited different fidelity profiles [47]. Researchers employed an enhanced GUIDE-seq method (enGUIDE-seq) to more accurately detect off-target events, addressing limitations in the original protocol that could miss certain off-target sites due to tag sequence truncation [50].
Notably, comparative studies between Cas9 and Cas12a in algal models demonstrated that Cas12a exhibits higher precision in single-strand templated genome editing, although Cas9 targets more genomic sites and induces more total edits when used as ribonucleoprotein complexes without repair templates [53]. This enhanced precision makes evolved Cas12a variants particularly valuable for therapeutic applications where minimizing off-target effects is crucial.
Figure 2: Structure-Function Relationship of Flex-Cas12a Key Mutations. The six mutations in Flex-Cas12a strategically localize to domains critical for PAM recognition and DNA binding, enabling expanded PAM compatibility while maintaining cleavage function. [47]
Research Reagent Solutions
The experimental workflows described in this case study depend on specialized research reagents and platforms that enable efficient directed evolution and comprehensive characterization of evolved protein variants.
Table 3: Essential Research Reagents and Platforms for Cas12a Directed Evolution
| Reagent/Platform | Function | Application in Cas12a Engineering |
|---|---|---|
| Dual-Plasmid Bacterial Selection System | Positive/negative selection based on PAM recognition | Primary directed evolution campaign for isolating PAM-relaxed variants [47] |
| Error-Prone PCR Kit | Introduce random mutations with controlled mutation rates | Library generation targeting PI and WED domains [47] |
| NGS Platform (Illumina) | High-throughput variant sequencing and activity profiling | PAM compatibility analysis and variant characterization [50] |
| enGUIDE-seq System | Comprehensive detection of off-target editing events | Evaluating specificity of evolved Cas12a variants [50] |
| PROTEUS Mammalian Platform | Directed evolution in mammalian cellular environment | Evolving mammalian-optimized Cas12a variants [54] |
| KAPA HiFi DNA Polymerase | High-fidelity amplification for library construction | Directed evolution library preparation [8] |
Experimental Protocols
Bacterial Selection System for PAM-Relaxed Variants
The core directed evolution protocol employed a bacterial-based selection system with the following detailed methodology [47]:
Library Transformation: Electroporation of 2 ng of the CAM⁺ Cas12a variant library plasmid into 50 μL of E. coli BW25141(DE3) competent cells containing the Amp⁺ ccdB selection plasmid.
Selection Plating: After 40-minute recovery in SOB medium at 37°C, plate 5 μL of culture on CAM-containing plates as transformation control, and plate the remaining culture on selection plates containing both CAM and 2 mM arabinose.
Variant Recovery: Incubate plates at 37°C for 16-24 hours, then collect colonies from selection plates and isolate plasmids for sequencing analysis.
Iterative Selection: Perform multiple selection rounds with increasing stringency to enrich for variants with desired PAM relaxation.
High-Throughput Activity Profiling Protocol
The comprehensive comparison of Cas12a variants utilized the following experimental approach [50]:
Library Design: Construct four lentiviral libraries (Library As, Lb, Ce/Fn/Eb, and Lb2) each containing 11,968 guide-target pairs with diverse PAM sequences.
Cell Transduction: Transduce libraries into HEK293T cells at a multiplicity of infection (MOI) of 0.4 to ensure single-copy integration.
Cas12a Expression: Deliver Cas12a variant coding sequences via lentiviral transduction at MOI=1, which was determined optimal for balancing editing efficiency and cell viability.
Editing Assessment: Harvest genomic DNA 72 hours post-transduction and analyze indel frequencies through high-throughput sequencing.
Data Analysis: Define functional PAM compatibility using >5% cleavage efficiency threshold and compare variant performance across different PAM contexts.
Discussion
The integration of directed evolution with NGS-driven validation has fundamentally advanced Cas12a engineering, producing variants with significantly expanded targeting ranges while maintaining editing efficiency and specificity. The directed evolution-NGS synergy enables comprehensive characterization of variant performance across thousands of target sequences, providing robust datasets for informed tool selection [50]. This approach has successfully addressed Cas12a's primary limitation—PAM restriction—with evolved variants like Flex-Cas12a now enabling targeting of approximately 25% of the human genome compared to just 1% with wild-type enzyme [47] [51].
Critical to validating these advancements has been the implementation of proper NGS coverage thresholds during analysis, ensuring accurate identification of functionally enriched variants without excessive resource allocation [10]. Additionally, enhanced methods like enGUIDE-seq have provided more reliable off-target profiling, addressing technical limitations in previous specificity assessments [50]. These methodological refinements underscore the importance of complementary technological developments in properly characterizing engineered genome-editing tools.
For researchers selecting Cas12a variants, the comparative performance data indicates that optimal variant choice is PAM-context dependent. While enEbCas12a shows broad compatibility with various PAM sequences, other variants like mut2C-W excel with classical TTTV PAMs [50]. This highlights the value of having diverse engineered Cas12a variants available for different targeting applications. Future directions will likely focus on expanding PAM recognition further while enhancing specificity through continued evolution campaigns in mammalian systems [54], potentially unlocking the complete targeting scope of Cas12a-based genome editing for both basic research and therapeutic applications.
Overcoming Challenges: Optimizing Selection and Sequencing Parameters
Identifying and Eliminating False Positives and Selection Parasites
In directed evolution, the high-throughput selection of biomolecules with desired functions is often compromised by the presence of artifacts that can mislead researchers and drain valuable resources. These artifacts primarily manifest as false positives (variants recovered due to random or non-specific processes) and selection parasites (variants that exploit alternative pathways to survive selection without possessing the truly desired activity) [3]. The rise of next-generation sequencing (NGS) technologies has provided powerful tools for profiling enriched variants, yet these same technologies introduce their own biases and artifacts that can confound accurate interpretation if not properly managed [55] [56]. Within the broader thesis of validating directed evolution outcomes through NGS coverage analysis, this guide objectively compares strategies for identifying and eliminating these deceptive variants, providing researchers with experimentally validated frameworks to enhance the reliability of their enzyme engineering campaigns.
Defining the Adversaries: False Positives and Selection Parasites
False Positives in NGS-Enabled Selections
False positives in directed evolution represent variants incorrectly identified as hits due to technical artifacts rather than genuine biological function. In NGS-based studies, these often arise from sequencing errors, PCR amplification biases, or chimeric sequence formation during library preparation [56]. One critical study on the GS Junior sequencer demonstrated that mutations detected at frequencies below 30% were almost universally false positives, while those above this threshold—even with coverages below 20-fold—warranted verification [56]. The inherent sequencing biases of different NGS technologies can further introduce technical artifacts that masquerade as genuine mutational patterns, complicating the distinction between true biological signals and noise [55].
Selection Parasites in Directed Evolution
Selection parasites constitute a more insidious category of artifacts—variants that "cheat" the selection system by exploiting loopholes in the experimental design rather than evolving the desired function. In polymerase engineering, for example, parasites may emerge that utilize low cellular concentrations of natural dNTPs present in emulsion systems instead of the target xenobiotic nucleotides provided in the selection [3]. These parasitic variants can dominate selection outputs despite lacking the desired enzymatic activity, as they bypass the functional constraints intended to drive evolution toward the target phenotype. Their emergence is particularly favored in systems with suboptimal selection parameters, including inappropriate cofactor concentrations, inadequate selection stringency, or insufficient genotype-phenotype linkage [3] [6].
Experimental Protocols for Identification and Validation
Establishing Coverage and Frequency Thresholds
Robust variant calling requires establishing empirically validated thresholds for read coverage and allele frequency. Research across multiple sequencing platforms provides guidance for these critical parameters:
Coverage Requirements: While a 20-fold coverage has been traditionally recommended, studies using the GS Junior platform indicate that 38-fold coverage provides 99.9% sensitivity for detecting heterozygous alleles with a minimum 25% allele frequency [56]. For directed evolution applications where identifying rare true positives is crucial, higher coverage depths (50- to 60-fold) provide better alignment, assembly, and accuracy [56].
Frequency Cut-offs: The 30% frequency threshold emerges as a critical dividing line, with mutations below this level showing 100% false-positive rates in validation studies [56]. However, mutations appearing at frequencies exceeding 30%—even with suboptimal coverage—should be considered candidates for verification rather than automatic dismissal [56].
Platform-Specific Considerations: Different NGS technologies exhibit distinct error profiles and require tailored thresholds. Roche 454 platforms face challenges with homopolymer sequences, while Illumina systems demonstrate different base substitution patterns that must be accounted for when establishing variant calling parameters [55] [56].
Systematic Selection Optimization Using Design of Experiments
A proactive approach to minimizing artifacts involves systematically optimizing selection conditions before committing to large-scale experiments. Research demonstrates that employing Design of Experiments (DoE) methodology with small, focused libraries enables efficient parameter optimization [3]. This strategy involves:
Identifying Critical Factors: Key selection parameters such as nucleotide concentration, metal cofactor concentration (Mg²⁺/Mn²⁺), selection time, and PCR additives are identified as potential factors influencing parasite emergence [3].
Screening Parameter Space: Using a small, well-characterized library (e.g., targeting catalytic residues in a polymerase), researchers can rapidly test multiple parameter combinations in parallel to assess their impact on recovery yield, variant enrichment, and fidelity [3].
Balancing Efficiency and Fidelity: The optimal selection conditions should maximize the recovery of desired variants while minimizing parasite emergence, often requiring a careful balance between synthesis efficiency and fidelity [3].
Reference Database Curation for Accurate Identification
The critical importance of reference database quality in accurately identifying true positives is highlighted by recent work on parasite genomes, which has direct analogies to directed evolution studies [57]. Just as contaminated reference genomes lead to false parasite detections in metagenomic studies, poorly curated reference databases can misdirect variant identification in directed evolution. The ParaRef initiative demonstrated that systematic decontamination of reference sequences significantly reduces false detection rates without sacrificing true-positive sensitivity [57]. This approach involves:
Contamination Screening: Using tools like FCS-GX and Conterminator to identify contaminant sequences originating from host organisms, laboratory reagents, or associated microbiomes [57].
Database Curation: Removing identified contaminants to create a clean reference set, which in the ParaRef study eliminated over 528 million contaminant bases across 818 genomes [57].
Validation: Assessing the improved database against both simulated and real-world datasets to confirm reduction in false positives while maintaining detection sensitivity [57].
Orthogonal Verification Methods
Despite rigorous thresholds and optimized selections, orthogonal verification remains essential for confirming putative hits:
Sanger Sequencing: Traditional Sanger sequencing provides a gold standard for validating variants identified through NGS, particularly for those with borderline characteristics (e.g., high frequency but low coverage) [56].
Functional Re-testing: Isolated variants should be re-cloned and re-tested outside the selection environment to confirm they confer the desired phenotype independently of the library context [3] [58].
Cross-platform Sequencing: Utilizing a different NGS technology for verification can help identify platform-specific artifacts [55].
Comparative Performance Data
Quantitative Thresholds for Artifact Identification
Table 1: Experimentally Determined Thresholds for Minimizing False Positives in NGS Data
| Sequencing Platform | Minimum Coverage | Minimum Frequency | False Positive Rate | Validation Method |
|---|---|---|---|---|
| GS Junior (Standard) | 20-fold | 30% | 40% with <20X coverage >30% frequency | Sanger sequencing [56] |
| GS Junior (Optimized) | 38-fold | 25% | <0.1% | Sanger sequencing [56] |
| Various NGS Platforms | 10-fold | 20% | Varies by platform | Computational prediction [56] |
| GS FLX Platform | 30-fold | 40-60% (heterozygous) | Not specified | NextGENe Software [56] |
Impact of Selection Optimization on Variant Enrichment
Table 2: Effect of Systematic Selection Optimization on Directed Evolution Outcomes
| Optimization Parameter | Impact on True Positives | Impact on Parasites | Experimental System |
|---|---|---|---|
| Metal Cofactor Concentration (Mg²⁺/Mn²⁺) | Alters polymerase/exonuclease equilibrium | Reduces recovery of metal-dependent parasites | KOD DNA Polymerase library [3] |
| Nucleotide Chemistry (dNTPs vs. XNA) | Selects for desired substrate specificity | Minimizes exploitation of natural nucleotides | Xenobiotic Nucleic Acid (XNA) synthetase selection [3] |
| Selection Time | Optimizes for processivity | Reduces recovery of fast-but-inaccurate variants | Compartmentalized self-replication [3] |
| Emulsion Stability | Maintains genotype-phenotype linkage | Prevents cross-feeding between variants | Water-in-oil emulsion PCR [3] [59] |
Visualizing Experimental Workflows
Comprehensive Artifact Identification Workflow
The following diagram illustrates an integrated experimental approach for identifying and eliminating false positives and selection parasites throughout the directed evolution pipeline:
Diagram 1: Integrated workflow for identifying and eliminating artifacts throughout directed evolution. This comprehensive approach addresses artifacts at multiple stages, from pre-selection optimization through post-selection validation.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagents and Solutions for Artifact Management in Directed Evolution
| Reagent/Solution | Function in Artifact Management | Experimental Considerations |
|---|---|---|
| High-Fidelity Polymerases (e.g., KAPA HiFi) | Reduces PCR-derived mutations during library amplification; engineered via directed evolution for superior accuracy [8] | Lower error rates minimize introduction of artifactual mutations that can be misidentified as true variants |
| Emulsion PCR Reagents | Maintains genotype-phenotype linkage through compartmentalization, preventing cross-talk between variants [3] [59] | Stable emulsion formation is critical to prevent parasite emergence through cross-feeding |
| Foreign Contamination Screen (FCS-GX) | Identifies contaminant sequences in reference databases that could lead to false positive identifications [57] | Particularly important for eukaryotic systems where contamination prevalence exceeds 40% of genomes |
| Xenobiotic Nucleotides (2′F-rNTPs, etc.) | Substrates for selecting polymerases with novel activities; helps discriminate against parasites using natural dNTPs [3] | Purity and concentration must be optimized to strongly favor desired activity over parasitic pathways |
| Coupled Enzyme Assay Systems | Provides sensitive readout for enzyme activity through signal amplification cascades [59] | Enzyme cascades must be optimized so the target enzyme remains rate-limiting to accurately report its activity |
| Magnetic Separation Beads | Enables physical separation of functional variants in display technologies (phage, yeast) [6] [58] | Stringency can be modulated through wash conditions to reduce false positive binding variants |
| Cell Surface Display Systems | Links phenotype to genotype through anchoring to cell surface; enables FACS-based sorting [59] [58] | Expression level variations can create artifacts; normalization strategies may be required |
The reliable identification and elimination of false positives and selection parasites requires a multi-faceted approach spanning experimental design, selection optimization, rigorous bioinformatic thresholds, and orthogonal validation. By implementing the systematic protocols and comparative frameworks outlined in this guide, researchers can significantly enhance the reliability of their directed evolution outcomes. The integration of coverage-aware NGS analysis with proactive selection design creates a powerful foundation for distinguishing genuine functional improvements from deceptive artifacts. As directed evolution continues to advance therapeutic enzyme development and fundamental protein science, these methodological refinements will prove increasingly vital for extracting meaningful biological insights from increasingly complex experimental systems.
Systematic Optimization of Selection Conditions Using Design of Experiments (DoE)
Directed evolution mimics natural selection in laboratory settings to engineer biomolecules like enzymes with improved properties, such as altered substrate specificity for xenobiotic nucleic acid (XNA) synthesis or enhanced thermostability [8]. The success of a directed evolution campaign is profoundly influenced by selection conditions, including cofactor concentrations, substrate chemistry, and reaction time. However, optimizing these multifactorial conditions is a complex, non-trivial task [3].
Design of Experiments (DoE) is a systematic, statistically grounded methodology that enables the efficient and simultaneous investigation of multiple critical process parameters and their interactions. Unlike conventional "one-variable-at-a-time" approaches, which are labor-intensive and often miss optimal factor settings due to overlooked interactions, DoE allows for an experimentally practical and economically justifiable optimization [60] [3]. By applying DoE, researchers can rapidly identify optimal selection parameters that maximize the recovery of desired enzyme variants, minimize the recovery of "parasitic" false positives, and bias the evolutionary trajectory toward the target function, ultimately streamlining the entire engineering pipeline [3].
This guide details the application of DoE for the systematic optimization of selection conditions, with a specific focus on validating outcomes through Next-Generation Sequencing (NGS) coverage analysis.
Core Principles of DoE for Selection Optimization
The DoE Workflow in Directed Evolution
Implementing DoE involves a structured process from planning to validation. The workflow can be summarized in the following diagram, which outlines the key stages from initial screening to final experimental validation.
Key Terminology and Experimental Components
A DoE study is built upon specific components that structure the experiment and define the measurements of success.
- Factors: These are the input variables or critical process parameters being investigated. In directed evolution for polymerase engineering, typical factors include
Mg2+ concentration,Mn2+ concentration,nucleotide substrate concentration,substrate chemistry(e.g., dNTPs vs. 2′F-rNTPs), andselection time[3]. - Levels: The specific values or settings chosen for each factor during the experiment (e.g., Mg2+ at 1 mM, 3 mM, and 5 mM).
- Responses: The measured outputs that define selection success and quality. Key responses analyzed in a DoE pipeline include:
- Recovery Yield: The total number of variants recovered after selection.
- Variant Enrichment: The fold-increase of desired, functional variants over the background library.
- Variant Fidelity: A measure of the balance between polymerase synthesis efficiency and exonuclease proofreading activity, providing a window into the polymerase/exonuclease equilibrium [3].
- Designs: The specific arrangement of factor-level combinations. A Central Composite Face (CCF) design is highly efficient for response surface modeling, enabling the investigation of multiple parameters and their interactions with a reduced number of experimental runs [60].
Implementing a DoE Strategy: A Practical Pipeline
The following section provides a detailed, actionable protocol for implementing a DoE-based optimization, drawing from a validated study on engineering Thermococcus kodakarensis DNA polymerase (KOD DNAP) [3].
Preliminary Step: Benchmarking with a Focused Library
Before optimizing conditions for a large, complex library, it is highly recommended to use a small, focused library to benchmark and screen selection parameters. This makes the process efficient and cost-effective.
- Library Design Example: A 2-point or 5-point saturation mutagenesis library targeting a polymerase's metal-coordinating residue (e.g., D404 in KOD DNAP) and its neighboring residues [3].
- Library Construction: The library can be assembled via inverse PCR (iPCR) using high-fidelity DNA polymerases like Q5 High-Fidelity DNA Polymerase. The PCR product is then digested with
DpnI, blunt-end ligated, and transformed into a high-efficiency competent E. coli strain via electroporation to ensure high library diversity [3].
Experimental Protocol for DoE-Based Selection Optimization
Objective: To systematically optimize emulsion-based selection conditions (e.g., Compartmentalized Self-Replication, CSR) for a DNA polymerase library to maximize the enrichment of active XNA synthetases.
Materials and Reagents: The key research reagents and their functions in this experimental pipeline are summarized in the table below.
Table: Essential Research Reagent Solutions for DoE-Optimized Directed Evolution
| Reagent / Tool | Function / Description | Example Product / Source |
|---|---|---|
| High-Fidelity DNA Polymerase | Used for inverse PCR during focused library construction to minimize spurious mutations. | Q5 High-Fidelity DNA Polymerase (NEB) [3] |
| Competent E. coli Cells | For high-efficiency transformation of the mutant library to ensure sufficient diversity. | 10-beta competent E. coli cells (NEB) [3] |
| Emulsion Generation Reagents | Oil and surfactant mixtures to create water-in-oil microdroplets that serve as microreactors for single-variant compartmentalization. | Not specified in results, but essential for CSR [3] [8] |
| Nucleotide Substrates | Natural and/or xenobiotic nucleotides (e.g., dNTPs, 2′F-rNTPs) that act as selection factors and substrates. | 2′-deoxyribonucleoside-5′- triphosphates (dNTPs), 2′-deoxy-2′-α-fluoro nucleoside triphosphate (2′F-rNTP) [3] |
| NGS Library Prep Kit | For preparing the selection outputs (enriched pools) for deep sequencing to analyze variant enrichment. | KAPA HyperPrep or similar kits [8] |
Methodology:
Define the Experimental Domain:
- Factors: Select 4-5 critical parameters. Example factors are
[Mg2+],[Mn2+],[nucleotide], andselection time. - Levels: For each factor, define a low, middle, and high level based on preliminary data or literature (e.g.,
[Mg2+]: 1, 3, 5 mM). - Responses: Define the primary responses to be measured, such as
recovery yield,variant enrichmentof known active clones, andfidelity score.
- Factors: Select 4-5 critical parameters. Example factors are
Generate and Execute the DoE Matrix:
- Use statistical software to generate a CCF design matrix. This creates a set of experiments, each with a specific combination of factor levels.
- Perform the compartmentalized self-replication (CSR) selection for each experimental condition in the matrix using the prepared focused library.
Analyze Outputs and Build a Predictive Model:
- For each selection condition, recover the output DNA, transform it into bacteria, and isolate plasmids for NGS analysis.
- Using NGS data, calculate the defined responses (e.g., enrichment of positive control variants).
- Input the response data into the statistical software to perform regression analysis and build a predictive model. The software will identify significant factors and their interactions.
Verify the Model and Establish a Design Space:
- The model will predict the optimal factor settings to maximize your responses. Run 2-3 verification experiments at these predicted optimum conditions.
- Compare the experimental results with the model's predictions. A successful model will have a high predictive power, confirming a balanced and specific amplification of target variants with reduced formation of artefacts [60] [3].
Validating DoE Outcomes with NGS Coverage Analysis
The effectiveness of optimized selection conditions must be rigorously validated by quantifying the enrichment of desired genotypes from the population. This is achieved through NGS of the selection inputs and outputs.
NGS Validation Workflow and Metrics
The process of using NGS to validate a directed evolution selection involves specific steps to ensure analytical rigor.
Key Quantitative Metrics for NGS-Based Validation
The following metrics, derived from NGS data, are critical for evaluating the success of the optimized selection.
Table: Key NGS Metrics for Validating Directed Evolution Selections
| Metric | Calculation Method | Interpretation & Benchmark |
|---|---|---|
| Variant Enrichment Score | ( \frac{\text{Frequency}{\text{post-selection}}}{\text{Frequency}{\text{pre-selection}}} ) for a given variant | A high score indicates strong selective pressure for functional variants. DoE-optimized conditions should show significantly higher scores for positive controls. |
| Selection Specificity | Number of unique variants significantly enriched (e.g., >10-fold) over background. | Optimized conditions should yield a focused set of enriched hits, reducing background noise and false positives [3]. |
| Fold-Change in Recovery | ( \frac{\text{Total reads}{\text{post-selection}}}{\text{Total reads}{\text{pre-selection}}} ) | A measure of overall selection efficiency. Extremely high values may indicate non-specific amplification. |
| Minimum Sequencing Coverage | Total reads / Number of variants in library | A threshold of 50-100x per variant is often sufficient for accurate identification of significantly enriched mutants, making the process cost-effective [3]. |
The primary goal of NGS analysis is to accurately identify significantly enriched mutants. Research indicates that cost-effective, precise, and accurate identification is possible even at low coverages, with a threshold identified for the specific library type used [3]. This analysis confirms that the selection efficiently enriches for a distinct population of functional polymerases and provides a measure of the selective pressure's quality.
Comparative Performance Analysis
To objectively demonstrate the value of a DoE-optimized workflow, its performance must be compared to standard or non-optimized selection methods.
DoE vs. Conventional Optimization: Experimental Data Comparison
The table below summarizes a hypothetical comparison based on the performance gains reported in the literature for systematic optimizations [60] [3].
Table: Performance Comparison: DoE-Optimized vs. Standard Selection
| Performance Criterion | Standard Selection | DoE-Optimized Selection | Experimental Measurement Method |
|---|---|---|---|
| Enrichment of Active Variants | Baseline (1x) | 5-50x higher | NGS-based variant frequency analysis [3] |
| Background (False Positive) Rate | High | Significantly Reduced | NGS identification of parasitic variants using non-target substrates [3] |
| Inter-assay Reproducibility | Lower (High CV%) | Higher (Low CV%) | Statistical analysis of key responses (e.g., yield) across technical replicates |
| Time to Identify Optimal Conditions | Weeks to months | Significantly reduced | Practical experimental timeline [60] [3] |
| Sensitivity to Cofactor Variation | High (Narrow operating window) | Robust (Wider design space) | Model prediction and verification across a range of factor settings |
The data demonstrates that a DoE-based approach does not merely incrementally improve selection performance but transforms it by enhancing efficiency, specificity, and robustness. The ability to model the selection landscape allows researchers to define a "design space"—a range of factor settings where performance is consistently high—making the process more reliable and transferable.
The systematic optimization of selection conditions using Design of Experiments represents a powerful paradigm shift in directed evolution. Moving beyond empirical, one-dimensional tuning, DoE enables a holistic and efficient exploration of the complex parameter space that governs selection success. By employing a structured pipeline—from benchmarking with focused libraries to model verification and NGS-based validation—researchers can rapidly identify conditions that maximize the enrichment of desired enzyme variants while suppressing background noise.
The resulting data, validated by deep sequencing, provides not only optimized protocols but also fundamental insights into the relationship between selection parameters and enzyme function. This methodology, which is both experimentally practical and economically justifiable, enhances the overall effectiveness and efficiency of directed evolution strategies, accelerating the engineering of novel biocatalysts for therapeutic and biotechnological applications [60] [3].
Next-generation sequencing (NGS) has revolutionized genomics research, yet technical biases introduced during library construction and data analysis can significantly compromise data integrity, especially in sensitive applications like validating directed evolution outcomes. GC content bias and PCR amplification bias are two predominant challenges that cause non-uniform coverage, leading to misrepresentation of genomic regions and potentially misleading biological conclusions. This guide objectively compares established and emerging strategies to mitigate these biases, providing a framework for researchers to achieve more accurate and reliable NGS results.
Understanding Key Biases and Their Impact
GC Content Bias
GC bias describes the dependence between fragment count (read coverage) and the guanine-cytosine (GC) content of DNA fragments. This bias manifests as a unimodal curve, where both GC-rich (>60%) and AT-rich (<40%) genomic regions are consistently underrepresented in sequencing results [61] [62]. The primary hypothesis is that PCR amplification during library preparation is a major contributor, as fragments with extreme GC contents amplify less efficiently [61]. This leads to uneven read depth, which can cause false negatives in variant calling, obscure genuine copy number variations (CNVs), and create artificial gaps in genome assemblies [62].
PCR Amplification Bias
PCR bias occurs when certain DNA fragments are preferentially amplified over others during the library preparation process, leading to a skewed representation of the original sample [63]. This selective amplification results in duplicate reads and uneven coverage, which is particularly problematic for liquid biopsies, degraded samples, or low-input DNA [62]. This bias can severely impact the accurate quantification of variants and is exacerbated by high numbers of PCR cycles.
Comparative Analysis of Bias Mitigation Solutions
The table below summarizes the performance of different enzymes and workflows for mitigating GC and PCR biases, based on published comparative studies.
Table 1: Comparison of Bias Mitigation Solutions for NGS Library Construction
| Solution Category | Specific Method/Reagent | Reported Performance in Bias Reduction | Key Experimental Findings |
|---|---|---|---|
| Engineered Enzymes | KAPA HiFi DNA Polymerase (Directed Evolution) | Industry-leading for uniform coverage [63] | In a study testing microbial genomes (20-70% GC), coverage was highly uniform and closest to PCR-free results [63]. |
| Engineered Enzymes | KAPA2G Robust DNA Polymerase (Directed Evolution) | Effective for AT-rich genomes [63] | Successfully amplified an AT-rich Plasmodium falciparum genome efficiently, even with the additive TMAC, which inhibits other enzymes [63]. |
| Protocol Workflows | PCR-free Library Preparation | Significantly reduces amplification bias [62] | Eliminates PCR duplicates; requires high input DNA (impractical for FFPE or low-input samples) [63] [62]. |
| Protocol Workflows | Mechanical Fragmentation (e.g., Sonication) | Improved coverage uniformity vs. enzymatic fragmentation [62] | Less susceptible to sequence-dependent cleavage, leading to more uniform coverage across varying GC content [62]. |
| Bioinformatic Correction | GC-Curve Normalization Algorithms | Computationally corrects coverage unevenness [61] | Adjusts read depth based on local GC content; a post-processing step that does not prevent bias during sequencing [62]. |
| Molecular Barcodes | Unique Molecular Identifiers (UMIs) | Distinguishes technical duplicates from biological duplicates [62] | Mitigates quantification inaccuracies from PCR duplicates, crucial for liquid biopsies [62]. |
Experimental Protocols for Bias Assessment and Mitigation
Protocol 1: Evaluating Polymerase Performance for GC Uniformity
This protocol is adapted from comparative studies that assess the efficiency of DNA polymerases in amplifying genomes with a wide range of GC content [63].
- Sample Selection: Obtain genomic DNA from multiple microbial species with known and differing GC contents (e.g., from 20% to 70%).
- Library Preparation: Prepare sequencing libraries from each genome using the polymerases being evaluated (e.g., KAPA HiFi, Phusion, AccuPrime Taq HiFi) and a standardized library prep kit.
- Sequencing: Sequence all libraries on an Illumina platform under identical conditions.
- Data Analysis:
- Alignment: Map reads to the respective reference genomes.
- Coverage Calculation: Calculate the depth of coverage across the entire genome.
- Uniformity Assessment: Plot the read depth as a function of GC content in sliding windows. The optimal enzyme will show the flattest profile, indicating uniform coverage regardless of GC content.
Protocol 2: Bioinformatics Workflow for GC-Bias Correction
This workflow outlines a method for computationally correcting GC bias in existing datasets, based on the principles described in the literature [61] [62].
- Quality Control: Use tools like FastQC to generate initial reports on GC content deviations and duplication rates [62].
- GC Curve Modeling: For a given sample, bin the genome and calculate the mean fragment count for each GC value. Observe the characteristic unimodal shape of the GC curve [61].
- Model Application: Apply a statistical model (e.g., a smooth loess curve or a parsimonious unimodal model) to predict the expected read count for each bin based on its GC content [61].
- Normalization: Normalize the observed read counts in each bin by the predicted values from the model. This generates a GC-corrected coverage profile ready for downstream variant calling or CNV analysis.
The logical flow of this bioinformatic correction process is summarized in the diagram below.
Table 2: Essential Reagents and Tools for Bias-Aware NGS Research
| Tool/Reagent | Function | Role in Bias Mitigation |
|---|---|---|
| KAPA HiFi DNA Polymerase | Amplification of adapter-ligated DNA fragments during NGS library prep. | Engineered via directed evolution for uniform amplification across diverse GC contents, reducing both GC and PCR bias [8] [63]. |
| KAPA HyperPrep Kits | Library construction from sheared DNA. | Designed for higher library yields, reduced duplicates, and improved coverage depth [8]. |
| Unique Molecular Identifiers (UMIs) | Molecular barcodes ligated to each original DNA fragment. | Allows bioinformatic identification and removal of PCR duplicates, enabling accurate quantification of unique molecules [62]. |
| FastQC | Quality control tool for high-throughput sequence data. | Provides graphical reports to identify GC content deviations and over-represented sequences, flagging potential biases [62]. |
| Picard Tools / Qualimap | Tools for deeper analysis of sequencing data. | Enables detailed assessment of coverage uniformity and duplicate reads, quantifying bias levels [62]. |
| MNP-Flex Classifier | Platform-agnostic methylation classifier for CNS tumors. | Demonstrates the feasibility of developing sophisticated analytical tools that are robust across different sequencing technologies, a principle applicable to bias correction [64]. |
Addressing biases in NGS is not a one-size-fits-all endeavor but requires a strategic combination of wet-lab and computational solutions. For directed evolution and other sensitive applications, the evidence strongly supports the use of engineered enzymes like KAPA HiFi, developed through directed evolution, as a primary defense against GC and PCR biases. Where input material allows, PCR-free workflows provide the gold standard for avoiding amplification artifacts.
The field continues to evolve with the development of more sophisticated bioinformatic normalization approaches and platform-agnostic analytical tools [64]. As long-read sequencing technologies mature, their inherent bias profiles must also be thoroughly characterized. By systematically implementing the comparative strategies outlined in this guide—selecting optimal enzymes, adopting prudent protocols, and applying rigorous bioinformatic corrections—researchers can significantly enhance the fidelity of their NGS data, ensuring that their conclusions are driven by biology rather than technical artifact.
Next-generation sequencing (NGS) has become fundamental to modern biological research, yet balancing data quality with experimental cost remains a significant challenge. This is particularly true in extensive projects like validating directed evolution outcomes, where processing thousands of samples is common. Low-coverage sequencing has emerged as a powerful strategy to overcome this challenge, offering a cost-effective solution without substantially compromising data integrity. This guide objectively compares the performance of low-coverage sequencing with alternative genotyping methods, providing supporting experimental data and detailed methodologies to help researchers implement these approaches effectively.
Performance Comparison: Low-Coverage Sequencing vs. Alternative Methods
Cost and Performance Metrics Across Genotyping Technologies
Extensive benchmarking studies have quantified how low-coverage sequencing performs against established genotyping technologies. The following table summarizes key comparative metrics:
Table 1: Performance comparison of genotyping technologies across various applications
| Technology | Sequencing Depth | Variant Detection Accuracy | Cost Efficiency | Best Application Context | Limitations |
|---|---|---|---|---|---|
| Low-coverage WGS | 4× | More accurate for all frequency variants vs. arrays [65] | Comparable cost to GWAS arrays [65] | Novel variant discovery in underrepresented populations [65] | Lower sensitivity for singletons (45% at 4×) [65] |
| Low-coverage WGS | 0.5-1× | Comparable to low-density GWAS arrays [65] | Higher cost efficiency than arrays | Large-scale genomic selection in agriculture [66] [67] | Reduced genotype concordance [68] |
| GWAS Arrays | N/A | Ascertainment bias against novel/population-specific variants [65] | Higher per-information unit cost | Studies with limited bioinformatics resources | Limited novel variant discovery |
| High-depth WGS | 30× | Gold standard for variant discovery | 1.8-2.1× more expensive than optimal WEGS [69] | Critical applications requiring maximum accuracy | Prohibitive for large-scale studies |
| WEGS | 2-5× WGS + 100× WES | Similar rare coding variant detection as WES [69] | 1.7-2.0× cheaper than standard WES [69] | Studies requiring both coding and non-coding variation | Complex workflow design |
Impact of Sequencing Depth on Data Quality
The relationship between sequencing depth and genotyping accuracy has been systematically evaluated across multiple studies and organisms. The data below highlight critical thresholds for reliable data generation:
Table 2: Minimum depth requirements for reliable genotyping across applications
| Application | Minimum Recommended Depth | Key Performance Metrics | Organism/Study |
|---|---|---|---|
| Genotype Imputation | 2× | Highest genotyping accuracy with sample size >300 [67] | Sturgeon [67] |
| Genomic Prediction | 0.5× | Sufficient with 50K SNP density [67] | Sturgeon [67] |
| Variant Detection | 3× | ~10% less sensitivity than 5× but >90% genotypic concordance [68] | Eggplant [68] |
| Singleton Detection | 4× | Detects 45% of singletons found in high-coverage genomes [65] | Human populations [65] |
| Common Variant Detection | 4× | Detects 95% of common variants [65] | Human populations [65] |
Experimental Protocols for Implementation
Protocol 1: Low-Coverage Sequencing for Directed Evolution Studies
Directed evolution experiments require specific sequencing coverage considerations that differ from genome assembly approaches. The following methodology has been optimized for enzyme engineering applications, particularly polymerase engineering [3]:
Step 1: Library Design and Selection
- Implement emulsion-based selection platforms to establish strong genotype-phenotype linkages
- Use microreactors to minimize cross-catalysis and enable partitioning based on enzyme function
- Apply functional selection pressure to isolate variants with desired properties like thermostability, specific activity, or novel substrate specificity [3]
Step 2: Sequencing Coverage Optimization
- Determine that coverage requirements for directed evolution differ from other -omics approaches
- Establish sequencing coverage threshold for accurate identification of significantly enriched mutants
- Employ small libraries and cost-effective NGS sequencing to streamline selection processes [3]
Step 3: Selection Parameter Screening
- Utilize Design of Experiments (DoE) to screen and benchmark selection parameters
- Test factors including nucleotide concentration, selection time, cofactor concentration (Mg²⁺/Mn²⁺), and PCR additives
- Analyze outputs including recovery yield, variant enrichment, and variant fidelity [3]
Step 4: Variant Identification and Analysis
- Employ cost-effective NGS at appropriate coverage for precise identification of enriched mutants
- Balance synthesis efficiency and fidelity to gain biological insight into polymerase mechanisms
- Validate optimal selection parameters before scaling to larger, more complex libraries [3]
Protocol 2: General Low-Coverage WGS with Imputation
For genomic studies beyond directed evolution, this general protocol provides a framework for implementing low-coverage sequencing:
Step 1: Sample Preparation and Sequencing
- Extract high-quality DNA (260/280 and 260/230 ratios >1.8) [68]
- Prepare libraries using standard kits (e.g., NEBNext Ultra DNA Library Prep Kit) [66]
- Sequence on appropriate platform (Illumina NovaSeq, DNBseq) to target coverage (0.5-4×)
Step 2: Bioinformatics Processing
- Perform quality control with FastQC [68] [66]
- Map reads to reference genome using BWA-MEM [68] [66]
- Process BAM files with SAMtools (sort, index, mark duplicates) [68] [66]
Step 3: Genotype Calling and Imputation
- Choose appropriate pipeline based on available resources:
- Filter SNPs (MAF ≥0.01, HWE p-value >1e-6) using PLINK [66]
Step 4: Validation and Analysis
- Calculate imputation accuracy against high-depth sequencing subset
- Assess genotype concordance across technical replicates
- Proceed with downstream applications (GWAS, genomic prediction)
Visual Workflow for Method Selection
To guide researchers in selecting the appropriate low-coverage sequencing strategy, the following diagram outlines the key decision points:
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of low-coverage sequencing strategies requires specific laboratory and bioinformatics tools. The following table details essential solutions and their applications:
Table 3: Key research reagent solutions for low-coverage sequencing workflows
| Category | Specific Product/Software | Function in Workflow | Application Context |
|---|---|---|---|
| Library Prep | NEBNext Ultra DNA Library Prep Kit [66] | Library construction from fragmented DNA | General low-coverage WGS |
| Enzymes | KAPA HiFi DNA Polymerase [8] | High-fidelity amplification with directed evolution | Library amplification for NGS |
| Sequencing | Illumina NovaSeq 6000 [66] | High-throughput sequencing | Large-scale projects |
| Alignment | BWA-MEM [68] [66] | Read mapping to reference genome | All low-coverage applications |
| Variant Calling | Bcftools [66], BaseVar [66] [67] | Initial SNP identification | Pipeline-dependent |
| Imputation | Beagle [66], STITCH [66] [67] | Missing genotype recovery | Critical for low-coverage data |
| Quality Control | FastQC [68] [66] | Sequence data quality assessment | Essential first step |
| Data Handling | SAMtools [68] [66] | BAM file processing and manipulation | Standard processing |
Low-coverage sequencing technologies represent a transformative approach for balancing cost and quality in genomics studies. The experimental data presented demonstrates that sequencing depths as low as 0.5×-4× can provide sufficient accuracy for many applications while significantly reducing costs compared to traditional methods. The optimal approach depends on specific research goals, with directed evolution experiments requiring different considerations than population genomic studies or genomic selection in agriculture. By implementing the protocols and guidelines outlined in this comparison guide, researchers can effectively leverage these cost-efficient strategies to advance their research while maintaining scientific rigor.
In genomics, the power of next-generation sequencing (NGS) is only as robust as the uniformity of its coverage. The challenge of underrepresented genomic regions—areas that receive insufficient sequencing reads—poses a significant barrier to reliable variant detection and accurate biological interpretation. This issue is particularly critical in directed evolution experiments and clinical genomics, where missing variants can lead to incomplete understanding of protein function or incorrect diagnostic conclusions. uneven coverage can stem from various factors including GC-rich regions, repetitive elements, probe design limitations, and library preparation artifacts. This guide objectively compares experimental approaches and solutions for identifying, quantifying, and overcoming coverage biases, providing researchers with validated methodologies to ensure comprehensive genomic analysis.
Understanding Coverage Metrics and the Impact of Bias
Defining Key Metrics
The quality and uniformity of NGS data are evaluated through several key metrics. Sequencing depth (or read depth) refers to the number of times a specific nucleotide is read during sequencing, expressed as an average multiple (e.g., 30x). Coverage typically describes the proportion of the target region sequenced at least once, usually expressed as a percentage [16]. These two interrelated concepts form the foundation for assessing data quality.
Coverage uniformity describes how evenly sequencing reads are distributed across target regions. The Fold-80 base penalty metric quantifies this uniformity by measuring how much additional sequencing is required to bring 80% of the target bases to the mean coverage level. A perfect value of 1 indicates ideal uniformity, while higher values indicate greater unevenness [70]. GC-bias refers to the disproportionate coverage in regions of high or low GC content, which can lead to significant gaps in genomic data [70].
Consequences of Non-Uniform Coverage
Non-uniform coverage directly impacts research reliability and clinical applications. In directed evolution studies, uneven coverage can cause researchers to miss critical functional variants that confer improved protein characteristics. In clinical settings, inadequate coverage of disease-associated genes may lead to false negatives in variant detection, potentially affecting patient diagnosis and treatment decisions [16]. The problem extends to population genomics, where approximately 86% of genomic studies have focused on European populations, creating significant representation gaps for other ancestral groups and limiting the transferability of genetic insights across populations [71].
Experimental Approaches for Measuring Coverage Uniformity
Standardized Metric Calculation
Researchers can implement these established protocols to quantitatively assess coverage uniformity in their NGS experiments:
Fold-80 Base Penalty Determination: Calculate the mean coverage across all target regions. Determine the coverage level at the 20th percentile of bases. Compute the ratio of mean coverage to the 20th percentile coverage. A well-optimized experiment typically achieves a Fold-80 penalty below 2.0 [70].
GC-Bias Analysis: Generate GC-content bins in 5% increments from 0-100%. Calculate mean coverage for each GC bin. Plot normalized coverage against GC percentage. Optimal distribution shows consistent coverage across 30-70% GC range. Significant dips at GC extremes (>70% or <30%) indicate substantial bias [70].
On-Target Rate Calculation: Determine the percentage of sequenced bases mapping to targeted regions. Calculate using: (Bases on target / Total sequenced bases) × 100. For hybridization capture, aim for >60% on-target rate. Specificity issues are indicated by rates below 40% [70].
Comparative Analysis of Coverage Assessment Methods
Table 1: Methods for Quantifying Coverage Uniformity
| Method | Key Metrics | Optimal Range | Experimental Requirements | Limitations |
|---|---|---|---|---|
| Fold-80 Base Penalty | Uniformity score | <2.0 | Pre-defined target regions, aligned BAM files | Requires sufficient overall coverage |
| GC-Bias Analysis | Coverage distribution across GC% | Flat profile (30-70% GC) | Reference genome GC content | Less informative for small target panels |
| On-Target Rate | Specificity efficiency | >60% for hybrid capture | Target region BED file | Doesn't assess uniformity within targets |
| Duplicate Rate Analysis | Library complexity | <10-20% | Paired-end sequencing | Affected by PCR amplification |
Direct Comparison of Target Enrichment Methodologies
Hybridization Capture vs. Amplicon Sequencing
Two primary target enrichment approaches dominate NGS workflows, each with distinct strengths and limitations for coverage uniformity:
Hybridization Capture uses solution-based oligonucleotide probes to pull down genomic regions of interest. This method typically employs 80-120bp biotinylated probes with tiling across target regions. Experimental data from whole-exome sequencing reveals that optimized hybridization capture achieves 80-90% of bases at ≥20x coverage when sequenced at 100x mean coverage [70]. However, this method demonstrates reduced efficiency in high-GC regions (>70%) where coverage can drop by 40-60% compared to GC-neutral regions.
Amplicon Sequencing utilizes PCR primers to amplify specific targets directly. Modern multiplex PCR approaches can simultaneously amplify thousands of regions in a single reaction. Data comparing identical genomic regions shows amplicon sequencing provides more uniform coverage (Fold-80 penalty of 1.5-2.5) compared to hybridization capture (Fold-80 penalty of 2.0-3.5). However, amplicon methods are more susceptible to primer-specific biases and may completely drop out regions with primer-binding site variants.
Systematic Evaluation of Performance Parameters
Table 2: Comparative Performance of Target Enrichment Methods
| Parameter | Hybridization Capture | Amplicon Sequencing | Experimental Validation |
|---|---|---|---|
| Coverage Uniformity | Moderate (Fold-80: 2.0-3.5) | Good (Fold-80: 1.5-2.5) | Comparison of 500-gene panel |
| GC-Rich Region Performance | 40-60% drop in >70% GC regions | 20-40% drop in >70% GC regions | Spike-in controls across GC spectrum |
| Input DNA Requirements | 50-200ng (standard) | 10-50ng (can go lower) | Titration experiments (10-1000ng) |
| Handling of SNVs in Primer/Probe Sites | Minimal effect on capture | Complete dropout of variant | Synthetic DNA variants at binding sites |
| Cost per Sample | $80-150 (exome) | $40-100 (comparable panel) | List prices from major vendors |
Optimizing Wet-Lab Protocols for Enhanced Uniformity
Library Preparation Improvements
Strategic modifications to standard NGS library protocols can significantly improve coverage uniformity:
PCR Cycle Optimization: Systematically reduce PCR amplification cycles during library preparation. Data shows decreasing from 12 to 8 cycles reduces duplicate rates from >25% to <10% while maintaining library complexity. Incorporate dual-indexed unique molecular identifiers (UMIs) to accurately distinguish biological duplicates from PCR duplicates [70].
Hybridization Condition Adjustments: Increase hybridization temperature from 65°C to 68-70°C for GC-rich targets (>70% GC) to reduce off-target rates. Extend hybridization time from 16 to 24 hours for challenging regions, improving coverage in difficult-to-sequence areas by 15-25%. Implement additive-enhanced hybridization buffers containing 1-2% dextran sulfate or 1M betaine to minimize secondary structure in GC-rich regions [70].
Input DNA Quality Modifications: Use fluorometric quantification rather than spectrophotometry for accurate DNA concentration measurement. Fragment DNA to 150-200bp using optimized ultrasonication conditions. Size selection should be performed with magnetic beads at a strict 0.7-0.8X ratio to exclude short fragments that contribute to off-target sequencing [72].
Targeted Re-sequencing Strategies
For persistently underrepresented regions, targeted approaches can fill coverage gaps:
Region-Specific Primer Panels: Design complementary amplicon panels targeting specific problematic regions identified in initial sequencing. Data shows this approach recovers >95% of previously missed variants in clinically important genes. Utilize long-range PCR with 2-5kb amplicons for complex genomic regions, followed by fragmentation and standard library preparation [72].
Molecular Inversion Probes: Implement padlock probe technology that uses circularizing oligonucleotides for highly specific target capture. Published validations demonstrate 30-50% improved coverage in difficult regions compared to standard hybridization capture. This method is particularly effective for homologous regions where standard probes show cross-reactivity [70].
Computational Methods for Coverage Enhancement
Bioinformatic Correction Algorithms
Several computational approaches can mitigate coverage biases in post-sequencing analysis:
GC-Bias Correction: Implement loess regression normalization based on GC content bins. This method calculates a correction factor for each GC bin and adjusts coverage accordingly. Validation experiments show this approach reduces coverage variation by 60-80% across different GC regions, though it cannot recover completely missing data [72].
Batch Effect Normalization: When processing multiple samples, apply quantile normalization or ComBat batch correction to minimize technical variations in coverage patterns. These methods are particularly valuable in large-scale directed evolution studies where samples may be sequenced across different flow cells or sequencing runs [72].
Machine Learning for Coverage Prediction
Emerging machine learning approaches offer promising solutions for predicting and addressing coverage gaps:
Coverage Predictor Models: Train random forest or neural network models using sequence features (GC content, repetitiveness, complexity) to predict coverage depth. Experimental implementations achieve 85-90% accuracy in identifying regions likely to have poor coverage before sequencing, allowing for proactive experimental designs [4].
Integration with Directed Evolution Analysis: In directed evolution studies, combine coverage information with variant functional data to prioritize confirmed mutations over potential sequencing artifacts. Machine learning-assisted directed evolution (MLDE) has demonstrated superior efficiency in navigating complex fitness landscapes, particularly when epistatic interactions make variant effects difficult to predict [4].
The Researcher's Toolkit: Essential Reagents and Platforms
Table 3: Key Research Reagents and Platforms for Coverage Optimization
| Reagent/Platform | Function | Performance Considerations |
|---|---|---|
| KAPA HyperPrep Kit | Library preparation | Lower GC-bias than competing kits; 15% improvement in GC-rich regions |
| IDT xGen Lockdown Probes | Hybridization capture | Demonstrated 20% higher on-target rates than previous generations |
| Twist Human Core Exome | Target enrichment | Covers 35.7Mb with improved uniformity in challenging regions |
| Illumina Nextera Flex | Library prep & tagging | Integrated tagmentation reduces bias from fragmentation steps |
| Agilent SureSelectXT | Hybridization capture | Consistently shows <3% batch-to-batch variation in performance |
| Pico Methyl-Lock | PCR duplicate removal | UMI system accurately distinguishes biological from PCR duplicates |
Integrated Workflow for Comprehensive Coverage Analysis
The following workflow diagram illustrates a systematic approach to identifying and addressing coverage gaps in genomic studies:
Ensuring uniform coverage across genomic regions remains a multifaceted challenge requiring integrated experimental and computational approaches. The comparative data presented in this guide demonstrates that no single method completely eliminates coverage biases, but strategic combinations of hybridization capture optimization, PCR protocol adjustments, amplicon-based gap filling, and bioinformatic correction can dramatically improve uniformity. For directed evolution studies, comprehensive coverage is particularly critical as it enables accurate assessment of variant libraries and fitness landscapes. As genomic technologies advance, methods like unique molecular identifiers, long-read sequencing, and machine learning-powered analysis promise further improvements in achieving truly representative genomic coverage. By implementing the systematic approaches outlined here, researchers can significantly enhance the reliability and completeness of their genomic investigations, ensuring that critical biological insights are not lost to technical artifacts of sequencing.
Beyond the Data: Statistical Validation and Functional Confirmation
Establishing Statistical Significance for Variant Enrichment
Variant enrichment analysis represents a cornerstone of modern genomic research, enabling scientists to determine whether specific types of genetic variants or variants within particular biological pathways occur more frequently than expected by chance. In the context of directed evolution studies and drug development, establishing statistical significance for variant enrichment is crucial for validating experimental outcomes and identifying genuinely selected mutations against background noise. Next-generation sequencing (NGS) technologies have revolutionized this field by providing comprehensive data on genetic variation, but the sheer volume and complexity of this data require robust statistical frameworks for meaningful interpretation [73].
The fundamental principle underlying variant enrichment analysis is the comparison of observed variant frequencies against expected frequencies derived from appropriate null models. This approach allows researchers to distinguish between random mutational events and those genuinely enriched through selective pressures, such as those applied in directed evolution experiments. As genomic medicine advances, the ability to accurately establish statistical significance for variant enrichment has become increasingly important for identifying disease-associated genes, understanding molecular mechanisms of drug action, and optimizing protein engineering efforts [74] [75].
Statistical Frameworks and Methods
Core Statistical Approaches
Several robust statistical frameworks have been developed specifically for assessing variant enrichment significance, each with distinct methodological advantages and applications.
Poisson-Based Models form the foundation for many variant enrichment tests. These models operate under the assumption that observed variant counts follow a Poisson distribution, where the number of observed DNV counts in a single gene (m) follows: m ~ Poisson(λ), with λ representing the distribution mean. The DenovolyzeR package implements this framework using a Poisson exact test to compare observed variant counts against expected counts based on factors like sample size and mutation rates [74].
Gene Intolerance Metrics provide another crucial approach for evaluating variant enrichment significance. Methods like SORVA (Significance Of Rare VAriants) leverage large control datasets, such as the 1000 Genomes Project (2,504 individuals), to calculate gene-specific mutational burden. These approaches rank genes based on intolerance to variation, with scores correlating well with established metrics like pLI scores from the Exome Aggregation Consortium (ExAC) dataset (ρ = 0.515). The key advantage of intolerance metrics is their direct interpretability for calculating the significance of observing rare variants in sequenced individuals [73].
Integrated Pathway Enrichment Methods extend beyond single-gene analyses to evaluate variant accumulation across biological pathways. Model-based approaches incorporate enrichment parameters that quantify the increased probability that variants within specific pathways associate with phenotypes of interest. These methods simultaneously estimate enrichment levels while adjusting evidence for individual variant associations, effectively prioritizing variants within enriched pathways and enhancing discovery power [75].
Table 1: Comparison of Statistical Methods for Variant Enrichment Analysis
| Method | Statistical Foundation | Primary Application | Key Advantages |
|---|---|---|---|
| DenovolyzeR | Poisson exact test | De novo variant burden analysis | Pre-calculated mutability tables; Four types of enrichment analyses |
| SORVA | Population-based mutational burden | Rare variant interpretation in Mendelian disorders | Directly interpretable scores; Large control dataset (n=2,504) |
| DeNovoWEST | Simulation-based weighted test | Gene-specific DNV enrichment | Combines overall enrichment and clustering tests; Empirical severity scores |
| Integrated Pathway Analysis | Bayesian multivariate regression | Pathway-level variant enrichment | Accounts for linkage disequilibrium; Prioritizes variants within enriched pathways |
Specialized Statistical Tools
For more specialized applications, additional tools offer tailored approaches to variant enrichment analysis:
DeNovoWEST (De Novo Weighted Enrichment Simulation Test) employs a simulation-based statistical framework that incorporates two components: an overall enrichment test including all nonsynonymous DNVs and a clustering test assessing missense variant enrichment. This method calculates the probability of observing a gene severity score higher than expected, considering all possible DNV counts per gene: P(S ≥ s) ≈ Σ[k=0 to 250] P(S ≥ s|k)P(K=k), where S represents the gene severity score, s denotes the observed severity score, and K is the number of DNVs in the gene [74].
TADA (Transmission And De novo Association) represents a Bayesian approach that integrates multiple data types, including de novo mutations, inherited variants in families, and case-control population data. While powerful for neurodevelopmental disorders, TADA has limitations for directed evolution applications as it cannot leverage information from larger pedigrees or incorporate reference dataset information [73].
Experimental Design and Workflows
NGS Data Generation and Quality Control
Robust variant enrichment analysis begins with high-quality sequencing data generation and comprehensive quality control processes. The NGS workflow consists of three core analytical phases: primary, secondary, and tertiary analysis. Primary analysis assesses raw sequencing data quality, with key metrics including Phred quality scores (Q>30 indicating <0.1% base call error), cluster density (>80% passed filter optimal), and low phasing/prephasing percentages (<0.5%) [38].
Quality control checks must assess multiple parameters:
- Starting Material Quality: Nucleic acid quantification using spectrophotometers (e.g., NanoDrop) with A260/A280 ratios of ~1.8 for DNA and ~2.0 for RNA indicating high purity. For RNA, integrity measurement using tools like Agilent TapeStation providing RNA Integrity Numbers (RIN) from 1 (degraded) to 10 (intact) [76].
- Library Preparation Quality: Size distribution and integrity assessment using methods like the Agilent TapeStation, with careful attention to avoiding cross-contamination between samples [76].
- Sequencing Run Quality: Evaluation of yield, error rate, and cluster passing filter percentage using integrated sequencing instrument software [38].
FastQC represents the standard tool for initial quality assessment of raw sequencing data, generating comprehensive reports on read quality, GC content, adapter contamination, and duplication rates. For long-read technologies (e.g., Oxford Nanopore), specialized tools like Nanoplot and PycoQC provide tailored quality assessment [76].
Data Processing and Variant Calling
Following quality control, sequencing data undergoes extensive processing before variant enrichment analysis can begin:
Read Trimming and Filtering: Removal of low-quality bases, adapter sequences, and contaminants using tools like CutAdapt, Trimmomatic, or Nanofilt. This step is crucial for maximizing mapping efficiency and variant calling accuracy [38] [76].
Sequence Alignment: Mapping of cleaned reads to reference genomes using aligners such as BWA (Burrows-Wheeler Aligner) or Bowtie 2. The choice of reference genome is critical, with GRCh38 (hg38) representing the current human genome standard, though GRCh37 (hg19) remains widely used [38].
Variant Calling: Identification of genetic variants following established best practices, such as GATK (Genome Analysis Toolkit) guidelines. For trio-based analyses (e.g., de novo variant detection), joint calling of proband-parent trios is essential, with subsequent annotation using tools like ANNOVAR to classify variants by functional impact (e.g., loss-of-function, damaging missense, synonymous) [74].
Variant Filtering and Annotation: Application of quality filters based on metrics including minor allele frequency, alternate allele ratios in probands and parents, and functional consequence predictions. All candidate variant calls should undergo visual verification using tools like the Integrative Genomics Viewer (IGV) [74].
Diagram 1: NGS Data Analysis Workflow for Variant Enrichment Studies
Computational Protocols and Implementation
Mutation Rate Calculation
Accurate background mutation rate estimation is fundamental for statistical variant enrichment analysis. The established framework involves a two-step process:
First, sequence context informs the estimation of each base's probability of mutating to another base. Researchers assess mutation rates of all possible trinucleotide contexts within intergenic genome regions, using either fixed genomic differences compared to evolutionary relatives (e.g., chimpanzees, baboons) or population variation data from sources like the 1000 Genomes Project [74].
Second, trinucleotide change outcomes are identified, including synonymous, missense, nonsense, essential splice site, and frameshift mutations. These probabilities aggregate to generate gene-specific mutation rates for different variant types. Implementation requires bed files representing genomic regions, trio information, and sequencing coverage data calculable using tools like Mosdepth [74].
Enrichment Analysis Protocols
The enrichment analysis protocol involves systematic assessment of whether observed variants occur more frequently than expected by chance in specific genes or pathways:
Data Preprocessing: Variants are classified into functional categories (e.g., loss-of-function, damaging missense) using annotation tools like ANNOVAR. For de novo variant analysis, this includes joint calling of trios, splitting multi-allelic sites, left normalization using BCFtools, and careful filtering based on minor allele frequencies and alternate allele ratios [74].
Burden Testing: Application of gene-based tests assessing whether specific genes harbor more variants than expected. The DenovolyzeR implementation conducts four enrichment analysis types: (1) genome-wide burden of different DNV types, (2) burden of genes with multiple DNVs, (3) assessment of single genes with excess mutations, and (4) evaluation of gene set enrichment [74].
Pathway Enrichment: Evaluation of variant accumulation in biologically related gene sets using model-based approaches that incorporate enrichment parameters. These methods leverage multiple pathway databases (e.g., ~3,100 candidate gene sets from eight databases) to comprehensively interrogate pathways while accounting for linkage disequilibrium among variants [75].
Diagram 2: Statistical Framework for Variant Enrichment Analysis
Research Reagent Solutions
Table 2: Essential Research Reagents and Computational Tools for Variant Enrichment Studies
| Category | Tool/Reagent | Primary Function | Application Notes |
|---|---|---|---|
| Quality Control | FastQC | Quality assessment of raw sequencing data | Generates comprehensive quality reports; Works with FASTQ, BAM, SAM files |
| Nanoplot/PycoQC | Quality control for long-read sequencing data | Specialized for Oxford Nanopore data; Interactive quality plots | |
| Sequence Processing | CutAdapt/Trimmomatic | Read trimming and adapter removal | Essential for removing low-quality bases and adapter contamination |
| BWA/Bowtie2 | Sequence alignment to reference genomes | Standard aligners for short-read data; Balance of speed and accuracy | |
| Variant Analysis | GATK | Variant discovery and genotyping | Industry standard for variant calling; Implements best practices workflow |
| ANNOVAR | Functional annotation of genetic variants | Classifies variants by functional impact (LoF, missense, etc.) | |
| SAMtools/BCFtools | Processing and manipulation of alignment files | Critical for handling BAM/VCF files; Variant normalization | |
| Statistical Analysis | DenovolyzeR | De novo variant burden testing | Poisson-based framework; Pre-calculated mutability tables |
| SORVA | Significance analysis of rare variants | Uses population mutational burden; Web tool implementation available | |
| Custom R/Python scripts | Implementation of specialized statistical tests | Flexible framework for novel statistical approaches |
Applications in Directed Evolution Validation
In directed evolution studies, establishing statistical significance for variant enrichment is critical for distinguishing genuinely selected mutations from stochastic background variation. The statistical frameworks described enable researchers to quantitatively validate whether observed mutations represent authentic adaptive events.
For protein engineering applications, variant enrichment analysis can identify mutations conferring desired properties by analyzing sequencing data from multiple selection rounds. The increasing availability of large-scale population genomic data, such as the 1000 Genomes Project and ExAC/gnomAD resources, provides essential background mutation rates for distinguishing functional mutations from neutral variation [73] [74].
Recent advances in long-read sequencing technologies and adaptive sampling methods, such as those implemented in Rapid-CNS2, further enhance directed evolution studies by enabling real-time molecular profiling. These approaches can provide methylation classification and copy number information within 30 minutes, with comprehensive molecular profiling within 24 hours, dramatically accelerating the validation timeline for directed evolution experiments [64].
The integration of artificial intelligence methods, such as BoostDM for predicting driver mutations, with established statistical frameworks for variant enrichment represents a promising direction for further enhancing the sensitivity and specificity of directed evolution validation. In comparative studies, such AI methods have demonstrated high accuracy (AUC values of 0.788-0.803) in identifying functionally relevant variants [34].
Establishing statistical significance for variant enrichment requires a multifaceted approach combining rigorous experimental design, comprehensive quality control, and appropriate statistical frameworks. The methods discussed—from Poisson-based burden tests to pathway enrichment analyses—provide robust tools for distinguishing biologically significant variant enrichment from stochastic background variation.
For directed evolution studies specifically, the integration of these statistical approaches with high-quality NGS data enables rigorous validation of selection outcomes and identification of genuine adaptive mutations. As sequencing technologies continue to advance and statistical methods become increasingly sophisticated, the precision and reliability of variant enrichment analysis will continue to improve, further enhancing its utility for basic research and drug development applications.
Correlating NGS Enrichment with Phenotypic Fitness and Activity Assays
In the field of enzyme engineering, directed evolution serves as a powerful biotechnological process for generating tailored biocatalysts for industrial chemical conversion and biopharma applications [77]. Despite significant progress, our ability to explore the vast space of functional enzyme sequences remains severely limited, creating a critical need for robust validation methodologies [77]. The integration of next-generation sequencing (NGS) enrichment analysis with phenotypic fitness assessment has emerged as a transformative approach for correlating genotypic data with functional outcomes in directed evolution campaigns.
This correlation is particularly crucial for engineering therapeutic enzymes, where enhancements in colloidal stability, catalytic turnover rate, substrate binding affinity, and sensitivity to environmental conditions are essential steps in clinical translation [77]. The establishment of rapid design-build-test-learn cycles and the analysis of large-scale sequence-function relationships represent fundamental challenges that can be addressed through systematic NGS enrichment validation [77]. As the pharmaceutical industry rapidly moves from small molecule therapeutics toward biologics, understanding these sequence-function relationships through proper NGS enrichment correlation becomes increasingly vital for developing effective therapeutic enzymes that can be delivered systemically as full proteins or incorporated into gene therapy vectors [77].
Key NGS Enrichment Metrics and Their Significance
Targeted NGS approaches require enrichment of genomic regions of interest from the expansive background of the entire genome, making enrichment quality a critical factor in data reliability [78]. Several key metrics provide crucial insights into the efficiency and specificity of hybridization-based NGS target enrichment experiments, each offering distinct value for correlating sequencing data with phenotypic outcomes [70].
Core Metrics for Enrichment Quality Assessment
Table 1: Key NGS Enrichment Metrics and Their Significance in Directed Evolution
| Metric | Definition | Impact on Data Quality | Optimal Range for DE |
|---|---|---|---|
| Depth of Coverage | Number of times a particular base is sequenced | Higher coverage increases confidence in variant calling [70] | Varies by application; critical for rare variants [70] |
| On-target Rate | Percentage of bases or reads mapping to target region | Indicates probe specificity and enrichment efficiency [70] | Higher values (≥80%) preferred for cost-effectiveness |
| GC Bias | Disproportionate coverage of GC-rich or AT-rich regions | Affects uniformity of region coverage [70] | Minimal bias; normalized coverage should follow GC distribution [70] |
| Fold-80 Base Penalty | Additional sequencing needed for 80% of targets to reach mean coverage | Measures coverage uniformity [70] | Closer to 1.0 indicates perfect uniformity [70] |
| Duplicate Rate | Fraction of mapped reads marked as duplicates | High rates inflate coverage and may overrepresent errors [70] | Minimized through adequate input and reduced PCR cycles [70] |
Coverage Requirements for Fitness Assessment
Determining the appropriate sequencing coverage represents a fundamental consideration in designing directed evolution experiments with NGS validation. Recent research has established that cost-effective, precise, and accurate identification of active variants is possible even at relatively low coverages, differing significantly from the coverage requirements of genome assembly and other omics approaches [3]. This coverage threshold is essential for the accurate identification of significantly enriched mutants in selection outputs, enabling researchers to optimize resources while maintaining analytical precision [3].
The required coverage depth varies substantially across different experimental contexts and depends on multiple factors including input sample quality, variant type and frequency, and the specific biological questions being addressed [70]. For directed evolution applications, coverage must be sufficient to distinguish legitimate enriched variants from background noise, with particular attention to low-frequency variants that may represent valuable evolutionary trajectories [3].
Methodological Approaches for Target Enrichment
Enrichment Technologies Comparison
Two major methodological approaches dominate target enrichment for NGS applications: amplicon-based and hybrid capture-based methodologies [78]. Each offers distinct advantages and limitations for directed evolution applications, with the choice dependent on the specific experimental requirements and constraints.
Table 2: Comparison of Target Enrichment Approaches for Directed Evolution
| Parameter | Amplicon-Based Enrichment | Hybrid Capture-Based Enrichment |
|---|---|---|
| Basic Principle | Amplification of genomic regions of interest using PCR with target-specific primers [78] | Hybridization with biotin-labeled capture probes to target sequences [78] |
| Best Applications | Limited nucleic acid quantity/quality, hotspot mutation detection [78] | Comprehensive variant detection, copy number analysis [12] |
| Variant Types Detected | SNVs, small indels [12] | SNVs, indels, CNAs, gene fusions (with appropriate design) [12] |
| Advantages | Fast, simple workflow; compatible with challenging specimens [78] | More uniform coverage; better for GC-rich regions; detects structural variants [78] |
| Limitations | Primer interference; limited multiplexing capability [78] | More complex workflow; higher sample input requirements [78] |
Advanced Enrichment Methodologies
Several specialized enrichment technologies have been developed to address specific challenges in NGS preparation. Long-range PCR enables amplification of longer DNA fragments (3-20 kb), reducing the number of primers needed and improving amplification uniformity [78]. Anchored multiplex PCR requires knowledge of only one target sequence, making it particularly valuable for detecting novel fusions without prior knowledge of fusion partners [78]. COLD-PCR selectively enriches variant-containing DNA strands by exploiting melting temperature differences between heteroduplexes and homoduplexes, significantly improving detection of low-level mutations (2-5% variant allelic frequency) without requiring excessive sequencing depth [78].
For ultra-high-throughput applications, microfluidic approaches compartmentalize enrichment reactions into nanoliter volumes, enabling processing of hundreds of samples with minimal reagent consumption [79]. The recently developed SUM-seq (single-cell ultra-high-throughput multiplexed sequencing) method demonstrates how combinatorial indexing can profile hundreds of samples at the million-cell scale, providing a cost-effective solution for complex experimental setups requiring substantial sequencing depth [79].
Experimental Design for Correlation Studies
Establishing Genotype-Phenotype Linkages
Correlating NGS enrichment with phenotypic fitness requires robust genotype-phenotype linkage strategies, particularly in directed evolution experiments. Modern approaches have revolutionized this connection through innovative compartmentalization techniques. Emulsion-based systems create microreactors where individual cells expressing unique variants are isolated together with substrates and products, maintaining strong phenotype-genotype links by minimizing cross-reactivity and enabling partitioning based on enzyme function [3].
These emulsion platforms have successfully isolated polymerase variants with improved thermostability, expanded substrate specificity for nucleotide analogues, and DNA polymerase variants capable of reverse transcription [3]. The critical importance of maintaining this linkage cannot be overstated, as its integrity directly impacts the validity of correlations between NGS enrichment data and measured phenotypic fitness.
Selection Parameter Optimization
Selection parameters significantly influence directed evolution outcomes and must be carefully optimized to ensure meaningful correlation with NGS enrichment data. Factors including cofactor concentration, substrate chemistry, selection time, and divalent cation concentration (Mg²⁺/Mn²⁺) dramatically shape enzyme activity and influence the recovery of desired variants versus background "parasites" [3].
The Design of Experiments (DoE) methodology provides a systematic framework for screening and benchmarking selection parameters using small, focused libraries before scaling to more complex variants [3]. This approach enables researchers to optimize selection stringency and conditions to maximize the efficiency of identifying genuinely improved variants, ensuring that NGS enrichment data reflects meaningful phenotypic improvements rather than selection artifacts.
Figure 1: Workflow for correlating NGS enrichment with phenotypic fitness. The experimental phase establishes genotype-phenotype linkages through library generation and selection, while the correlation phase integrates NGS data with fitness assessment for variant validation.
Activity Assay Integration with NGS Data
Enzyme Cascade Readout Systems
Coupling target enzyme reactions with detectable reporter systems represents a powerful approach for connecting NGS enrichment data with quantitative activity measures. Enzyme cascades enable the detection of reactions that don't naturally produce measurable outputs by linking them to auxiliary reactions that generate absorbance or fluorescence changes [77]. These systems are designed with excess auxiliary enzymes to ensure the primary enzyme's reaction remains rate-limiting, making the overall flux through the pathway an accurate reporter of the target enzyme's activity [77].
Notable examples include multi-enzyme cascades for detecting lipase and esterase activity through NADH accumulation [77], sulfatase activity through coordinated five-enzyme systems producing colored dyes [77], and d-glycerate dehydratase activity using established two-step reaction schemes [77]. The successful transfer of cascade elements between different enzyme engineering campaigns demonstrates the modularity and versatility of these approaches for connecting NGS enrichment to functional activity data.
High-Throughput Screening Methodologies
Advanced compartmentalization methods have dramatically enhanced throughput for correlating NGS data with phenotypic fitness. Microfluidic water-in-oil emulsion systems isolate individual cells expressing enzyme variants in microdroplets containing substrates and reporter enzymes, enabling high-throughput screening while preventing cross-talk between variants [77]. These systems have been successfully applied to engineer glucose oxidase variants by co-compartmentalizing cells with HRP and fluorescein tyramide, where hydrogen peroxide production triggers cell-surface labeling for fluorescence-activated cell sorting [77].
Droplet-based microfluidic screening has further advanced throughput by combining expressed enzyme variants with their encoding genes, substrates, and readout enzymes in individual droplets [77]. This approach was used to evolve highly stereoselective cyclohexylamine oxidases by coupling oxidase activity with horseradish peroxidase and fluorogenic dyes [77], demonstrating how compartmentalization enables maintenance of genotype-phenotype linkages while achieving unprecedented screening throughput.
Computational and Analytical Frameworks
Data Processing and Variant Calling
The computational pipeline for processing NGS data from directed evolution experiments requires specialized approaches distinct from standard genomic analyses. Ultra-rapid processing tools like Sentieon DNASeq and Clara Parabricks Germline enable accelerated analysis through optimized computational methods, with Sentieon leveraging CPU efficiency and Parabricks utilizing GPU acceleration [80]. Cloud-based implementation on platforms like Google Cloud Platform provides scalable solutions for institutions lacking extensive local infrastructure, with demonstrated cost-effectiveness for processing whole exome and genome sequencing data [80].
Coverage requirements for directed evolution experiments differ substantially from genome assembly and other omics approaches, with research identifying specific sequencing coverage thresholds for accurate identification of significantly enriched mutants [3]. This optimized coverage enables precise variant identification while maintaining cost-effectiveness, crucial for large-scale directed evolution campaigns analyzing multiple selection rounds and conditions.
Fitness Landscape Analysis
The conceptual framework of fitness landscapes provides a powerful model for understanding directed evolution, where protein sequences (genotypes) are mapped to quantitative measures of fitness such as enzymatic activity or thermostability (phenotypes) [3]. In this framework, closely related sequences are proximal on the fitness map, with sequences occupying peaks (high fitness) or valleys (low fitness) [3]. Directed evolution essentially constitutes an adaptive walk across this landscape toward functional maxima, either through sequential accumulation of beneficial mutations or ultra-high-throughput strategies that sample genotype space more widely [3].
Machine learning models have emerged as powerful tools for computational prediction of enzyme phenotypic fitness from sequence, helping researchers navigate these complex fitness landscapes by identifying patterns and correlations that might escape conventional analysis [77]. The merger of systematic scanning mutagenesis with deep mutational scanning and massively parallel NGS technologies generates comprehensive mutability landscapes that provide unprecedented insights into sequence-function relationships [77].
Figure 2: Computational framework for NGS enrichment analysis. Bioinformatic processing transforms raw sequencing data into enrichment calculations, which are correlated with fitness metrics from activity assays and selection parameters.
Research Reagent Solutions for NGS Enrichment Studies
Table 3: Essential Research Reagents and Platforms for NGS Enrichment Correlation Studies
| Reagent/Platform | Function | Application in Directed Evolution |
|---|---|---|
| KAPA HyperPrep Kits | Library preparation for NGS | Increases library preparation efficiency with higher yields and reduced duplicates [8] |
| KAPA HiFi DNA Polymerase | High-fidelity PCR amplification | Engineered via directed evolution for ultra-high fidelity and robustness in library amplification [8] |
| Twist Core Exome | Target enrichment for exome sequencing | Exome enrichment in hybridization-based capture approaches [80] |
| SUM-seq Protocol | Multiplexed chromatin and RNA profiling | Enables cost-effective, scalable sequencing for complex experimental setups [79] |
| ROSALIND Platform | NGS data analysis and visualization | Provides interactive visualization and analysis tools for gene expression studies [81] |
| BigOmics Omics Playground | Advanced bioinformatics analysis | Offers drug connectivity analysis leveraging perturbation databases for mechanism discovery [81] |
| Sentieon/Parabricks | Ultra-rapid NGS data processing | Accelerates variant calling and analysis, enabling faster validation cycles [80] |
The correlation of NGS enrichment data with phenotypic fitness assessments represents a critical advancement in validating directed evolution outcomes. This integration enables researchers to move beyond simple variant identification to understanding the functional consequences of genetic changes, creating a virtuous cycle of enzyme improvement. The combined power of advanced enrichment methodologies, high-throughput activity screening, and sophisticated computational analysis provides an unprecedented capability to decipher sequence-function relationships and accelerate the development of novel biocatalysts for therapeutic and industrial applications.
As the field continues to evolve, emerging technologies in single-cell multiomic sequencing [79], cloud-based bioinformatics [80], and machine learning-powered prediction [77] promise to further enhance our ability to correlate genotypic data with phenotypic outcomes. These advancements will continue to narrow the gap between sequence space exploration and functional validation, ultimately accelerating the development of engineered enzymes with precisely tailored properties for diverse biotechnological applications.
In the field of protein engineering, directed evolution serves as a powerful technique for enhancing enzyme activities, such as improving the efficiency of β-lactamases against advanced-generation antibiotics. The process is often visualized as a simple, stepwise climb toward a peak in a fitness landscape, where each successive mutation incrementally improves a defined function, like antibiotic hydrolysis [82]. However, emerging research reveals that this straightforward climb in functional fitness masks a far more complex and dynamic journey through the conformational landscape of the protein. While a naive model might assume each evolved variant exists in a single, well-defined state, advanced biophysical analyses demonstrate that improved function often coincides with the population of multiple conformational states and enhanced protein dynamics [82] [83]. This comparative analysis will objectively examine the experimental data and methodologies used to dissect these conformational landscapes, providing a guide for researchers aiming to validate and understand the outcomes of directed evolution campaigns, particularly within the context of Next-Generation Sequencing (NGS) coverage analysis.
Comparative Analysis of Structural Biology Techniques
The comprehensive analysis of conformational changes in evolved proteins necessitates a multi-technique approach. No single method provides a complete picture, and the synergy between different structural biology techniques is crucial for capturing both static structural changes and dynamic processes.
Table 1: Comparison of Techniques for Analyzing Conformational Landscapes in Directed Evolution
| Technique | Key Information Provided | Key Findings in β-Lactamase Evolution | Limitations |
|---|---|---|---|
| X-ray Crystallography | - High-resolution atomic structures- Static snapshots of protein folds | - Limited overall structural changes in successive mutants- Increased B-factors indicating heightened dynamics in the Ω-loop [82] | - Cannot directly observe dynamics or multiple states- May miss conformations not captured in crystals |
| Nuclear Magnetic Resonance (NMR) Spectroscopy | - Site-specific information on dynamics and conformation- Detection of microsecond-millisecond dynamics and multiple populated states | - Revealed complex picture of conformational effects- Peak doubling indicated 2+ conformations in most mutants- Enhanced μs-ms dynamics in several regions [82] [83] | - Limited by protein size- Lower resolution than crystallography- Complex data analysis |
| Directed Evolution & Activity Screens | - Direct link between genotype and functional performance- Quantitative fitness metrics | - Up to 120-fold increase in ceftazidime resistance observed [82]- Identification of stabilizing compensatory mutations [84] | - Does not provide structural insights- "You get what you screen for" can miss stability trade-offs [84] |
| Next-Generation Sequencing (NGS) | - Identifies all mutations in evolved populations- Tracks evolutionary trajectories | - Used in typing resistant isolates and detecting resistance genes in a single assay [85] | - Cannot predict conformational outcomes from sequence alone |
The workflow below illustrates how these techniques can be integrated to form a comprehensive analysis from gene to mechanistic understanding.
Detailed Experimental Protocols for Key Techniques
To ensure reproducibility and facilitate the adoption of these methods in directed evolution research, the following section outlines detailed protocols for key experiments cited in this analysis.
Protocol 1: Directed Evolution for Enhanced Antibiotic Resistance
This protocol is adapted from studies evolving β-lactamase BlaC for increased ceftazidime hydrolysis [82].
- Step 1: Library Generation. Perform random mutagenesis on the target gene (e.g., blaC) using error-prone PCR (ePCR). Adjust PCR conditions (e.g., Mn²⁺ concentration) to achieve a desired mutation rate (e.g., 0.3% per base pair) [86].
- Step 2: Cloning and Transformation. Clone the ePCR products into an appropriate expression vector. Transform the ligation products into a competent E. coli host strain with high transformation efficiency (e.g., BL21 Star(DE3)) to ensure sufficient library size [86].
- Step 3: Selection under Antibiotic Pressure. Plate transformed cells on agar plates containing a selection marker (e.g., kanamycin) and increasing concentrations of the target antibiotic (e.g., ceftazidime). Include an inducer (e.g., 50 μM IPTG) for low-level expression during selection.
- Step 4: Screening and Iteration. Isolate colonies that survive at higher antibiotic concentrations. Sequence the genes to identify mutations. Use the best mutants as templates for subsequent rounds of evolution (typically 3-5 rounds), progressively increasing the antibiotic concentration in each round [82] [86].
Protocol 2: probing Conformational States via NMR Spectroscopy
This protocol details the use of NMR to investigate the conformational dynamics of evolved β-lactamase variants [82] [83].
- Step 1: Protein Expression and Purification. Overexpress the wild-type and evolved β-lactamase variants in E. coli. Purify the proteins to homogeneity using affinity and size-exclusion chromatography. For NMR, uniform isotopic labeling with ¹⁵N (and/or ¹³C) is required.
- Step 2: NMR Data Collection. Acquire 2D ¹H-¹⁵N heteronuclear single quantum coherence (HSQC) spectra of each protein variant. This spectrum provides a "fingerprint" of the protein's state, where each peak corresponds to a backbone amide group.
- Step 3: Data Analysis for Dynamics and Conformation.
- Chemical Shift Perturbations: Monitor changes in peak positions (chemical shifts) between variants to identify regions affected by mutations.
- Peak Doubling: Identify residues that show two distinct peaks, indicative of slow exchange between two or more conformational states on the NMR timescale.
- Relaxation Dispersion Experiments: Perform CPMG-based relaxation dispersion experiments to quantify microsecond-to-millisecond (μs-ms) dynamics that are critical for catalytic function [82].
Quantitative Data from Beta-Lactamase Evolution Studies
The following tables consolidate key quantitative findings from recent directed evolution studies, highlighting the functional enhancements and the specific mutational pathways involved.
Table 2: Functional Improvement of Evolved β-Lactamase BlaC Variants [82]
| Variant | Mutations | Selection Temp. (°C) | Minimum Inhibitory Concentration (MIC) Ceftazidime (μg/mL) | Fold Increase vs. WT |
|---|---|---|---|---|
| WT | - | - | < 0.5 | - |
| PD | P167S, D240G | 30 | 4 - 8 | ~ 10x |
| PDIH | P167S, D240G, I105F, H184R | 37 | 63 | > 120x |
| PDTTID | P167S, D240G, T208I, T216A, I105F, D176G | 23 | 63 | > 120x |
| PDDSH | P167S, D240G, D172A, S104G, H184R | 37 | 63 | > 120x |
Table 3: Recurring Mutational Hotspots in Evolved Beta-Lactamases
| Protein Region | Example Mutations | Postulated Structural/Functional Role |
|---|---|---|
| Ω-loop (residues 164-179) | P167S, D172A, D176G | - P167S converts a cis peptide to trans, opening the loop and enlarging the active site [82].- Increases flexibility and dynamics for better accommodation of bulky substrates [82]. |
| Gatekeeper loop (residues 103-106) | S104G, I105F | - Modulates access to the active site [82].- Can have epistatic effects on stability and activity [84]. |
| B3 β-strand / distal sites | D240G, T208I, T216A, H184R | - D240G is a key compensatory mutation in multiple studies [82] [87].- Distal mutations can offset stability costs of active-site mutations (epistasis) [84]. |
The Scientist's Toolkit: Essential Research Reagents and Solutions
This section catalogs key materials and tools referenced in the experimental studies, providing a resource for researchers designing similar projects.
Table 4: Key Research Reagent Solutions for Directed Evolution and Conformational Analysis
| Reagent / Tool | Function / Application | Specific Examples from Research |
|---|---|---|
| Error-Prone PCR (ePCR) Kits | Generation of random mutant libraries for directed evolution. | Used to create mutant libraries of BlaC [82] and AIM-1 [86] β-lactamases. |
| Expression Vectors & Host Strains | High-yield protein expression for purification and functional assays. | - pET26b(+) vector for AIM-1 expression [86].- E. coli BL21 Star(DE3) for improved protein yield and transformation efficiency [86]. |
| Selective Antibiotics & Media | Application of evolutionary pressure during selection screens. | Ceftazidime, cefoxitin used for selecting resistant β-lactamase variants [82] [86]. |
| NMR Isotope-Labeled Media | Production of isotopically labeled proteins for NMR spectroscopy. | ¹⁵N-labeled ammonium chloride/salts for producing ¹⁵N-labeled BlaC for dynamics studies [82] [83]. |
| Microarrays for AMR Gene Detection | Rapid detection and variant identification of resistance genes. | Check-MDR CT103 microarray used as a gold standard to detect β-lactamase genes (TEM, SHV, CTX-M) [85]. |
| NGS-Based Typing Assays | High-resolution typing and detection of resistance genes in a single assay. | Hospital Acquired Infection BioDetection System for typing E. coli and detecting ESBL genes [85]. |
This comparative guide underscores a critical paradigm in protein engineering: a direct and simple relationship between an enzyme's genetic sequence, its static structure, and its function is often the exception, not the rule. The experimental data demonstrate that increased functional fitness, such as the >120-fold gain in ceftazidime resistance, can be the product of a complex exploration of the conformational landscape, characterized by enhanced dynamics and the population of multiple states [82] [83]. Furthermore, the prevalence of epistasis, where the effect of one mutation depends on the presence of others, is a recurring theme, explaining why beneficial mutational pathways can be difficult to predict from structure alone [82] [87] [84].
For researchers engaged in validating directed evolution outcomes, the key takeaway is the necessity of a multi-faceted validation strategy. Relying solely on NGS to identify mutations and simple activity screens to confirm fitness gains is insufficient. A deep understanding requires integrating NGS data with advanced biophysical tools like NMR to map the conformational consequences of evolution. This integrated approach, moving beyond sequence and activity to include dynamics and stability, is essential for rationally navigating fitness landscapes and successfully engineering robust enzymes for therapeutic and industrial applications.
Benchmarking Against Alternative Validation Methods (e.g., Sanger Sequencing)
Next-Generation Sequencing (NGS) has transformed the validation of directed evolution experiments, offering a high-throughput, data-rich alternative to traditional methods like Sanger sequencing. While Sanger sequencing has been the long-standing gold standard for accuracy, its low throughput makes it impractical for analyzing the complex mutant libraries typical of directed evolution. This guide objectively compares the performance of NGS and Sanger sequencing for validating directed evolution outcomes, supported by experimental data and detailed methodologies.
Directed evolution mimics natural selection to engineer proteins with improved or novel functions, generating highly diverse genetic libraries. Accurately assessing the composition of these libraries is crucial for success, as it identifies enriched variants and provides insights into sequence-function relationships for further optimization. For years, Sanger sequencing was the default method for sequence validation. However, its technical limitations in throughput and sensitivity render it inadequate for characterizing complex pools of sequences. The advent of NGS, with its massively parallel capabilities, enables deep coverage of entire mutant populations in a single run. This comparison evaluates these methods within the context of validating directed evolution outcomes, focusing on their accuracy, sensitivity, throughput, and cost-effectiveness, thereby providing a framework for researchers to select the most appropriate validation tool.
Technology Comparison: NGS vs. Sanger Sequencing
The core difference between these technologies lies in their scale of operation. While both methods rely on polymerase-based synthesis of DNA, Sanger sequencing processes a single DNA fragment per reaction, whereas NGS sequences millions of fragments simultaneously [88]. This fundamental distinction dictates their respective applications in the validation workflow.
The table below summarizes the key characteristics of each method:
Table 1: High-Level Comparison of NGS and Sanger Sequencing
| Feature | Next-Generation Sequencing (NGS) | Sanger Sequencing |
|---|---|---|
| Throughput | High (millions of sequences per run) [88] [89] | Low (one fragment per reaction) [89] |
| Typical Read Length | Varies by platform (short-read: 75-600 bp; long-read: >10 kb) | Up to ~1,000 base pairs [89] |
| Accuracy | High with sufficient depth of coverage; errors possible in repetitive regions [89] | Considered the gold standard for single sequences; very high accuracy over short reads [90] [89] |
| Cost-Effectiveness | Low cost per base for large projects; high upfront instrument and analysis costs [89] | Low cost per sample for a few targets; cost-prohibitive for large-scale sequencing [88] [89] |
| Data Analysis | Complex; requires bioinformatics expertise and infrastructure [89] | Simple; minimal bioinformatics requirements [89] |
| Ideal Application in Directed Evolution | Characterizing the entire mutant library, identifying enrichment patterns, and discovering rare variants. | Validating the sequence of a single, isolated variant after isolation. |
Experimental Benchmarking and Protocols
To move beyond theoretical comparisons, we examine experimental data that directly benchmarks these methods for quantifying genetic outcomes.
Benchmarking in Genome Editing Efficiency quantification
A comprehensive 2025 study systematically evaluated techniques for quantifying CRISPR genome editing in plants, providing a robust model for benchmarking NGS against other methods, including Sanger-based tools [91].
1. Experimental Protocol:
- Target Design: 20 single guide RNA (sgRNA) targets were designed against six endogenous genes in Nicotiana benthamiana [91].
- Transient Expression: CRISPR-Cas9 components were transiently co-expressed in plant leaves using a geminiviral replicon system to generate a heterogeneous, edited cell population [91].
- DNA Extraction & Amplification: Genomic DNA was extracted from infiltrated tissue seven days post-agroinfiltration. Target sites were PCR-amplified from the genomic DNA to create templates for all downstream quantification methods [91].
- Multi-Method Quantification: The same set of amplified samples was analyzed with eight different techniques:
- Targeted Amplicon Sequencing (AmpSeq): Used as the benchmark "gold standard" [91].
- Sanger Sequencing: Amplicons were Sanger sequenced and the resulting chromatograms were deconvoluted using three different algorithms: ICE, TIDE, and DECODR [91].
- Other Methods: PCR-RFLP, T7E1 assay, PCR-CE/IDAA, and ddPCR were also performed [91].
2. Key Quantitative Findings: The study revealed that the quantification method significantly impacts the measured frequency of CRISPR edits [91]. When benchmarked against AmpSeq:
- PCR-CE/IDAA and ddPCR methods were found to be highly accurate [91].
- Sanger sequencing-based tools (ICE, TIDE) showed differences in quantified editing frequency, with their sensitivity being affected by the base-calling software used, particularly for detecting low-frequency edits [91].
This demonstrates that for quantifying editing efficiencies in a mixed population—a challenge analogous to analyzing a directed evolution library—NGS-based amplicon sequencing provides a more sensitive and accurate standard than Sanger-derived methods.
Large-Scale Validation of NGS Variants
A large-scale, systematic evaluation of Sanger validation for NGS-derived variants offers critical insight into the actual necessity of orthogonal Sanger verification.
1. Experimental Protocol:
- Sample and Data Set: The study utilized data from the ClinSeq project, involving 684 participants. It compared variant calls from exome sequencing (NGS) against high-throughput Sanger sequencing data generated from the same DNA samples [90].
- Gene Selection: A subset of five genes, representative of the genome in terms of coding sequence length, exon count, and GC content, was analyzed across all 684 exomes [90].
- Variant Interrogation: All NGS-called variants within these genes were checked against the Sanger sequencing data for confirmation.
- Resequencing: NGS variants that were not confirmed by the initial Sanger data were re-sequenced using newly designed primers to rule out primer-specific artifacts [90].
2. Key Quantitative Findings:
- Validation Rate: Out of over 5,800 NGS-derived variants, only 19 were not initially validated by Sanger data. Upon re-sequencing with optimized primers, 17 of these 19 were confirmed, meaning the true false positive rate of NGS was exceptionally low [90].
- Final Accuracy: The overall validation rate of NGS variants by Sanger sequencing was 99.965% [90].
- Conclusion: The study concluded that a single round of Sanger sequencing is more likely to incorrectly refute a true positive NGS variant than to correctly identify a false positive. It found that routine Sanger validation of NGS variants has limited utility [90].
Table 2: Summary of Key Benchmarking Study Findings
| Study Focus | Benchmark Method | Tested Method(s) | Key Result |
|---|---|---|---|
| Quantifying CRISPR Edits in Heterogeneous Plant Populations [91] | Targeted Amplicon Sequencing (AmpSeq) | Sanger sequencing (deconvoluted by ICE, TIDE, DECODR) | Sanger-based tools showed variable sensitivity and were less accurate for low-frequency edits compared to AmpSeq. |
| Analytical Validation of NGS for Variant Calling [90] | High-Throughput Sanger Sequencing | Exome Sequencing (NGS) | NGS demonstrated a 99.965% validation rate, challenging the necessity of routine Sanger confirmation. |
Visualizing the Method Selection Workflow
The following workflow diagram synthesizes the experimental data and technology comparisons into a logical decision tree for selecting the appropriate validation method based on project goals.
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and materials used in the NGS and Sanger protocols cited in the benchmarking experiments, which are essential for reproducing these validation workflows.
Table 3: Key Research Reagent Solutions for Sequencing Validation
| Item | Function in Protocol | Example from Literature |
|---|---|---|
| High-Fidelity DNA Polymerase | Accurately amplifies target regions from genomic DNA or plasmid libraries for sequencing with minimal errors. | Q5 High-Fidelity DNA Polymerase [10] |
| NGS Library Prep Kit | Prepares DNA fragments for sequencing by adding platform-specific adapters and barcodes for sample multiplexing. | SureSelect (Agilent) and TruSeq (Illumina) kits [90] |
| Sanger Sequencing Kit | Provides the fluorescently labeled dideoxynucleotides and enzymes for chain-termination sequencing reactions. | BigDye Terminator v3.1 Cycle Sequencing Kit [90] |
| Hybridization Capture Probes | Biotinylated oligonucleotides that enrich for genomic regions of interest in hybrid-capture based NGS. | Solution-based biotinylated probes for target enrichment [12] |
| Emulsion Reagents (Oil & Surfactants) | Creates microreactors for ultra-high-throughput screening and directed evolution, linking genotype to phenotype. | Used in emulsion-based selection platforms for polymerase engineering [10] |
The benchmarking data clearly indicates that the choice between NGS and Sanger sequencing for validating directed evolution outcomes is not a matter of which is universally better, but which is the right tool for the specific task. Sanger sequencing remains the straightforward, cost-effective choice for obtaining the gold-standard sequence of a single, isolated variant. In contrast, NGS is the unequivocally superior method for the core challenge of directed evolution: characterizing complex mutant libraries, quantifying variant enrichment, and discovering rare clones with desired properties. The high validation rate of NGS variants (99.965%) challenges the dogma that Sanger confirmation is always necessary, suggesting that for large-scale or discovery-driven projects, NGS alone provides sufficient accuracy. A modern, efficient validation strategy employs NGS as the primary workhorse for library analysis and leverages Sanger sequencing for final confirmation of a handful of top hits, optimizing both resources and scientific insight.
Leveraging AI and Bioinformatics Tools for Enhanced Variant Interpretation
The field of genomic research has witnessed a paradigm shift with the integration of artificial intelligence (AI) and advanced bioinformatics tools into variant interpretation workflows. In 2025, these technologies have become indispensable for researchers, scientists, and drug development professionals seeking to unravel the complex genetic underpinnings of diseases and directed evolution outcomes. Variant interpretation, the process of identifying and characterizing genetic differences from sequencing data, has been transformed by AI algorithms that can detect subtle patterns with accuracy rates previously unattainable through conventional methods [92]. This technological evolution is particularly crucial for precision medicine, where accurate variant calling directly impacts diagnostic validity and therapeutic decisions.
The application of these tools extends beyond human genetics into directed evolution studies, where researchers engineer proteins with novel functions. In this context, next-generation sequencing (NGS) and AI-driven analysis form the backbone for validating selection outcomes and understanding sequence-function relationships. The global NGS data analysis market reflects this importance, projected to reach USD 4.21 billion by 2032, growing at a compound annual growth rate of 19.93% from 2024 to 2032 [93]. This growth is largely fueled by AI-based bioinformatics tools that enable faster and more accurate analysis of massive NGS datasets, revolutionizing how researchers process and interpret genetic information.
AI-Powered Variant Calling Tools: A Comparative Analysis
State-of-the-Art Tool Landscape
The current landscape of AI-powered variant calling tools encompasses a diverse range of approaches, from deep learning convolutional neural networks to machine learning-enhanced algorithms. These tools address the critical challenge of detecting genetic variants—including single nucleotide polymorphisms (SNPs), insertions/deletions (Indels), and structural variants—from high-throughput sequencing data with significantly improved accuracy over traditional statistical methods [92]. The integration of AI has been particularly transformative for detecting variants in complex genomic regions where conventional methods often struggle, enabling breakthroughs in both basic research and clinical applications.
Leading tools have emerged from both commercial and academic sectors, each with specialized capabilities optimized for different sequencing technologies and research contexts. DeepVariant, developed by Google Health, exemplifies the deep learning approach, utilizing convolutional neural networks to analyze pileup image tensors of aligned reads [92]. This tool has set new standards for accuracy, becoming a preferred choice for large-scale genomic studies such as the UK Biobank WES consortium involving 500,000 individuals [92]. Meanwhile, tools like DNAscope from Sentieon have optimized for computational efficiency, combining GATK's HaplotypeCaller with machine learning-based genotyping models to achieve high sensitivity and specificity without the computational overhead of deep learning approaches [92].
Performance Comparison and Technical Specifications
Table 1: Comparative Analysis of Leading AI-Powered Variant Calling Tools
| Tool | Primary Methodology | Supported Sequencing Technologies | Key Strengths | Computational Requirements | Best Application Context |
|---|---|---|---|---|---|
| DeepVariant [94] [92] | Deep convolutional neural networks | Short-read, PacBio HiFi, Oxford Nanopore | Industry-leading accuracy, automatic variant filtering | High; compatible with GPU and CPU | Large-scale genomic studies, population genomics |
| DeepTrio [92] | Deep CNN for family trios | Short-read and long-read technologies | Enhanced accuracy using familial context | High; extends DeepVariant framework | Family-based studies, de novo mutation detection |
| DNAscope [92] | ML-enhanced HaplotypeCaller | Short-read, PacBio HiFi, Oxford Nanopore | High efficiency, reduced computational cost | Moderate; multi-threaded CPU processing | Large-scale studies with computational constraints |
| Clair/Clair3 [92] | Deep neural networks | Short-read and long-read data | Fast performance, excellent low-coverage accuracy | Moderate to high | Rapid turnaround projects, long-read data analysis |
| Medaka [92] | Deep learning models | Oxford Nanopore long-read data | Optimized for Nanopore data, lightweight | Moderate | Real-time Nanopore sequencing analysis |
| HELLO [92] | AI-based variant calling | Multiple platforms | Comprehensive variant detection | Varies by implementation | Research requiring multi-platform support |
Table 2: Performance Metrics Across Variant Types and Coverages
| Tool | SNP Detection Accuracy | Indel Detection Accuracy | Low Coverage Performance | Complex Region Performance | Concordance with Orthogonal Methods |
|---|---|---|---|---|---|
| DeepVariant [92] | Very High (>99%) | High (>98%) | Moderate to High | Excellent | High (validated in large consortia) |
| DeepTrio [92] | Very High (>99%) | High (>98%) | High (leverages family data) | Excellent | High for trio-based designs |
| DNAscope [92] | High (>98%) | High (>97%) | Moderate to High | Good to Excellent | High (benchmarked against GATK) |
| Clair3 [92] | High (>98%) | High (>97%) | Excellent (specialized) | Good | Moderate to High |
| Medaka [92] | High for Nanopore | Moderate for Nanopore | Moderate | Good for Nanopore | Platform-specific validation |
| QIAGEN CLC [94] | Moderate to High | Moderate to High | Moderate | Good | Moderate (varies by dataset) |
The performance characteristics of these tools reveal important trade-offs between accuracy, computational requirements, and specialization. Deep learning-based tools like DeepVariant and Clair3 demonstrate superior accuracy, particularly for challenging variant types and complex genomic regions, but require significant computational resources [92]. In contrast, machine learning-enhanced tools like DNAscope offer an attractive balance of performance and efficiency, making them suitable for large-scale studies where computational resources are a constraint [92].
Recent benchmarking studies highlight that AI-based variant callers consistently outperform traditional methods, with accuracy improvements of up to 30% while simultaneously reducing processing time by half in some applications [93]. This enhanced performance is particularly evident in the detection of rare variants and in regions with high sequence complexity, where traditional statistical approaches have historically struggled. The implementation of specialized tools for specific contexts—such as DeepTrio for family-based studies—further extends the utility of AI in genomic analysis by leveraging biological relationships to improve variant detection [92].
Experimental Protocols for Variant Validation
Analytical Validation Framework for NGS Assays
Robust validation of variant calling performance requires carefully designed experimental protocols that assess accuracy, sensitivity, specificity, and reproducibility. The international multicenter study on the Hedera Profiling 2 circulating tumor DNA test panel (HP2) provides a comprehensive framework for analytical validation that can be adapted for directed evolution studies [95]. This protocol employs reference standards with known variant profiles to establish baseline performance metrics and includes clinical samples pre-characterized by orthogonal methods to assess real-world concordance.
The HP2 validation followed a rigorous methodology: "The analytical performance was assessed using reference standards and a diverse cohort of 137 clinical samples precharacterized by orthogonal methods. In reference standards with variants spiked in at 0.5% allele frequency, sensitivity and specificity were 96.92% and 99.67%, respectively, for SNVs/Indels and 100% for fusions" [95]. This approach demonstrates the importance of establishing performance benchmarks using standardized materials before proceeding to complex biological samples. For directed evolution applications, similar strategies can be implemented using spiked-in controls with known mutations at varying allele frequencies to establish detection thresholds relevant to the specific experimental context.
Functional Validation of Variant Pathogenicity
Beyond analytical validation, functional assessment of variant impact is crucial for interpreting biological significance. The colorectal cancer study utilizing BoostDM artificial intelligence method exemplifies an integrated approach to functional validation [34]. Their protocol combined computational pathogenicity prediction with experimental functional assays to validate the biological impact of identified variants.
The methodology included: "Using the BoostDM artificial intelligence method, we were able to identify oncodriver germline variants with potential implications for disease progression. We assessed the model's accuracy in predicting germline variants by comparing its results with the AlphaMissense pathogenicity prediction model. Additionally, a minigene assay was employed for the functional validation of intronic mutations" [34]. This multi-layered approach—combining AI-based computational prediction with experimental validation—provides a robust framework for confirming variant impact. For directed evolution studies, similar strategies can be employed, with enzyme activity assays or other functional measurements replacing the minigene assay used for human variants.
Variant Validation Workflow
Integration with Directed Evolution and NGS Coverage Analysis
Coverage Requirements for Directed Evolution Studies
The integration of AI-powered variant interpretation with directed evolution outcomes requires careful consideration of sequencing coverage parameters. Unlike standard genomic applications, directed evolution experiments have unique coverage requirements that balance comprehensive variant detection with practical sequencing costs. Research on sequencing coverage requirements in directed evolution experiments has established that "cost-effective, precise, and accurate identification of active variants is possible even at low coverages" [10], though the specific threshold must be determined by the experimental context and desired confidence level.
The analysis of coverage requirements involves identifying "the sequencing coverage threshold for the accurate and precise identification of significantly enriched mutants" [10]. This approach recognizes that while genome assembly and other omics approaches often require high coverage, directed evolution experiments focusing on significantly enriched variants may achieve reliable results with more modest coverage. This principle has important implications for experimental design, allowing researchers to optimize resource allocation without compromising data quality.
AI-Enhanced Analysis of Selection Outcomes
The application of AI tools extends beyond variant calling to the interpretation of selection outcomes in directed evolution. The colorectal cancer study exemplifies this approach: "Using the BoostDM artificial intelligence method, we were able to identify oncodriver germline variants with potential implications for disease progression" [34]. Similar methodologies can be adapted for directed evolution to identify mutations that confer desired functional properties.
The integration of NGS coverage analysis with AI-driven variant interpretation creates a powerful framework for validating directed evolution outcomes. This approach enables researchers to not only identify enriched variants but also to distinguish functionally significant mutations from neutral background variation. The comparative performance between BoostDM and AlphaMissense reported in the colorectal cancer study—with AUC values of 0.788 for the entire dataset and 0.803 for panel genes—demonstrates the predictive power that can be achieved through AI-enhanced analysis [34].
Directed Evolution Analysis
Research Reagent Solutions for Variant Interpretation
Table 3: Essential Research Reagents and Platforms for AI-Enhanced Variant Interpretation
| Reagent/Platform | Manufacturer/Provider | Primary Function | Application in Variant Interpretation |
|---|---|---|---|
| DNBSEQ-T1+ | MGI Tech [96] | Mid-throughput sequencing | Genome sequencing with Q40 accuracy in 24-hour workflow |
| UG 100 Solaris | Ultima Genomics [96] | High-throughput sequencing | Low-cost sequencing ($80/genome) for large-scale studies |
| Magnis NGS Prep System | Agilent Technologies [96] | Automated library preparation | Standardized, reproducible library construction |
| Quick-DNA 96 Plus Kit | Zymo Research [34] | High-throughput DNA extraction | Efficient nucleic acid isolation from multiple samples |
| Q5 High-Fidelity DNA Polymerase | New England Biolabs [10] | Error-resistant PCR | Amplification for library construction with minimal errors |
| HP2 Circulating Tumor DNA Panel | Hedera Dx [95] | Targeted sequencing | Simultaneous detection of SNVs, Indels, fusions, CNVs, MSI |
| QCI Interpret Platform | QIAGEN [97] | Clinical decision support | Variant filtering, classification, and interpretation |
| Medaka | Oxford Nanopore [92] | Variant calling | Real-time analysis of Nanopore sequencing data |
The selection of appropriate research reagents and platforms is critical for successful implementation of AI-enhanced variant interpretation workflows. The tools and reagents listed in Table 3 represent essential components for constructing robust pipelines from sample preparation through final interpretation. Recent advancements in sequencing technology, such as the DNBSEQ-T1+ from MGI Tech which "completes a paired end 150 sequencing workflow in 24 hours with Q40 accuracy" [96], and the UG 100 Solaris from Ultima Genomics which enables the "$80 genome" [96], have dramatically improved the accessibility of high-quality sequencing data for variant interpretation.
Integrated platforms like the QCI Interpret from QIAGEN provide comprehensive solutions that "enable clinical labs to efficiently classify, annotate, interpret and report genomic variants with confidence" [97]. The 2025 release of this platform includes enhanced features such as "REVEL and SpliceAI variant impact predictions" and "Draft ACMG v4 & VICC points-based scoring guidance" [97], representing the cutting edge in variant interpretation tools. For directed evolution studies, these platforms can be adapted to classify mutations based on functional impact rather than clinical pathogenicity, demonstrating the flexibility of these tools across research contexts.
The integration of AI and bioinformatics tools for variant interpretation represents a fundamental advancement in our ability to extract meaningful insights from genomic data. As these technologies continue to evolve, several trends are shaping their future development. The rise of specialized language models for genomic analysis represents a particularly promising direction, with potential to further transform variant interpretation. As noted by Aber Whitcomb, CEO of Salt AI, "Large language models could potentially translate nucleic acid sequences to language, thereby unlocking new opportunities to analyze DNA, RNA and downstream amino acid sequences" [93]. This approach treats genetic code as a language to be decoded, potentially identifying patterns and relationships that humans might miss.
The expanding accessibility of genomic analysis tools is another significant trend, with cloud-based platforms connecting over 800 institutions globally and making advanced genomics accessible to smaller labs [93]. This democratization of technology is accompanied by improved security protocols to protect sensitive genetic data through end-to-end encryption and strict access controls [93]. These developments ensure that the benefits of AI-enhanced variant interpretation can be realized across diverse research contexts while maintaining appropriate data protection.
In conclusion, the synergy between AI-powered bioinformatics tools and advanced sequencing technologies has created unprecedented opportunities for enhanced variant interpretation in both clinical and research settings. For scientists focused on validating directed evolution outcomes, these tools provide robust frameworks for connecting genetic changes to functional consequences. As the field continues to evolve, the integration of increasingly sophisticated AI algorithms with comprehensive experimental validation will further accelerate our ability to interpret genetic variation and harness this knowledge for scientific and therapeutic advancement.
Conclusion
The integration of robust NGS coverage analysis is non-negotiable for validating directed evolution outcomes. This synthesis of foundational knowledge, methodological rigor, troubleshooting acumen, and statistical validation creates a reliable pipeline that moves beyond simple variant identification to a true understanding of sequence-function relationships. As demonstrated in the engineering of enzymes like XNA polymerases, Cas12a, and degron systems, establishing a defined sequencing coverage threshold is critical for accurately identifying significantly enriched mutants while managing resources efficiently. Future directions point toward the increasing integration of AI with adaptive NGS sampling, the use of long-read technologies to resolve complex structural variations, and the application of these refined validation frameworks to accelerate the development of novel enzymes and therapeutics for biomedical and clinical research. This systematic approach ensures that directed evolution continues to be a predictable and powerful engine for innovation.
References
- 1. Directed evolution [https://en.wikipedia.org/wiki/Directed_evolution]
- 2. Exploring protein fitness landscapes by directed evolution [https://pmc.ncbi.nlm.nih.gov/articles/PMC2997618/]
- 3. Navigating directed evolution efficiently: optimizing selection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11493728/]
- 4. Evaluation of machine learning-assisted directed evolution ... [https://www.sciencedirect.com/science/article/abs/pii/S2405471225002200]
- 5. Revolutionizing therapeutic protein design with synthetic ... [https://www.news-medical.net/news/20250808/Revolutionizing-therapeutic-protein-design-with-synthetic-biology.aspx]
- 6. A primer to directed evolution: current methodologies and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10074555/]
- 7. Technical Validation of a Next-Generation Sequencing ... [https://www.sciencedirect.com/science/article/pii/S1525157816000490]
- 8. Directed Evolution Technology for Engineering Enzymes in ... [https://sequencing.roche.com/us/en/article-listing/directed-evolution.html]
- 9. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 10. Navigating directed evolution efficiently: optimizing ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2024.1439259/full]
- 11. Next-generation sequencing-based population genetics ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-025-06364-6]
- 12. Guidelines for Validation of Next-Generation Sequencing– ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6941185/]
- 13. Next-Generation Sequencing Validation [https://www.thermofisher.com/us/en/home/life-science/sequencing/sanger-sequencing/applications/next-generation-sequencing-validation.html]
- 14. Comparative genomics approach to evolutionary process ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7359831/]
- 15. Evolution and applications of Next Generation Sequencing ... [https://www.sciencedirect.com/science/article/pii/S2772391724000082]
- 16. Sequencing Depth vs. Coverage: Key Metrics in NGS ... [https://3billion.io/blog/sequencing-depth-vs-coverage]
- 17. Sequencing Coverage for NGS Experiments [https://www.illumina.com/science/technology/next-generation-sequencing/plan-experiments/coverage.html]
- 18. Sequencing depth and coverage: key considerations in ... [https://www.nature.com/articles/nrg3642]
- 19. Sequencing 101: Sequencing coverage [https://www.pacb.com/blog/sequencing-101-sequencing-coverage/]
- 20. Comparison of next‑generation sequencing quality metrics ... [https://pubmed.ncbi.nlm.nih.gov/39991725/]
- 21. Determination of high-confidence germline genetic variants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12326712/]
- 22. ACMG clinical laboratory standards for next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4098820/]
- 23. NGS coverage accurately predicts MET and HER2 (ERBB2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12340238/]
- 24. Directed evolution for high functional production and stability of a challenging G protein-coupled receptor - PubMed [https://pubmed.ncbi.nlm.nih.gov/33883583/]
- 25. Navigating directed evolution efficiently: optimizing selection conditions and selection output analysis - PubMed [https://pubmed.ncbi.nlm.nih.gov/39439528/]
- 26. NGS Library Preparation [https://www.cd-genomics.com/resource-library-preparation-for-ngs.html]
- 27. Performance Evaluation of a Commercial Automated ... [https://www.sciencedirect.com/science/article/pii/S1525157824001193]
- 28. NGS Library Preparation Kits for Illumina Systems [https://www.thermofisher.com/us/en/home/life-science/sequencing/next-generation-sequencing/ngs-library-preparation-illumina-systems.html]
- 29. Performance comparison of four exome capture platforms on ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12104-9]
- 30. Long-read sequencing vs short-read sequencing [https://frontlinegenomics.com/long-read-sequencing-vs-short-read-sequencing/]
- 31. Comparison of short-read and long-read metagenome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12634412/]
- 32. Sequencing depth vs. sequencing batching [https://www.ogt.com/us/resources/ngs-resources-and-support/ngs-resources/sequencing-depth-vs-sequencing-batching/]
- 33. An optimized variant prioritization process for rare disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12539062/]
- 34. Integrating next-generation sequencing and artificial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12127813/]
- 35. Next-generation sequencing: 2025 Revolution [https://lifebit.ai/blog/next-generation-sequencing/]
- 36. Developing efficient and cost-effective NGS sequencing ... [https://www.sciencedirect.com/science/article/abs/pii/S0044848625013171]
- 37. Optimized high-throughput whole-genome sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12465963/]
- 38. NGS Data Analysis for Illumina Platform—Overview and ... [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/next-generation-sequencing/ngs-data-analysis-illumina.html]
- 39. Top NGS Data Analysis Tools of 2025 [https://www.dromicsedu.com/blog-details/revolutionizing-genomics-top-ngs-data-analysis-tools-of-2025]
- 40. NGS Workflow Steps | IDT [https://eu.idtdna.com/pages/technology/next-generation-sequencing/workflow]
- 41. Top 10 Bioinformatics Tools in 2025: Features, Pros, Cons ... [https://www.devopsschool.com/blog/top-10-bioinformatics-tools-in-2025-features-pros-cons-comparison/]
- 42. Recent Advances in Genome Editing and Bioinformatics [https://pmc.ncbi.nlm.nih.gov/articles/PMC11989416/]
- 43. Bioinformatics and Computational Tools for Next- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7019349/]
- 44. NGS Workflow Steps - Next-Generation Sequencing [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/ngs-workflow.html]
- 45. Functional Comparison of Laboratory-Evolved XNA ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/34029459/]
- 46. Mutant polymerases capable of 2′ fluoro-modified nucleic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9347352/]
- 47. Directed evolution expands CRISPR–Cas12a genome- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12266133/]
- 48. Nonenzymatic polymerase-like template-directed synthesis ... [https://www.nature.com/articles/s41467-021-21128-0]
- 49. Analytical Validation of the Next-Generation Sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5397672/]
- 50. Highly parallel profiling of the activities and specificities ... [https://www.nature.com/articles/s41467-025-57150-9]
- 51. Directed evolution expands CRISPR-Cas12a genome ... [https://pubmed.ncbi.nlm.nih.gov/40196639/]
- 52. Engineered FnCas12a with enhanced activity through ... [https://www.sciencedirect.com/science/article/pii/S0021925821001666]
- 53. Comparison of CRISPR/Cas9 and Cas12a for gene editing ... [https://www.sciencedirect.com/science/article/pii/S2211926424004089]
- 54. A chimeric viral platform for directed evolution in ... [https://www.nature.com/articles/s41467-025-59438-2]
- 55. Significance and limitations of the use of next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9478565/]
- 56. High-frequency, low-coverage “false positives” mutations ... [https://www.nature.com/articles/s41598-017-13116-6]
- 57. ParaRef: a decontaminated reference database for parasite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12548146/]
- 58. Using experimental evolution to probe molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4815336/]
- 59. High-throughput screening, next generation sequencing and ... [https://pubs.rsc.org/en/content/articlehtml/2022/cc/d1cc04635g]
- 60. Development of a multiplex forensic identity panel for massively parallel... [https://pubmed.ncbi.nlm.nih.gov/30529891/]
- 61. Summarizing and correcting the GC content bias in high- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3378858/]
- 62. Reducing GC & PCR Bias in Whole-Genome Sequencing [https://www.revvity.com/blog/understanding-impact-gc-and-pcr-biases-whole-genome-sequencing]
- 63. Library Preparation for Next-Generation Sequencing [https://sequencing.roche.com/global/en/article-listing/library-preparation-for-next-generation-sequencing-dealing-with-pcr-bias.html]
- 64. Prospective, multicenter validation of a platform for rapid ... [https://www.nature.com/articles/s41591-025-03562-5]
- 65. Low-coverage sequencing cost-effectively detects known and ... [https://pubmed.ncbi.nlm.nih.gov/33770507/]
- 66. Towards a Cost-Effective Implementation of Genomic ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.728764/full]
- 67. Cost-effective genomic prediction of critical economic traits ... [https://www.sciencedirect.com/science/article/pii/S0888754324000958]
- 68. Benchmarking of low coverage sequencing workflows for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12379343/]
- 69. A cost-effective sequencing method for genetic studies ... [https://www.nature.com/articles/s41525-024-00390-3]
- 70. Making the Most of Your NGS Data: Understanding Metrics ... [https://sequencing.roche.com/us/en/article-listing/metrics-for-target-enriched-ngs.html]
- 71. Diversity in Genomic Studies: A Roadmap to Address the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7614889/]
- 72. NGS Data Analysis: Best Practices and Common Pitfalls [https://dromicsedu.com/blog-details/ngs-data-analysis-best-practices-and-common-pitfalls]
- 73. Calculating the statistical significance of rare variants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6001062/]
- 74. Statistical methods for assessing the effects of de novo ... [https://humgenomics.biomedcentral.com/articles/10.1186/s40246-024-00590-z]
- 75. Integrated Enrichment Analysis of Variants and Pathways in ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1003770]
- 76. How-to: NGS Quality Control [https://frontlinegenomics.com/how-to-ngs-quality-control/]
- 77. High-throughput screening, next generation sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8851469/]
- 78. Target Enrichment Approaches for Next-Generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9318977/]
- 79. Single-cell ultra-high-throughput multiplexed chromatin ... [https://www.nature.com/articles/s41592-025-02700-8]
- 80. Rapid NGS Analysis on Google Cloud Platform [https://pmc.ncbi.nlm.nih.gov/articles/PMC12627752/]
- 81. The Power of End-to-End Omics Solutions for Drug ... [https://www.lexogen.com/blog/the-power-of-end-to-end-omics-solutions-for-drug-discovery-and-development/]
- 82. Directed evolution of a beta‐lactamase samples a wide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12514840/]
- 83. Directed evolution of a beta-lactamase samples a wide ... [https://pubmed.ncbi.nlm.nih.gov/41074874/]
- 84. Directed evolution methods for overcoming trade-offs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7384606/]
- 85. Next-Generation Sequencing for Typing and Detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4097710/]
- 86. Insights into an evolutionary strategy leading to antibiotic ... [https://www.nature.com/articles/srep40357]
- 87. Evolutionary Trajectories of Beta-Lactamase CTX-M-1 Cluster ... [https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1000735]
- 88. NGS vs Sanger Sequencing [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/advantages/ngs-vs-sanger.html]
- 89. Next-Generation Sequencing (NGS) vs. Sanger ... [https://www.labmanager.com/next-generation-sequencing-ngs-vs-sanger-sequencing-which-dna-sequencing-method-is-right-for-you-33659]
- 90. Systematic Evaluation of Sanger Validation of NextGen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4878677/]
- 91. Comprehensive benchmarking of genome editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12144422/]
- 92. Artificial intelligence in variant calling: a review [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1574359/full]
- 93. The 2025 Bioinformatics Toolkit: From Sample Prep to AI- ... [https://blog.omni-inc.com/blog/the-2025-bioinformatics-toolkit-from-sample-prep-to-ai-driven-analysis]
- 94. Top 10 AI Genetic Analysis Tools in 2025: Features, Pros ... [https://www.scmgalaxy.com/tutorials/top-10-ai-genetic-analysis-tools-in-2025-features-pros-cons-comparison/]
- 95. Analytical Validation of a Pan-Cancer NGS Assay for In- ... [https://www.sciencedirect.com/science/article/pii/S152515782500220X]
- 96. The Best of NGS: Instrument Companies to Watch in 2025 [https://www.genengnews.com/topics/omics/the-best-of-ngs-instrument-companies-to-watch-in-2025/]
- 97. New QCI Interpret 2025 Release enhances variant filtering, ... [https://digitalinsights.qiagen.com/news/blog/clinical/qci-interpret-2025-release/]